
INTRODUCTION
Triple-negative breast cancer (TNBC), defined by lack of expression of estrogen receptor, progesterone receptor, and HER2 gene amplification, accounts for approximately 15% of breast cancers [1]. TNBC is a heterogeneous group of tumors with more aggressive clinical features [2]. The Cancer Genome Atlas (TCGA) analysis demonstrated that frequent loss-of-function and gain-of-function alterations in TNBC involving genes associated with the DNA damage repair and phosphatidylinositol 3-kinase (PI3K) pathways [3]. Homologous recombination repair (HRR) is a predominantly error-free DNA double-strand break repair mechanism [4]. Key components of the HRR pathway include the tumor-suppressor proteins BRCA1 and BRCA2 [4]. DNA damage repair pathways are attractive therapeutic targets in TNBC with germline or somatic HRR dysfunction [1, 2]. We previously reported experience with the polyADP ribose polymerase (PARP) inhibitor (PARPi) olaparib for women with germline BRCA mutation [5]. We now examine the ability to extend those findings to women with TNBC who do not have intrinsic HRR dysfunction.
Approximately 15% of TNBC harbor a germline mutation in BRCA1 or BRCA2; up to 80% of BRCA1 mutation-associated and 35% of BRCA2 mutation-associated breast cancer has the TNBC phenotype [6, 7]. TNBC may also have HRR deficiency based on other molecular alterations. Recent data suggest approximately 10% of young or familial TNBC patients with no BRCA1 or BRCA2 mutation carry inherited deleterious mutations in other breast cancer predisposition genes, particularly in genes involved in HRR such as PALB2, BARD1, RAD51C, RAD51D, and BRIP1 [8-10]. In addition, BRCA1 promoter hypermethylation has been identified in one-third of 377 TNBCs [11]. Deficient HRR leads to activation of alternate DNA repair pathways including the base excision repair and non-homologous end-joining pathways, that require PARP. Increased PARP-1 expression and/or activity in tumor cells have been demonstrated in TNBC [12, 13]. HRR dysfunction sensitizes cells to PARPi that lead to further chromosomal instability, cell cycle arrest, and apoptosis [14, 15]. The PARPi class has shown clinical potential in TNBC [5, 16-18], with a response rate (RR) of 54% in patients with advanced TNBC with germline BRCA mutations [18].
Subsets of sporadic TNBC, particularly the basal-like phenotype, share pathologic and molecular features, such as p53 mutation, genomic instability, and sensitivity to DNA cross-linking agents cancers, with germline BRCA mutation-associated breast cancers [1]. Platinum agents, such as carboplatin or cisplatin, have clinical activity in TNBC patients with germline BRCA mutation [19-21]. Increased in vivo activity was reported with the combination of olaparib and cisplatin compared to single agent alone in a BRCA1-deficient breast cancer mouse model [22]. We previously reported the combination of olaparib and carboplatin tolerable and active in breast and ovarian cancer patients with germline BRCA mutation, in which four of four TNBC patients had either complete or partial responses [5]. However, there are limited data on the activity of PARPi in combination with chemotherapy in patients with metastatic sporadic TNBC. We hypothesized that the addition of olaparib to carboplatin could be safely given and would yield clinical benefit in subsets of sporadic TNBC. Our translational aim was to examine potential biomarkers of response to the PARPi and carboplatin combination.
RESULTS
Patients
Patient accrual is shown in the Consort diagram (N = 28; Supplementary Figure 1). Patient characteristics are detailed in Table 1A. Twenty patients had previous negative commercial germline BRCA mutational analysis and eight had a BRCAPro score < 10% calculated on the basis of ascertained detailed family history.
Dose optimization
Patients received olaparib on days 1-7 and carboplatin on day 1 or 2 of a 21-day cycle (Table 1B). One of 6 patients at AUC4 had dose-limiting toxicity (DLT) of grade 4 thrombocytopenia requiring platelet transfusion during the 2-cycle observation period. Increase to carboplatin AUC5 resulted in DLT of grade 4 thrombocytopenia lasting > 7 days in 2 of 2 treated patients, and grade 3 symptomatic hyponatremia with cognitive dysfunction in 1 of 2 patients. The recommended phase 2 dose is olaparib 400mg every 12 hours days 1-7/21 with carboplatin AUC4 day 1.
Table 1A: Patient characteristics (N = 28)
* All except one patient had been exposed to anthracyclines and/or taxanes-containing regimens, 21/28 (75%) patients were treated with > 3 prior therapy.
** Three patients had previously received platinum-based therapy upon development of recurrent disease; two patients who were not previously platinum sensitive (prior exposure 8 and 12 months prior to study enrollment) developed progressive disease after two cycles of combination olaparib and carboplatin therapy.
Table 1B: Dose levels and clinical response
Dose Level [DL] |
Schedule and Dose |
DLT |
Best response*** (Duration of response) |
|
Olaparib oral capsule, bid |
Carboplatin IV q 3 wk |
|||
DL 1 (N= 4)* |
400mg, days 1-7 |
AUC3, day 1 or 2 |
1 CR (69+mo), 1 PR (5mo), 1 SD (4mo), 1 PD (1.5mo) * |
|
DL 2 (N= 9)** |
400mg, days 1-7 |
AUC4, day 1 or 2 |
1/6 treated |
5 SD (median 3mo), 4 PD (median 1.5mo) |
DL 3 (N= 2) |
400mg, days 1-7 |
AUC5, day 1 or 2 |
2/2 treated |
1 NE (off due to toxicity), 1 PD (1.5mo) |
Expansion cohort (N= 13) |
400 mg, days 1-7 |
AUC4, day 1 or 2 |
4 PR (median 4mo), 6 SD (median 3.5mo), 3 PD (median 1.5mo) |
|
* One patient in DL 1 was replaced because she missed half of the olaparib doses during cycle 1.
