
Introduction
The transcription factor Sox2 is widely recognized for its critical roles during mammalian embryogenesis. Although Sox2 was first shown to regulate the transcription of FGF4 in mouse embryonal carcinoma cells [1], its importance was firmly established with the discovery that knocking out both alleles of Sox2 results in embryonic lethality in mice. Sox2 null embryos reach the blastocyst stage, but do not survive after implantation [2]. Shortly thereafter, knocking down Sox2 in mouse embryonic stem cells (ESC) was shown to disrupt their self-renewal and induce differentiation [3]. One year later, interest in Sox2 rose dramatically with the paradigm-shifting discovery by Takahashi and Yamanaka demonstrating conversion of mouse embryonic fibroblasts into induced pluripotent stem (iPS) cells by ectopic expression of Sox2 along with Oct4, Klf4, and cMyc [4].
The excitement surrounding the key roles of Sox2 in ESC and iPS cells, which are themselves tumorigenic, soon led to the search for SOX2 in cancer. Within a few years after the discovery of iPS cells, numerous reports of SOX2 expression in human cancer had already appeared. This soon turned into an avalanche of studies examining SOX2 in human cancer. The search terms “SOX2 and cancer” generate over 1,600 hits in the PubMed database and over 11,000 hits in PubMed Central. Since 2006, SOX2 has been implicated in growth, tumorigenicity, drug resistance, and metastasis in at least 25 different cancers, including cancers of the ovary, lung, skin, brain, breast, prostate, and pancreas (Tables 1-2). In the majority of these cancers, SOX2 has been reported to have increased expression or gene amplification in tumor tissue; however, the effects of SOX2 on tumorigenicity, prognosis, and drug resistance in human cancer have only begun to be explored. Nonetheless, it is evident from the impressive body of work published thus far that SOX2 is a major player in cancer and a potential therapeutic target.
In this review, we provide an overview of SOX2 in cancer and focus on two broad topics. The first part of the review discusses the expression and functions of SOX2 in cancer and specifically focuses on five main topics: 1) expression and amplification of SOX2 in cancer, 2) SOX2 expression and cancer prognosis, 3) SOX2 expression by cancer stem cells (tumor-initiating cells), 4) SOX2 and drug resistance, and 5) tight control of SOX2 expression in cancer. This last topic addresses what we believe is a defining feature of SOX2: its levels in actively proliferating tumor cells appear to be optimized to maximize tumor growth; namely, too little or too much SOX2 inhibits tumor cell proliferation. In the second part of the review, we delve into the exquisite levels of regulation used to ensure both proper expression and function of SOX2. Specifically, we focus on four main topics: 1) transcriptional regulation of SOX2, 2) regulation of SOX2 expression by microRNAs, 3) regulation of SOX2 expression by long non-coding RNAs, and 4) post-translational modifications of SOX2. In the final section of this review, Conclusions and Future Perspectives, we discuss several important questions that remain unanswered concerning the roles of SOX2 in cancer.
More generally, this review draws attention to four major themes. First, SOX2 plays important roles in many cancers. In at least three cancers, SOX2 has been shown to be expressed by the cancer stem cell/tumor-initiating cell population of the tumor. Second, there is growing evidence that SOX2 also influences the responses of tumor cells to drugs used in the treatment of cancer. Third, small changes in the levels of SOX2 alter tumor cell physiology. In several cancers, SOX2 expression increases during tumor progression, but, paradoxically, experimentally increasing SOX2 expression on its own in tumor cells with the aid of an inducible promoter leads to a reduction in growth. An explanation for this paradoxical finding is discussed. Fourth, a multitude of mechanisms have been shown to control the expression and function of SOX2. However, additional work will be needed to determine which of these mechanisms are utilized in specific cancers to fine tune SOX2 expression and function.
Finally, we recognize that other important topics are not included in this review. Specifically, we have not provided an overview of the downstream targets of SOX2 or the convergence of cell signaling and SOX2 expression. Similarly, we have not discussed the roles of SOX2 during embryogenesis and its roles in adult tissues. Readers interested in these subjects are directed to other recent reviews [5-7].