**First three patients in DL 2 were replaced due to rapid disease progression including new brain metastases and new chest wall diseases within two cycles. One DLT was observed among six patients on DL 2 and two patients on DL 3.
*** Overall response rate without an exceptional responder: 19% (5 of 27 patients), Disease control rate (CR+PR+SD > 4 months): 4% (12 of 27 patients)
Abbreviations: bid: twice daily, wk: week, mo: months, CR: complete response, PR partial response, SD: stable disease, PD: progressive disease, NE: non-evaluable
Adverse events
All patients had at least one treatment-emergent adverse event, summarized in Table 2A. Common ( > 10% of patients) non-hematologic events included nausea, fatigue, headache, gastroesophageal reflux, and skin rash. These were mainly mild to moderate in severity, self-limiting, and manageable with standard treatments. Hematologic toxicity was the most common adverse event (Table 2B). Grade 3 and 4 neutropenia was observed in 10 of 28 (36%) but no episodes of febrile neutropenia were observed. 15 of 28 (54%) patients had treatment delays due to hematologic toxicities, and received pegfilgrastim or filgrastim to prevent recurrent delays, starting at cycle 2 (2 patients; non-dose limiting toxicity cohort), cycle 3 (3 patients), cycle 4 (2 patients), cycle 5 (4 patients), cycle 6 (2 patients) and cycle 7 (2 patients). It was continued during all subsequent combination treatment cycles. Grade 3 and 4 anemia was seen in 3 of 28 patients (11%). Six patients (21%) received red blood cell transfusions and 3 (11%) received darbepoetin starting with cycle 3, 4 or 8, respectively. Grade 3 and 4 thrombocytopenia occurred in 3 of 28 patients (11%). Two patients required carboplatin dose reduction due to grade 4 thrombocytopenia lasting more than one week. Four patients (14%) discontinued carboplatin before the planned eight cycles because of myelosuppression. One patient in dose level 3 had both grade 4 thrombocytopenia and grade 3 cognitive dysfunction related to grade 3 hyponatremia; she withdrew from study after one cycle of treatment. MRI was negative for brain metastases, and the electrolyte imbalance and somnolence improved with hydration, pain medication adjustment, and study drug discontinuation. This event was considered possibly related to the study treatment.
Clinical activity
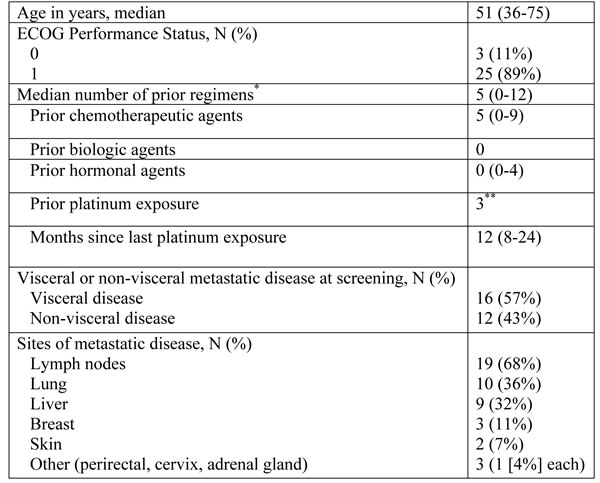
The clinical activity results are summarized in Table 1B and are shown in Figures 1A and 1B. 27 patients were evaluable for response determination. Four patients discontinued treatment during cycle 1 or 2, due to drug toxicity (1, not evaluable for response), disease progression with development of brain metastases (2 patients) or of new chest wall lesions (1 patient). The latter three were counted as disease progression. One patient on the dose escalation cohort achieved a durable complete response (CR; 69+ months), 5 had partial response (PR; median 4 months, range 4-7 months) for an objective RR (ORR) of 22% (6/27). Seven patients had stable disease (SD; median 5.5 months, range 4-9 months) yielding a disease control rate of 48% (13/27). Three patients received a carboplatin regimen prior to study entry. The patient with a 24-month carboplatin-free window achieved SD for 9 months. The two patients who had a prior carboplatin regimen, had disease progression at first imaging reassessment.