EXPRESSION AND FUNCTION OF SOX2 IN CANCER
SOX2 expression and amplification in cancer
SOX2 expression has been reported at both the RNA and protein levels for many cancers. Data available from The Cancer Genome Atlas indicates that SOX2 mRNA is elevated in many cancers, relative to normal tissue. For example, SOX2 is reported to be elevated in > 85% of glioblastoma multiforme samples compared to normal patient controls [8]. Interestingly, hypomethylation of the SOX2 promoter was detected in over 250 glioblastoma specimens compared to normal patient controls [8]. In tumors such as glioblastoma, ovarian, esophageal, lung, oral, prostate, and sinonasal carcinoma, SOX2 has been shown to be amplified in some subsets of patient tumors [8-19]. One study found SOX2 to be amplified in 26% of serous ovarian cancers [9], and the SOX2 locus (3q26.33) was amplified in ~8% of glioblastoma cases [8], indicating that an increase in copy number is part of the puzzle regarding SOX2 expression in cancer.
For most cancers, SOX2 expression has also been documented at the protein level by immunohistochemistry [8, 10, 14-17, 20-32]. For example, in a study of breast cancer patients, SOX2 was strongly detected by immunohistochemistry in the nucleus of breast carcinoma cells compared to weak or no SOX2 staining in normal, non-tumorigenic mammary epithelial issue [20, 33]. Although SOX2 expression has been reported in many cancers [8-29, 32-36], the percent of SOX2-positive cells within SOX2-positive tumors has not been consistently reported. Additionally, in many studies, reference to “normal” tissue often was to unrelated, non-tumorigenic tissue rather than matched adjacent tissue.
In the case of ovarian cancer, both the percent of SOX2-positive tumors and the percent of SOX2-positive cells within these tumors have been reported [26]. Interestingly, the percent of SOX2-positive cells differs between different ovarian tumor subtypes [26]. In over 50% of cases of serous cystadenocarcinoma, SOX2 was expressed in > 75% of the cells of the tissue examined, whereas in only 5% of cases of less severe serous cystadenoma was SOX2 expressed in > 75% of the cells. Similar expression patterns were observed with mucinous epithelial lesion [26]. Interestingly, this variable expression of SOX2 across cells within the same tumor has been observed in multiple cancers [37, 38] and may influence their physiology, as discussed below in the section “SOX2 and Tumor-Initiating Cells/Cancer Stem Cells”. Finally, it should be stressed that comparisons of SOX2 expression between tumor cells and normal tissues may be misleading, because tumor cells arise from a small subset of cells in a tissue and the expression of SOX2 in this subset of cells may differ from that in the remainder of the tissue.
For several cancers, the levels of SOX2 expression at different stages of the cancer have been examined [25]. In pancreatic ductal adenocarcinomas (PDAC), SOX2 is rarely expressed cases of in pre-malignant pancreatic intraepithelial neoplasia, but its expression has been reported to increase to ~60% in cases of poorly differentiated and neurally invasive components [28]. Similarly, studies of glioblastoma, esophageal, breast, and prostate cancers have reported that SOX2 levels increase with increasing tumor grade [14, 29, 33, 39, 40], and for prostate cancer the percentage of SOX2-positive cells correlates with Gleason score [41]. In the case of ovarian epithelial carcinoma, SOX2 expression was reported to increase from ~55% of normal ovarian epithelia samples expressing some SOX2 (in a relatively low percent of cells) to over 90% of serous and mucinous cystadenocarcinomas samples expressing SOX2 and in a much higher percent of the cells [26]. Interestingly, in the case of gastric cancer, reports regarding the levels of SOX2 expression during tumor progression are conflicting. In one study, SOX2 mRNA was reported to be significantly elevated compared to adjacent benign tissues [34]. In contrast, other studies reported lower SOX2 expression in gastric cancer and its metastatic lesions compared to matched, normal gastric mucosa [30, 31, 42, 43]. Notably, SOX2 expression also appears to vary with different mucosal subgroups in gastric cancer [30, 44]. Thus, for several cancers, there is a need to more carefully determine how SOX2 levels change during tumor progression. Recognizing how SOX2 expression is altered between normal and tumorous tissues is important for understanding molecular changes necessary for tumor initiation and progression.
SOX2 expression: prognosis and survival
In addition to determining how SOX2 levels change during tumor progression, it is essential to determine whether SOX2 levels correlate with clinical prognosis for cancer patients. Studies reported thus far indicate that high SOX2 levels correlate with poor prognosis for patients with many different cancers, including breast, colorectal, esophageal, ovarian, prostate, and some lung tumors, as well as nasopharyngeal and sinonasal carcinoma (Table 1) [27, 38, 40, 45-48]. Furthermore, a higher incidence of recurrence was correlated with SOX2 amplification in sinonasal carcinomas [19], and rectal cancer patients with elevated SOX2 displayed significantly shorter disease-free survival following chemoradiotherapy [45]. Studies in esophageal, hepatocellular, oral/tongue and some lung cancers have also found a correlation between elevated SOX2 and decreased survival [27, 40, 48-52]. In addition to survival and recurrence, in the majority of cancers examined thus far, high SOX2 expression has been linked to the infiltrative and metastatic capacity of tumor cells [31, 50, 53-56]. For example, in the cases of colorectal cancer and prostate cancer, SOX2-expressing tumors have been shown to correlate with increased distant and lymphatic metastases [29, 53]. Similarly, in esophageal squamous cell carcinomas, tumors in which more than 50% of the cells express SOX2 were correlated with increased lymphatic and vascular invasion, poor differentiation, and incomplete surgical resection [55].
Consistent with many reports linking SOX2 expression to increases in metastasis, a strong connection between SOX2 and epithelial-mesenchymal transition (EMT) has been established in many tumor types, including colorectal, esophageal (ESCC), laryngeal, pancreatic, lung (NSCLC), gastric, breast, and prostate cancer [54, 57-63]. Additionally, high SOX2 expression has been linked to increases in migration and invasion [54, 57, 58]. The link between SOX2 and EMT in some cancers was shown by directly altering the levels of SOX2. For example, knockdown of SOX2 in colorectal tumor cells induced mesenchymal-epithelial transition [54]. In other studies, changes in SOX2 expression and EMT were observed in response to changing other factors. For example, in pancreatic cancer, expression of the transcription factor NFATc1 was reported to drive EMT via SOX2-dependent transcription [59].
Although SOX2 is associated with poor prognosis in many cancers, high SOX2 levels may not be uniformly indicative of poor patient prognosis. For at least four cancers, including gastric cancer and squamous cell lung cancer, low SOX2 expression has been reported to correlate with poor prognosis (Table 1) [16, 31, 56, 64]. Moreover, as noted earlier, some studies of gastric cancer indicate that elevated SOX2 levels are linked to reduced lymph node and distant metastases. The reasons for the contrasting results for SOX2 levels in different cancers remain to be determined.
Disappointingly, for some cancers, in particular head and neck squamous cell carcinomas and ovarian cancer, there are conflicting reports regarding the levels of SOX2 expression and patient survival [9, 49, 52, 65]. In the case of head and neck squamous cell carcinomas, initial studies by Lee et al showed that SOX2 expression is correlated with poor prognosis and a nearly 5-fold higher risk of recurrence [49], but subsequent studies by Bayo et al determined that SOX2high tumors had a median progression-free survival of 51 months compared to SOX2low tumors (16 months) and that SOX2high tumors had a > 110 month improved overall survival compared to SOX2low tumors [65]. Questions also exist in the case of ovarian cancer. Belotte et al reported that tumors with SOX2 amplification had statistically significant improved survival [9]; however, an earlier study from Wang et al reported that high SOX2 levels in both primary and metastatic tumor components statistically correlated with significantly worse survival [52]. It is evident from the discussion in this section that there is a clear need for further investigation into the clinical implications of SOX2 expression, particularly how SOX2 levels influence tumor progression and patient survival.
SOX2 and tumor-initiating cells/cancer stem cells
SOX2 is not only expressed in many types of cancer, it has also been implicated in the tumor-initiating populations (proposed cancer stem cell population) of many of these tumors (Table 2). Many studies have used putative cancer stem cell markers, such as CD133, CD44, ABCG2, and side population via Hoechst efflux assay, to isolate and enrich for cells capable of forming tumors [tumor-initiating cells (TIC)] [13, 35, 66-72]. For example, in the case of an ovarian cancer cell line, the side population exhibited elevated levels of SOX2 mRNA and a higher percentage of TIC when assayed using a limiting cell dilution tumor assay, the gold standard for assessing the frequency of TIC within a tumor [69]. However, for most cancers, the link between SOX2 and their TIC has not been firmly established. For several tumor types, knockdown of SOX2 and/or ectopic expression of SOX2 have been used to implicate SOX2 in the biology of the TIC [38, 73-77]. For example, Lee et al and Santini et al determined that stable knockdown of SOX2 in limiting cell dilution tumor assays dramatically reduced tumor initiation/formation in both head and neck squamous cell carcinomas and melanomas, respectively [49, 78]. Conversely, others generated lung and ovarian tumor cells that stably overexpress SOX2 and reported an elevated number of TIC when these cells were tested in limiting cell dilution tumor assays [79, 80]. However, as discussed later in the review, there are concerns about the use of tumor cells engineered to stably overexpress SOX2. Additionally, we raise concerns about the use of putative stem cell markers for the isolation of TIC/cancer stem cells.