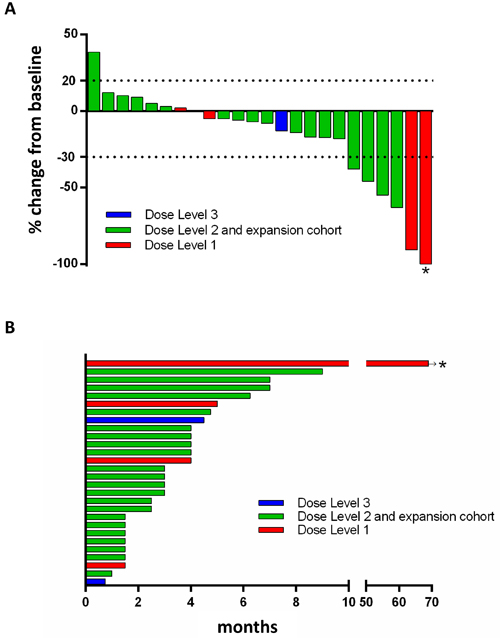
Figure 1: Waterfall plot (A) and duration on the study (B). A. Twenty four patients with baseline and subseqent imaging reassessment are shown. Best RECIST response is graphed for each patient. B. All 28 patients are shown in a swmmier plot. Four patients became off-treatment due to rapid clinical progression (n = 3) and toxicity (n = 1) prior to first reassessement scans. Color code defines dose level of treatment with arbitrary patient number assignment. * represents the exceptional responder with ongoing complete response. Dose level 1: olaparib 400mg bid days1-7 with carboplatin AUC3; dose level 2: olaparib 400mg bid days1-7 with carboplatin AUC4; dose level 3: olaparib 400mg bid days1-7 with carboplatin AUC5.
Translational studies
PAR levels and PARP1/XRCC1 polymorphisms
A significant decrease in the mean value of PAR concentrations was found 24 hours post-olaparib compared with their respective baselines ( 12.68 pg/mL [7.8-35.3 pg/mL] vs. 90.43 pg/mL [32.5-172.7 pg/mL], p = 0.002). This expected finding demonstates that olaparib achieved pharmacologically effective concentrations. Decrease in PAR incorporation by greater than 50% after one cycle of olaparib/carboplatin was observed in 9 patients and did not correlate with response or PFS.
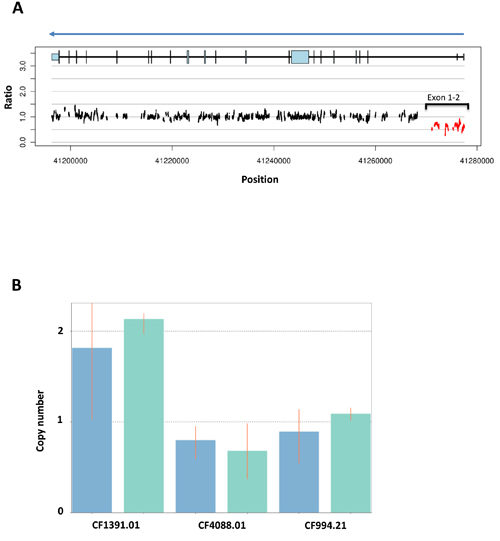
Figure 2: BROCA-HR deep sequencing result of the exceptional responder (A) and validation with qPCR for deletion of BRCA1 exons 1 and 2 (B). This patient was initially diagnosed with stage I TNBC (T1cNoMo) in 2007 at age 46, treated with lumpectomy, adjuvant radiation followed by 4 cycles of doxorubicin and cyclophosphamide at the local hospital. She was treated for recurrence in 2008 with 2 cycles of docetaxel and capecitabine, followed by 3 cycles of paclitaxel and gemcitabine, after which she underwent surgical excision of a remaining right parasternal mass. This was consolidated with external beam radiation. This was followed by supraclavicular and mediastinal nodal progression in Feb 2010, leading to enrollment in the present study dose escalation cohort. The patient had no known family history of breast, ovarian or prostate cancer. A. BROCA-HR readout for BRCA1 copy number variations (CNVs) based on read depth and split read alignment demonstrates reduced copy number at BRCA1 exons 1 and 2 for our exceptional responder. B. Taqman Copy Number Assay (Applied Biosystems, Carlsbad, CA) with CopyCaller Software v2.0 was used to confirm CNV within DNA obtained from PBMCs of the exceptional responder (CF4088.01). Copy number analysis with two different BRCA1 exon probes; blue columns represent a probe within BRCA1 exon 1 (BRCA1 within exon 1 Chr.17:41277232) and green columns represent a probe within an intergenic region between exons 1 and 2 (BRCA1 intron 1 Chr.17:41276450). CF1391.01 (left), a negative control without BRCA1 mutation, demonstrates the normal two copies, while the exceptional responder (CF4088.01, middle) and a positive control for a deletion of exons 1 and 2 (CF994.21, right) lack one copy, which is consistent for the two probes.
Reverse phase protein array (RPPA)
Protein expression or their post-translational modifications were assessed by RPPA [23]. The relationship between clinical response and pretreatment biopsy lysate expression of 218 proteins or posttranslational modifications of proteins was examined [23]. We explored protein quantity changes between patients who achieved a clinical benefit of ≥ 4 months vs. those who did not, and between patients with objective response (CR or PR) vs. no objective response (SD or progression). A false discovery rate (FDR) of 5% was used to control for multiple comparisons, yielding four proteins differentially expressed as a function of clinical benefit (Supplementary Table 1A). Additionally, we compared changes in protein quantity between the pre-cycle 2 biopsy and baseline, removing any changing ≤ 10%. No difference was found as a function of objective response. However, significant differences associated with clinical benefit were found for five proteins (FDR = 5%; Supplementary Table 1B). High starting quantities of cyclin D1, collagen VI and YAP-1 were identified in patients who did not derive clinical benefit as defined above. Increases in the three proteins over time were associated with clinical benefit.
Apoptosis
The mean apoptotic index (AI) at baseline was 52.1% (39.9- 80.6%) and increased to 60.3% (51.7- 74.6%) post-cycle 1 (p = 0.09). The AI fold or % change per patient did not correlate with response or PFS.