Arguably the most conclusive studies have linked SOX2 to TIC by isolating the SOX2-positive cell subpopulation rather than experimentally altering the levels of SOX2 within cells. This is important, because SOX2 is expressed heterogeneously throughout the cells of many tumors [37, 38, 65, 81]. Moreover, for some tumors, only a small percentage of the cells express SOX2. This is particularly evident for SOX2-positive tumor cell lines [37, 38, 82]. Thus far, the SOX2-positive cells isolated from heterogeneous populations were engineered to express either GFP that was knocked into the endogenous SOX2 gene (GFP coding sequences replaced single SOX2 exon) [37, 82] or GFP driven by a transgene under the control of the SOX2 promoter and enhancer [83]. In these three studies, SOX2-positive cells exhibited a higher frequency of TIC compared to the SOX2-negative cells of the same tumor cell population in a limiting cell dilution tumor assay. Furthermore, studies by Vanner et al showed that the rare SOX2-positive cells are members of a quiescent, slowly-cycling cancer stem cell population that repopulates the tumor when cytotoxic drugs are withdrawn [37]. Similarly, a recent study in bladder cancer has also shown that the quiescent label-retaining cancer stem cell population does not respond to cytotoxic therapy and is capable of repopulating the tumor following drug removal [84]. Such studies not only call attention to a role for SOX2 in the TIC population, but also that this slowly-cycling, SOX2-positive population may be responsible for repopulating the tumor after drug treatment is suspended.
Cancer stem cell markers - a cautionary note
As discussed earlier, there is compelling evidence that SOX2 is associated with the tumor-initiating population of at least three cancers and there is growing evidence for this association in many other cancers (Table 2). However, as we explain below, caution should be exercised when putative stem cell markers (e.g. CD133 and ALDH1) that change rapidly in response to cellular conditions are used to isolate cancer stem cells. To illustrate this point, we focus on CD133, which has been linked with SOX2 expression and cancer stem cells in many tumor cell types [85-91]. In 2003, CD133 was reported to serve as a marker for the tumor-initiating cells of brain tumors. Specifically, it was reported that the capacity for self-renewal of brain tumor cells in culture (non-adherent tumor spheres) resides in the CD133+ cell population and not in the CD133- population [92]. Subsequently, it was reported that transplantation of as few as 100 CD133+ glioblastoma cells or 1,000 CD133+ medulloblastoma cells were sufficient for tumor formation; whereas, 105 CD133- glioblastoma cells or 5X104 CD133- medulloblastoma cells were incapable of forming tumors [93]. The work with CD133 in brain tumors was followed by a series of studies supporting the value of CD133 as a marker for identifying and isolating tumor-initiating cells in several other cancers, including pancreas, prostate, lung, liver, and colon tumors [94-99]. However, the use of CD133 as a stem cell marker for several cancers, including pancreatic ductal adenocarcinoma and glioblastoma, has become controversial [22, 100]. In the case of glioblastoma, one study reported that both CD133- and CD133+ cells were able to induce tumors in nude mice, although tumors formed by the CD133- cells exhibited a lower level of proliferation [100].
We believe the conflicting results reported for CD133- cells, which has been noted by others [101], is due at least in part, if not entirely, to rapid changes in expression of CD133 in response to cell culture conditions. For example, the percentage of CD133+ DAOY medulloblastoma cells increases when they are switched from 20% to 2% oxygen [102] and there is a large increase in the expression of CD133 when CD133- cells are switched from serum-containing medium to serum-free medium [103]. Similarly, elevating SOX2 in medulloblastoma cells, which causes a major reduction in their growth, leads to over a 10-fold increase in the number CD133+ cells just one day after elevating SOX2 [104]. Additionally, we have observed that cell surface expression of CD133 on DAOY medulloblastoma cells increases (~50 to 90%) as cell density increases (Wilder and Rizzino, unpublished results). Importantly, we have also shown that CD133 expression rapidly reappears when freshly isolated CD133- glioblastoma cells are returned to cell culture [104]. Similarly, we have observed the reappearance of ALDH1+ cells when freshly isolated ALDH1- pancreatic ductal adenocarcinoma cells were returned to cell culture (Wilder and Rizzino, unpublished results). Clearly, if stem cell markers fluctuate in respond to cell culture conditions, it is not surprising that conflicting results regarding CD133 and ALDH1 have been reported.
If CD133 is not a bonafide marker of tumor-initiating cells, why has it been reported in many studies to enrich for putative cancer stem cells? Although the answer to this question remains to be determined fully, three findings suggest an explanation. First, CD133 has been shown to influence MAPK/ERK signaling in brain tumor cells [105]. Second, the 52-amino acid C-terminal cytoplasmic tail of CD133 can be phosphorylated by a Src-family member on two tyrosine residues (Y828 and Y852) in medulloblastoma cell lines [106]. Third, N-linked glycosylation of CD133 is decreased when colon tumor cells are treated with butyrate [107]. Thus, CD133 appears to be involved in signal transduction, and it may help cells sense and respond to changes in the extracellular environment. Interestingly, this is highly reminiscent of the discovery that the transmembrane glycoprotein MUC1, which was initially identified as a tumor-associated antigen, plays an important role in signal transduction. In this regard, the cytoplasmic C-terminal tail of MUC1 can be phosphorylated by different kinases, including EGFR, PDGFR-β, and MET. Remarkably, phosphorylation of the C-terminal tail of MUC1 by MET and PDGFR-β induces its translocation into the nucleus and increases the metastasis of pancreatic ductal adenocarcinoma cells, respectively [108-110].
Together, the studies discussed above, which point to the role of CD133 in signal transduction, along with the studies demonstrating rapid modulation of CD133 when cell culture conditions are altered, lead us to propose that CD133 is part of the essential stress response machinery that enables tumor cells to rapidly adapt to a range of highly detrimental conditions, such as hypoxia, when transplanted either subcutaneously or orthotopically. If, in fact, expression of CD133 and other stem cell markers provides protection against a range of harmful conditions, this could explain why cells that express these markers exhibit a higher frequency of tumor-initiating cells: they are primed to handle the stress. Importantly, if the expression of putative stem cell markers, such as CD133, provides protection against the stresses of transplantation, their use for isolation of cancer stem cells may lead to an overestimation of the number of TIC, especially when performing limiting cell dilution tumor assays. Thus, we suggest, wherever possible, that future efforts to associate SOX2 with cancer stem cells of a given type of tumor focus on those markers that do not change readily in response to changes in cellular condition. As discussed above (SOX2 and Tumor-Initiating Cells/Cancer Stem Cells), this problem was avoided in those studies where GFP had been knocked into the endogenous SOX2 locus, and GFP+ cells were isolated and tested for enrichment of tumor-initiating/cancer stem cells.
Table 1: SOX2 expression and patient prognosis
Cancer Type |
Amplified/ Increased Expression |
Decreased Expression |
Poor Prognosis/ High Tumor Grade |
Good Prognosis/ Low Tumor Grade |
Breast |
Chen et al., 2008 |
|
Chen et al., 2008 |
|
Rodriguez-Pinilla et al., 2007 |
|
Piva et al., 2014 |
|
|
Colorectal |
Long et al., 2009 |
|
Saigusa et al., 2009 |
|
|
|
Lundberg et al., 2014 |
|
|
|
|
Talebi et al., 2015 |
|
|
Embryonal (testicular germ cell) carcinoma |
Biermann et al., 2007 |
|
|
|
Esophageal |
Gen et al., 2010 |
|
Wang et al., 2009 |
|
Bass et al., 2009 |
|
|
|
|
Long et al., 2009 |
|
|
|
|
Ewing's sarcoma |
Ren et al., 2016 |
|
|
|
Gastric |
Tian et al., 2014 |
Chen et al., 2016 |
|
Zhang et al., 2010 |
|
Wang et al., 2015 |
|
Chen et al., 2016 |
|
|
Otsubo et al., 2008 |
|
Wang et al., 2015 |
|
|
Tsukamoto et al., 2005 |
|
|
|
|
Li X et al., 2004 |
|
|
|
Glioblastoma |
Alonso et al., 2011 |