Exceptional responder BROCA-HR testing
Germline HRR gene mutational analysis was done for the exceptional responder [24]. She had negative comprehensive commercial BRCA testing prior to enrollement on study (Myriad Genetic Laboratories; Salt Lake City, Utah) in 2008. A comprehensive test for gene rearrangements was not performed at that time as she did not meet Myriad-defined criteria [25]. BROCA-HR analysis identified one wild type germline BRCA1 allele and one BRCA1 allele containing a deletion of exons 1-2 (Figure 2A), which was confirmed by a TaqMan copy number assay (Figure 2B). This deletion resulted in a stop codon in the mRNA, encoding a truncated BRCA1 protein, defining this as a deleterious germline mutation in BRCA1.
Table 2A:Drug-related adverse events by maximum grade per patient (N = 28)
Adverse Event |
Grade 1 |
Grade 2 |
Grade 3 |
Grade 4 |
Grade 3/4 (%) |
Hematology |
|
|
|
|
|
Lymphocytopenia |
10 (36%) |
3 (11%) |
6 (21%) |
0 |
21% |
White Blood Count |
8 (29%) |
7 (25%) |
7 (25%) |
1 (4%) |
29% |
Neutropenia |
1 (4%) |
7 (25%) |
8 (29%) |
2 (7%) |
36% |
Thrombocytopenia |
14 (50%) |
3 (11%) |
0 |
3 (11%) |
11% |
Anemia |
9 (32%) |
10 (36%) |
3 (11%) |
0 |
11% |
Gastrointestinal disorders |
|
|
|
|
|
Nausea |
11 (39%) |
1 (4%) |
0 |
0 |
0% |
Vomiting |
3 (11%) |
1 (4%) |
0 |
0 |
0% |
Gastroesophageal reflux disease |
4 (14%) |
0 |
0 |
0 |
0% |
Constipation |
2 (7%) |
0 |
0 |
0 |
0% |
Diarrhea |
2 (7%) |
0 |
0 |
0 |
0% |
Chemistry |
|
|
|
|
|
Hyponatremia |
3 (11%) |
0 |
1 (4%) |
0 |
4% |
Hypomagnesemia |
1 (4%) |
0 |
0 |
0 |
0% |
Increased AST |
4 (14%) |
1 (4%) |
1 (4%) |
0 |
4% |
Increased ALT |
4 (14%) |
0 |
0 |
0 |
0% |
Other |
|
|
|
|
|
Fatigue |
8 (29%) |
5 (18%) |
0 |
0 |
0% |
Carboplatin allergic reaction |
0 |
0 |
0 |
0 |
0% |
Skin rash |
3 (11%) |
1 (4%) |
0 |
0 |
0% |
Weight Loss |
1 (4%) |
0 |
0 |
0 |
0% |
Headache |
5 (18%) |
0 |
0 |
0 |
0% |
Table 2B: Drug-related hematologic adverse events by dose level (N=28)
Adverse Event |
Grade 1 |
Grade 2 |
Grade 3 |
Grade 4 |
||||||||
DL1 |
DL2 |
DL3 |
DL1 |
DL2 |
DL3 |
DL1 |
DL2 |
DL3 |
DL1 |
DL2 |
DL3 |
|
Lympho-cytopenia |
1 (25%) |
9 (41%) |
0 |
1 (25%) |
2 (9%) |
0 |
1 (25%) |
3 (14%) |
2 (100%) |
0 |
0 |
0 |
Leukopenia |
2 (50%) |
6 (27%) |
0 |
1 (25%) |
6 (27%) |
0 |
1 (25%) |
4 (18%) |
2 (100%) |
0 |
1 (5%) |
0 |
Neutropenia |
1 (25%) |
0 |
0 |
1 (25%) |
6 (27%) |
0 |
0 |
6 (27%) |
2 (100%) |
0 |
2 (9%) |
0 |
Thrombo-cytopenia |
3 (75%) |
11 (50%) |
0 |
0 |
2 (9%) |
1 (50%) |
0 |
0 |
0 |
0 |
2 (9%) |
1 (50%) |
Anemia |
2 (50%) |
7 (32%) |
0 |
1 (25%) |
8 (36%) |
1 (50%) |
0 |
2 (9%) |
1 (50%) |
0 |
0 |
0 |
Abbreviations: DL: dose level
DL 1 (4 patients), DL 2 (22 patients) and DL 3 (2 patients)
DISCUSSION
Recurrent TNBC is not curable and constitutes a subset of breast cancer with an important unmet therapeutic need. This phase I/Ib study identified olaparib 400mg twice daily in capsule formulation for 7 days, administered with carboplatin AUC 4 every 3 weeks as tolerable with maxium supportive care, and providing modest activity in a group of heavily pretreated TNBC patients. Many patients required growth factor support on progressive cycles, despite this carboplatin dose. Our previously reported cohort of patients with BRCA mutation-associated breast or ovarian cancer identified the same olaparib dosage with an AUC5 of carboplatin as the MTD [5]. Prior treatment exposures were similar, although all BRCA mutated ovarian cancer patients had prior carboplatin. This difference in the tolerated carboplatin dose between these two groups is unexplained.