|
Annovazzi et al., 2011 |
|
Schmitz et al., 2007 |
|
Ma et al., 2008 |
|
|
Phi et al., 2008 |
|
|
|
|
Annovazzi et al., 2011 |
|
|
|
|
Head and neck squamous cell carcinoma |
Bourguignon et al., 2012 |
|
Lee et al., 2014 |
Bayo et al., 2015 |
Hepatocellular carcinoma |
|
|
Sun et al., 2013 |
|
Lung adenocarcinoma |
Sholl et al., 2010 |
|
Sholl et al., 2010 |
|
Lung cancer, non-small cell |
|
|
Chou et al., 2013 |
|
Lung cancer, small cell |
Güre et al., 2000 |
|
|
|
Rudin et al., 2012 |
|
|
|
|
Lung cancer, squamous cell |
Bass et al., 2009 |
|
|
Lu Y et al., 2010 |
Hussenet et al., 2010 |
|
|
|
|
Yuan et al., 2010 |
|
|
|
|
Sholl et al., 2010 |
|
|
|
|
Wilbertz et al., 2011 |
|
|
Wilbertz et al., 2011 |
|
Lung cancer, neuroendocrine |
Sholl et al., 2010 |
|
|
|
Melanoma |
Laga et al., 2011 |
|
Chen et al., 2013 |
|
Nasopharyngeal carcinoma |
|
|
Wang et al., 2012 |
|
Oral squamous cell carcinoma |
Freier et al., 2009 |
|
Du et al., 2011 |
|
Ovarian |
Belotte et al., 2015 |
|
Wang et al., 2014 |
Belotte et al., 2015 |
Ye et al., 2011 |
|
Zhang et al., 2012 |
|
|
Zhang et al., 2012 |
|
|
|
|
Pancreas |
Sanada et al., 2006 |
|
|
|
Prostate |
Sattler et al., 2000 |
|
Kregel et al., 2013 |
|
Jia et al., 2011 |
|
Jia et al., 2011 |
|
|
Sinonasal carcinoma |
Schrock et al., 2013 |
|
Schrock et al., 2013 |
|
SOX2 and drug resistance
Several recent studies have shown that exogenous elevation of SOX2 can promote resistance to chemotherapeutics currently being used clinically [29, 34, 38, 48, 49, 67, 75, 80, 111-114]. In a report from Bareiss et al, ovarian cancer cell lines that did not express SOX2 and that were sensitive to carboplatin, cisplatin, and paclitaxel became resistant following stable, ectopic expression of SOX2 [80]. Furthermore, in a SOX2-expressing ovarian cancer cell line, SOX2 knockdown using short hairpin RNAs (shRNA) provided susceptibility to these drugs, which was reversed upon re-expression of SOX2 ectopically [80]. Similar results were seen in breast cancer cell lines, as stable overexpression of SOX2 in MCF-7 cells promoted resistance to tamoxifen, while stable downregulation of SOX2 using shRNAs enhanced the sensitivity of MCF-7 cells to tamoxifen [38]. Additionally, stable overexpression of SOX2 in PC3 prostate cancer cells promoted evasion of apoptosis in cells treated with paclitaxel [113]. Furthermore, it has been shown that inducible-overexpression and inducible-knockdown of SOX2 in PDAC cell lines altered responses to small molecule inhibitors targeting MEK and AKT signaling. In this study, overexpression of SOX2 protected PDAC cells from the growth inhibitory effects of MEK and AKT inhibitors; whereas, knocking down SOX2 enhanced the growth inhibition in the presence of these drugs [114]. While SOX2 may be acting to protect tumor cells through antiapoptotic signaling or quiescent-like phenotypes [29, 37, 67, 75], SOX2 may also be promoting drug resistance through various ATP-binding cassette (ABC) transporters, including ABCG2, ABCC3, and ABCC6. In particular, ABCG2 has been shown in various tumors to be upregulated in the side population TIC [66, 69] and, in some tumors, is considered to be an additional cancer stem cell marker. Furthermore, studies in lung cancer, as well as head and neck squamous cell carcinoma, have shown that stable downregulation of SOX2 via shRNAs decreases ABCG2, which implicates this transporter in SOX2-related drug resistance [49, 76]. Recognizing and focusing on the role of SOX2 in drug resistance could greatly improve the treatment options for patients with a multitude of cancers, especially those with highly refractory tumors, as the ability to eradicate the TIC population is likely to be the only way to prevent recurrence.
SOX2 levels and tumor growth
Many studies have used stable overexpression and/or knockdown of SOX2 in tumor cell lines to better understand the roles of this transcription factor in cancer. Knockdown of SOX2 using either small interfering RNA (siRNA) or shRNA have been used in multiple studies [8, 11, 12, 25, 29, 32-34, 48, 52, 69, 111, 114-118]. Importantly, even partial reductions in SOX2 levels have been reported to significantly decrease cell viability, clonal growth, sphere formation, and tumorigenicity in multiple cancer types. Clearly, knockdown studies have established that SOX2 plays important roles in these cancers. However, SOX2 overexpression studies have generated conflicting results. For example, stable overexpression of SOX2 in the gastric tumor cell line N87 was reported to increase growth both in vitro and in vivo [34]. In contrast, stable overexpression of SOX2 in the gastric cell line MKN28 was reported to decrease growth both in vitro and in vivo. Currently, the reasons for the conflicting results are unclear. In both studies, SOX2 was substantially overexpressed in gastric tumor cell lines that endogenously express relatively little SOX2. Part of the explanation may be due to differences in the cell lines used. However, as discussed below, other factors related to experimental design may also be a contributing factor.
Conflicting reports from SOX2 overexpression studies have also been reported for breast, prostate, and pancreatic cancers. Stable overexpression in MCF-7 (breast), DU145 (prostate), and Patu8988t (PDAC) cells have been reported to increase growth in vitro [29, 33, 70]. However, there are reports showing that elevating SOX2 does not promote growth. In the case of three colorectal cancer cell lines, growth inhibition was observed during the initial five days when SOX2 was elevated. In the case of HT-29, growth is almost completely arrested when SOX2 was elevated [119]. Moreover, overexpression of SOX2 from a doxycycline inducible transgene demonstrated that overexpression of SOX2, where one can monitor the early, short-term consequences of elevating SOX2, does not increase cell proliferation. Inducibly elevating SOX2 (~5 to 7-fold) in glioblastoma (U87, U118), medulloblastoma (DAOY), breast carcinoma (MDA-MB-231), and prostate carcinoma (DU145) cell lines led to growth inhibition in each case. More recently, the same results were observed in three different PDAC cell lines [114]. Elevating SOX2 (~5 to 7-fold) in each of these cell lines with the aid of an inducible promoter led to growth inhibition in vitro. Furthermore, the effects of elevating SOX2 in vivo have also been examined. Initially, we determined that elevating SOX2 with the aid of an inducible promoter in one of these PDAC cell lines dramatically reduced tumor growth [114]. More recently, we have observed the same effect on tumor growth when SOX2 was elevated in a second PDAC cell line (Wuebben and Rizzino, unpublished results). Thus, the effect of elevating SOX2 in these tumor cell lines, in particular PDAC tumor growth, is growth inhibition. Going forward, it will be important to reexamine the effects of SOX2 in other cancers using inducible overexpression of SOX2, in particular where stable overexpression has been reported to increase tumor cell growth.
The contrasting results obtained from studying cells in which SOX2 was stably overexpressed versus inducible overexpression of SOX2 are likely to result from the fundamental difference in experimental design. Cell lines engineered for inducible overexpression of SOX2 were generated via drug selection of lentiviral transduced cells, which occurred at frequencies greater than 70%, before SOX2 levels were altered. In direct contrast, establishing cells lines that stably overexpress SOX2, which takes several weeks, involves drug selection at a time when SOX2 levels are also ectopically elevated. As a result, any cells that are unable to grow or grow more slowly due to elevated levels of SOX2 will be lost during the drug selection period as the faster proliferating cells expand. Consequently, the cells present in the drug selected population represent only a subpopulation of the parental cells.