The successful addition of PARPi to chemotherapy has been hampered by myelotoxicity, limiting either the dose or exposure duration of PARPi or of the concomitant chemotherapy [16, 17, 26-28]. Our phase I study in women with BRCA mutated breast or ovarian cancer found that continuous daily olaparib required more than halving the standard carboplatin dose due to early marrow toxicity [5]. We again found interactive hematologic toxicity in this non-BRCA mutation cohort, requiring a lower AUC of carboplatin than recommended in the BRCA mutation patients. These results are consistent with a recent phase I study of olaparib and cisplatin for patients with a variety of cancers which found hematologic toxicity requiring intermittent and lower dose olaparib (olaparib 50-100mg capsules days 1-5 or 1-10, with cisplatin dose every 21 days) [16]. A phase I study found that continuous olaparib with weekly paclitaxel was not tolerable in patients with sporadic TNBC, and was halted without identifying a recommended phase II dose [17]. The intermittent dosing of olaparib (200mg capsules on days 1-10) ameliorated some of the toxicity allowing its successful administration with paclitaxel (175 mg/m2 every 3 weeks) and carboplatin (AUC4 every 3 weeks) in previously untreated recurrent ovarian cancer patients [26]. The greater olaparib exposure may have been tolerable due to minimal prior treatment exposure and/or to the purported marrow-sparing effects of the addition of paclitaxel [26].
There are limited data on anti-tumor activity of PARPi/chemotherapy combinations in metastatic sporadic TNBC. Gelmon and colleagues found a 2 month PFS using single agent olaparib in unselected patients with TNBC [29]. The olaparib and paclitaxel study of Dent et al. treated unselected metastatic TNBC, yielding an ORR of 37% (7/19) in women who received no (15/19) or one prior cytotoxic therapy regimen for metastatic disease [17]. In addition, platinum agents (cisplatin or carboplatin) resulted in 20% ORR (13/66) in metastatic TNBC patients with BRCA wild type [30]; a lower ORR (12%) was seen in those who received platinum agents as a second-line therapy [30]. Our patients were heavily pretreated, 75% of them were treated with > 3 prior therapy. Approximately half of our patients had clinical benefit by prolonged stable disease and/or reduction in tumor size by olaparib and carboplatin. Although combination therapy consistently resulted in greater toxicity than olaparib alone, studies suggested some antitumor activity of different PARPi/chemotherapy combinations [16, 17, 31]. Assessment of the value of such combinations requires randomized trial design and may benefit from patient reported outcome and/or quality of life analyses. However, our data suggest that the identification of predictive biomarkers may be necessary before such large scale studies are undertaken.
Exceptional responder analyses provide windows into disease processes. Our exceptional responder had no family history of breast and/or ovarian cancer, and commercial testing identifed no deleterious germline events although large genomic rearrangement testing was not done per Myriad-defined criteria at that time [25, 32]. Large genomic rearrangements constitute approximately 8-14% of BRCA1 mutations and 1-4% of BRCA2 mutations [32, 33]. They are more likely to be identified with copy number assays, or whole exome and whole genome approaches, now more commonly applied. Jackson et al. reported rearrangements in 42 of 1300 (3.2%) patients referred for BRCA mutation testing and only 10 of the 42 had met Myriad criteria [34]. Our patient’s outcome of an over 5-year olaparib-maintained CR identifies her as an exceptional responder, remarkable even for patients with BRCA mutation-associated breast cancer. The median duration of response to either olaparib or olaparib/carboplatin is less than one year in BRCA mutated patients with locally advanced or metastatic breast cancer [5]. Secondary somatic mutations that restore BRCA protein function are a mechanism of platinum and PARPi resistance in ovarian cancer [35]. The large deletion found in our exceptional responder would not be amenable to such a restorative event, possibly explaining her long response duration. This finding suggests the need to review clinical outcome by mutation type and site.
A challenge remains to identify, develop, and validate effective predictive biomarkers to apply within and beyond this sporadic TNBC patient population [2]. RPPA has been widely employed as a large-scale proteomic analysis for target and biomarker discovery [36-38]. Our proteomic evaluation of paired cases did not confirm our prior findings of altered FOXO3a found in our BRCA mutation cohort [5]. New observations identified higher pretreatment expression of cyclin D1, collagen VI, and YAP-1 in nonresponders. Cyclin D1 and collagen VI have previously been described as negative prognostic biomarkers, and their overexpression has been suggested as a possible molecular driver in breast cancer [39-41]. However, in this study, increasing amounts of these proteins across one cycle of therapy was seen in patients who had clinical benefit. YAP-1 is a context-dependent tumor suppressor gene, where loss of expression in breast cancer prognosticates a poor outcome [42] [43-45], thus, potentially supporting the upregulated expression of YAP-1 in responding patients but not readily explaining why high levels prior to treatment would account for the lack of benefit.
Our study has some limitations. Our small sample size may introduce biases in estimating clinical benefit and our translational endpoints were exploratory. Although we controlled for multiple comparisons to reduce the incidence of type 1 errors, all findings will need to be examined and validated prospectively. The RPPA findings were not validated by other methods due to the limited remaining core biopsy samples and should be interpreted with caution. Additionally, we did not assess BRCA1 promoter hypermethylation by immunohistochemistry as part of this study, thus cannot address how many of our patients may have tumors with BRCA1 methylation. Analysis of HRR dysfunction was not prospectively planned, nor were we able to evaluate other patients within this cohort due to insufficient clinical samples, lack of appropriate informed consent with genetic counseling, and some patients have since died. It is possible that additional patients had underlying HRR issues contributing to their treatment response or lack thereof.
There are now a number of PARPi trials in locally advanced, metastatic or recurrent breast cancer. Our findings present an opportunity to further investigate this combination in a subgroup of sporadic TNBC patients, taking into consideration that better results may be observed in women with fewer prior treatments. Although our prospectively planned biomarkers did not correlate with clinical response, our exceptional responder results reconfirm the benefit of carboplatin with olaparib in TNBC with HRR dysfunction due to BRCA1 loss, and supports the consideration for wider spectrum mutational testing or homologous recombination deficiency score testing [46] in women with TNBC.
Figure 3: Study schema. Abbreviations: DL: dose level, PBMC; peripheral blood mononuclear cells, bid: twice daily
PATIENTS and METHODS
Patients
This study was approved by the Institutional Review Board of the Center for Cancer Research, National Cancer Institute. Patients with sporadic TNBC (defined by ER < 10%, PR < 10% and no HER2 gene amplification by FISH or HER2 negative by immunohistochemistry [0 or 1+]) had no identified deleterious germline BRCA mutation on prior testing, or a BRCAPro score < 10% [47] calculated on the basis of a detailed family history. Additional eligibility criteria included recurrent, refractory, or locally advanced, unresectable TNBC; measurable or evaluable disease during dose escalation, and biopsiable disease in the expansion cohort; ECOG performance status 0-2; age ≥ 18 years old; normal end organ function except grade 1 anemia, neutropenia, leukopenia, and AST/ALT; no tumor-related therapy for 4 weeks; no platinum therapy for at least 6 prior months irrespective of prior response and no history of NCI Common Terminology Criteria (CTCAE v3.0) grade 4 platinum allergy; no prior PARPi; no infection requiring antibiotics within 7 days; no brain metastases diagnosed or active within the past year. All patients provided written informed consent. ClinicalTrials.gov identifier: NCT01445418.
Drug administration and determination of MTD
This open label 3+3 dose escalation study examined the combination of olaparib 400mg capsules every 12 hours on days 1-7 with carboplatin AUC3, 4, or 5 on day 1 or 2 (dose levels 1-3), in every 21-day cycles (Figure 3). Carboplatin scheduling was adjusted to accommodate patient travel or when a progressive carboplatin concentration infusion program was required to obviate allergic reactions. No more than 8 cycles of combined therapy was given after which continuous daily olaparib monotherapy at a maintenance dose of 400mg capsules every 12 hours was given. DLTs (CTCAEv3.0) were defined as grade 3 or 4 nonhematologic and grade 4 hematologic adverse events related to study medications occurring during the first two cycles of therapy. The following were exceptions: grade 3 diarrhea, nausea, or vomiting must have been unresponsive to optimal medical management, and asymptomatic grade 3 reductions in electrolytes rapidly reversed with medical management. Grade 3 thrombocytopenia lasting for ≥ 7 days or requiring transfusion, and grade 4 neutropenia for ≥ 7 days or with fever, were dose-limiting. Complete blood counts and serum chemistries were monitored weekly during the DLT period.
Granisetron (days 1-7) and dexamethasone (days 1-4) were given as prophylactic antiemetics during each cycle of the combination therapy and discontinued during olaparib maintenance. Pegfilgrastim was indicated for use if the day 1 absolute neutrophil count [ANC] was less than 1500/mL necessitating a treatment delay, or in subsequent cycles if the day 1 ANC was 1500-1800/mL. It was not allowed during the first 2 cycles of the dose escalation phase. Once initiated, pegfilgrastim was continued during all combination treatment cycles. It was not used during olaparib maintenance therapy. Clinical response was assessed every two cycles by imaging using RECISTv1.0 criteria. Study treatment was discontinued for progression of disease, intercurrent illness, adverse events not recovering to ≤ grade 1 within 3 weeks, and patient preference.
Translational studies
The translational studies schema is shown in Figure 3. Peripheral blood mononuclear cells (PBMCs) were collected and separated within 4 hours, then stored in aliquots at -80oC until use. PBMC DNA poly(ADP-ribose; PAR) incorporation was measured using a commercial PAR immunoassay (Trevigen, Gaithersburg, MD) as previously described [48]. PBMC DNA was isolated and tested for polymorphisms of PARP1 A762V, XRCC1 R194W, and XRCC1 Q399R using a commercial DNA purification kit (Qiagen, Germantown, MD) as reported [5]. Paired tumor biopsies were collected in the expansion cohort patients as described [5]. Percutaneous biopsies were obtained by interventional radiologists under CT or ultrasound guidance using local anesthesia. Samples were processed in real time into optimal cutting temperature compound and stored at −80°C, then cut and stained immediately prior to use [49]. Optimal quality of tissue was defined as paired sequential biopsies with solid tissue areas containing at least 50% tumor cells and less than 25% necrosis [49]. Tissue area was measured and prepared [50] and RPPA was executed by the MD Anderson RPPA Core facility using their 218 antibody protocol including key proteins in DNA repair pathways [23]. Apoptotic cells were counted using the DeadEnd colorimetric TUNEL kit (Promega, Madison, WI) as described [5]. The apoptotic index was defined as the percentage of TUNEL-positive single cells in five high-power fields.
BROCA-HR mutational analysis
The patient with a 69+ month olaparib-maintained CR gave informed consent for germline mutation evaluation of 65 genes [24]. Whole blood DNA underwent massively parallel sequencing with BROCA-HR [51]. Copy number alterations in BRCA1 exon 1-2 were confirmed by a Taqman Copy Number Assay (Applied Biosystems, Carlsbad, CA) using CopyCaller Software v2.0 (Applied Biosystems).
Statistical analyses
A protocol-defined expansion cohort was accrued for exploratory biomarker analyses. For any given translational endpoint comparison, 10 paired biopsies were needed to provide 80% power to detect a difference between pre- and on-treatment values equal to one standard deviation of the difference (α2 = 0.05). Normalized linear values for RPPA proteins were analyzed against those who had clinical benefit (progression-free survival [PFS] ≥ 4 months) compared to those who did not using multiple paired t-tests (GraphPad Prism 6, La Jolla, CA). The Hochberg method was used to control the false discovery rate with Q = 0.05 [52]. Linear correlations between protein levels at initiation of treatment and PFS were examined using JMP 9.0 (SAS Institute Inc., Cary, NCI). The per-patient percent TUNEL change was compared using the Fisher’s exact test (Prism 6). The apoptosis index per patient, and baseline PBMC DNA PAR concentrations were correlated with response and PFS using the Fisher’s exact test. Correlation between polymorphisms in PARP1 and XRCC1 and PFS was tested using the log-rank test, and response using the Fisher’s exact test.
ACKNOWLEDGMENTS
We also thank Anne Noonan MD, Ciara O’Sullivan MD, Nicole Houston RN, Irene Ekwede RN, and Mireya Gomez for their contributions in clinic. This article is dedicated to Ms. Jinyoung Choi, who died recently from breast cancer.
CONFLICTS OF INTEREST
All authors maintain no conflicts of interest to report.
FUNDING
This clinical trial and translational studies were funded by the Intramural Program of the Center for Cancer Research, NCI, NIH (JML, #ZIA BC011525). The PAR assay was funded in part by NCI Contract No. HHSN261200800001E (JJ/ JD). The BROCA-HR testing was funded by the Department of Defense Ovarian Cancer Research Program OC120506 (ES). Olaparib was supplied to the Center for Cancer Research, NCI under a Cooperative Research and Development Agreement between the CCR/NCI and AstraZeneca. All authors reviewed the data and had access to all the data upon request. All authors reviewed the manuscript and made the final decision to submit for publication.
REFERENCES
1. Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clinical cancer research. 2014; 20:782-790.
2. Abramson VG, Lehmann BD, Ballinger TJ, Pietenpol JA. Subtyping of triple-negative breast cancer: implications for therapy. Cancer. 2015; 121:8-16.
3. Robertson L, Hanson H, Seal S, Warren-Perry M, Hughes D, Howell I, Turnbull C, Houlston R, Shanley S, Butler S, Evans DG, Ross G, Eccles D, et al. BRCA1 testing should be offered to individuals with triple-negative breast cancer diagnosed below 50 years. British journal of cancer. 2012; 106:1234-1238.
4. Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014; 25:32-40.
5. Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, Figg WD, Azad N, Wood BJ, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. Journal of the National Cancer Institute. 2014; 106:dju089.
6. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010; 363:1938-1948.
7. Podo F, Buydens LM, Degani H, Hilhorst R, Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J, Monleon D, Postma GJ, Schneiderhan-Marra N, Santoro F, et al. Triple-negative breast cancer: present challenges and new perspectives. Molecular oncology. 2010; 4:209-229.
8. LaDuca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, Chen E, Gau CL, Palmaer E, Shoaepour K, Shah D, Speare V, Gandomi S, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genetics in medicine. 2014; 16:830-837.
9. Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. Journal of clinical oncology. 2014; 32:2001-2009.
10. Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Hallberg E, Guidugli L, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. Journal of clinical oncology. 2015; 33:304-311.
11. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, Wesseling J, Rodenhuis S, Nederlof PM. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. British journal of cancer. 2013; 108:2172-2177.
12. Domagala P, Huzarski T, Lubinski J, Gugala K, Domagala W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast cancer research and treatment. 2011; 127:861-869.
13. Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer. 2010; 1:812-821. doi: 10.1177/1947601910383418.
14. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917-921.
15. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913-917.
16. Balmana J, Tung NM, Isakoff SJ, Grana B, Ryan PD, Saura C, Lowe ES, Frewer P, Winer E, Baselga J, Garber JE. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann Oncol. 2014; 25:1656-1663.
17. Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL, Carmichael J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast cancer research. 2013; 15:R88.
18. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010; 376:235-244.
19. Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, Fatima A, Gelman RS, Ryan PD, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. Journal of clinical oncology. 2010; 28:1145-1153.
20. Byrski T, Huzarski T, Dent R, Gronwald J, Zuziak D, Cybulski C, Kladny J, Gorski B, Lubinski J, Narod SA. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast cancer research and treatment. 2009; 115:359-363.
21. Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, Cybulski C, Marczyk E, Chrzan R, Eisen A, Lubinski J, Narod SA. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast cancer research. 2012; 14:R110.
22. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:17079-17084.
23. https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/antibody-information-and-protocols.html. Accessed March 10, 2017.
24. http://tests.labmed.washington.edu/BROCA. Accessed March 10, 2017.
25. https://webapps.myriad.com/lib/brac/BART-faq.pdf. Accessed March 10, 2017.
26. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Spacek J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet oncology. 2015; 16:87-97.
27. Zhang J, Wang L, Wang Z, Hu X, Wang B, Cao J, Lv F, Zhen C, Zhang S, Shao Z. A phase II trial of biweekly vinorelbine and oxaliplatin in second- or third-line metastatic triple-negative breast cancer. Cancer Biol Ther. 2015; 16:225-232.
28. Van der Noll R, Ang JE, Jager A, Marchetti S, Mergui-Roelvink M, De Bono JS, Lolkema M, Brunetto A, Arkenau HT, De Jonge MJ, Van der Biessen D, Tchakov I, Bowen K, et al. Phase I study of olaparib in combination with carboplatin and/or paclitaxel in patients with advanced solid tumors. Journal of clinical oncology. 2013; 31:suppl; abstr 2579.
29. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The lancet oncology. 2011; 12:852-861.
30. Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, Timms KM, Hartman AR, Borger DR, et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. Journal of clinical oncology. 2015; 33:1902-1909.
31. Rugo HS, Olopade OI, DeMichele A, Yau C, van ‘t Veer LJ, Buxton MB, Hogarth M, Hylton NM, Paoloni M, Perlmutter J, Symmans WF, Yee D, Chien AJ, et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N Engl J Med. 2016; 375:23-34.
32. Judkins T, Rosenthal E, Arnell C, Burbidge LA, Geary W, Barrus T, Schoenberger J, Trost J, Wenstrup RJ, Roa BB. Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer. 2012; 118:5210-5216.
33. Kwong A, Chen J, Shin VY, Ho JC, Law FB, Au CH, Chan TL, Ma ES, Ford JM. The importance of analysis of long-range rearrangement of BRCA1 and BRCA2 in genetic diagnosis of familial breast cancer. Cancer genetics. 2015; 208:448-454.
34. Jackson SA, Davis AA, Li J, Yi N, McCormick SR, Grant C, Fallen T, Crawford B, Loranger K, Litton J, Arun B, Vande Wydeven K, Sidani A, et al. Characteristics of individuals with breast cancer rearrangements in BRCA1 and BRCA2. Cancer. 2014; 120:1557-1564.
35. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. Journal of clinical oncology. 2011; 29:3008-3015.
36. Nishizuka SS, Mills GB. New era of integrated cancer biomarker discovery using reverse-phase protein arrays. Drug Metab Pharmacokinet. 2016; 31:35-45.
37. Kamel D, Brady B, Tabchy A, Mills GB, Hennessy B. Proteomic classification of breast cancer. Curr Drug Targets. 2012; 13:1495-1509.
38. Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, Shen Y, Mills GB, Minna JD, Heymach JV, Byers LA. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clinical cancer research. 2013; 19:6322-6328.
39. Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth RF, Wittenbel KD, Simpson JF, Page DL, Steeg PS. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nature medicine. 1995; 1:1257-1260.
40. Sutherland RL, Musgrove EA. Cyclin D1 and mammary carcinoma: new insights from transgenic mouse models. Breast cancer research. 2002; 4:14-17.
41. Karousou E, D’Angelo ML, Kouvidi K, Vigetti D, Viola M, Nikitovic D, De Luca G, Passi A. Collagen VI and hyaluronan: the common role in breast cancer. BioMed research international. 2014; 2014:606458.
42. Wang H, Du YC, Zhou XJ, Liu H, Tang SC. The dual functions of YAP-1 to promote and inhibit cell growth in human malignancy. Cancer metastasis reviews. 2014; 33:173-181.
43. Yu SJ, Hu JY, Kuang XY, Luo JM, Hou YF, Di GH, Wu J, Shen ZZ, Song HY, Shao ZM. MicroRNA-200a promotes anoikis resistance and metastasis by targeting YAP1 in human breast cancer. Clinical cancer research. 2013; 19:1389-1399.
44. Chen HY, Yu SL, Ho BC, Su KY, Hsu YC, Chang CS, Li YC, Yang SY, Hsu PY, Ho H, Chang YH, Chen CY, Yang HI, et al. R331W Missense Mutation of Oncogene YAP1 Is a Germline Risk Allele for Lung Adenocarcinoma With Medical Actionability. Journal of clinical oncology. 2015; 33:2303-2310.
45. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014; 158:171-184.
46. Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, Isakoff SJ, Ryan PD, Greene-Colozzi A, et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clinical cancer research. 2016; 22:3764-3773.
47. http://bcb.dfci.harvard.edu/bayesmendel/brcapro.php Accessed March 10, 2017.
48. Kinders RJ, Hollingshead M, Khin S, Rubinstein L, Tomaszewski JE, Doroshow JH, Parchment RE. Preclinical modeling of a phase 0 clinical trial: qualification of a pharmacodynamic assay of poly (ADP-ribose) polymerase in tumor biopsies of mouse xenografts. Clinical cancer research. 2008; 14:6877-6885.
49. Lee JM, Hays JL, Noonan AM, Squires J, Minasian L, Annunziata C, Wood BJ, Yu M, Calvo KR, Houston N, Azad N, Kohn EC. Feasibility and safety of sequential research-related tumor core biopsies in clinical trials. Cancer. 2013; 119:1357-1364.
50. Azad N, Yu M, Davidson B, Choyke P, Chen CC, Wood BJ, Venkatesan A, Henning R, Calvo K, Minasian L, Edelman DC, Meltzer P, Steinberg SM, et al. Translational predictive biomarker analysis of the phase 1b sorafenib and bevacizumab study expansion cohort. Molecular & cellular proteomics. 2013; 12:1621-31.
51. Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:12629-12633.
52. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological). 1995:289-300.