It is evident from the studies where SOX2 was elevated from an inducible transgene that many, if not most, SOX2-expressing tumor cell lines are growth inhibited when SOX2 is suddenly elevated. However, this does not mean that SOX2 levels cannot rise during cancer. In fact, several lines of evidence argue that SOX2 levels do rise during cancer. As discussed earlier, the SOX2 gene is amplified in several cancers, and SOX2 has been shown to be expressed in some tumors, but not in their surrounding tissue. Moreover, in some tumors, SOX2 expression has been shown to increase during tumor progression [14, 26, 29, 33, 39-41]. We suggest that two factors, which are not mutually exclusive, are likely to contribute to the apparent increase in SOX2 expression during tumor progression. First, there may be an increase in the number of SOX2-positive cells in the tumor population. If SOX2 is required for the tumor-initiating/cancer stem cell population, which is the case for at least some cancers, SOX2 levels may rise as the population of TIC increases during tumor progression. Second, increases in SOX2 levels in individual cells may contribute to tumorigenicity, but only when accompanied by corresponding changes in the expression of other genes that counterbalance the growth inhibitory effects of elevated SOX2. In this regard, two recent studies have shown that mutations in RB1 and p53, which occur in high risk prostate cancer patients, leads to significant elevation of SOX2 in an animal model of prostate cancer and in the androgen dependent prostatic tumor cell line LNCaP [120, 121]. Thus, the effects of SOX2 appear to be highly context-dependent, similar to other genes, notably TGFβ, which can act as a tumor suppressor or oncogene. Furthermore, we propose that identifying genes that permit SOX2 to contribute to tumorigenicity provides a novel strategy for identifying new therapeutic targets that can block the growth of SOX2-dependent tumors. In this regard, blocking the expression or function of genes that enable elevated SOX2 levels to promote tumor growth could convert the action of SOX2 from a growth promoter (oncogene) to a growth inhibitor (tumor suppressor gene). Given the difficulty of targeting transcription factors directly, this should be given serious consideration.
Exquisite control of SOX2 levels
The discussion in the previous section deals with an important property of SOX2 in cancer: its levels must be carefully controlled. This delicate balance was first identified in ESC. Both the knockdown of SOX2 and its overexpression block the self-renewal of ESC and lead to their differentiation [3, 122]. Remarkably, a 2-fold increase in SOX2, from an inducible transgene, is sufficient to rapidly induce the loss of self-renewal and trigger the differentiation of ESC [122]. More recently, we have shown that inducible overexpression of SOX2 in multiple tumor cells, including three PDAC cell lines, leads to growth inhibition [104, 114]. In the case of PDAC cells, knockdown as well as the overexpression of SOX2 in PDAC cell lines strongly reduces their growth both in vitro and in vivo [114]. Accordingly, we propose that SOX2 levels in actively proliferating tumor cells are optimized to maximize tumor growth: too little or too much SOX2 decreases tumor growth and alters cell behavior [104, 114].
The need to carefully control the levels of SOX2 raises a fundamental question, why do small changes in the levels of SOX2 exert such a profound change in cellular function? This question has been addressed in earlier review articles, which discussed why small changes in SOX2 levels disrupt the self-renewal and pluripotency of ESC [6, 123]. Therefore, only the main points are summarized here. Unbiased proteomic screens of the Sox2-interactome and genome wide binding studies of Sox2 in mouse ESC demonstrate that Sox2 is part of a highly interdependent transcriptional network that is interconnected at multiple levels [124]. Moreover, proteomic analysis of ESC undergoing differentiation indicate that the SOX2-interactome changes when ESC initiate differentiation [124, 125]. Analysis of the Sox2-interactome in ESC demonstrates that Sox2 physically associates with many other master regulators in ESC, including Oct4, and these master regulators associate with many of the same nuclear proteins. This creates a highly integrated transcriptional network. As predicted, knockdown of proteins that associate with Sox2 and other master regulators in ESC leads to the loss of self-renewal and pluripotency [125-127]. More surprising was the finding that Sox2 associates with > 50% of the genes that code for Sox2-associated proteins [124]. Furthermore, SOX2 associates with proteins in ESC that have prominent roles in signal transduction and DNA repair and replication. This is not only true in ESC, SOX2 has also been shown to associate with a diverse array of functionally distinct proteins in brain tumor cells [128, 129]. Thus, it is not surprising that small changes in SOX2 levels exert such profound effects over cell physiology. Given the need for careful regulation of SOX2, its expression must be controlled at many levels. In the next section, we describe an exquisite array of mechanisms that are used to precisely control the expression and function of SOX2.
Table 2: SOX2 expression in tumor-initiating cells, drug resistance, and tumor cell growth
Cancer Type |
Cancer Stem Cells/ Tumorigenicity |
Drug Resistance |
Alter Growth |
Bladder |
Hepburn et al., 2012 |
Hepburn et al., 2012 |
|
Breast |
Piva et al., 2014 |
Piva et al., 2014 |
Leis et al., 2012 |
Simões et al., 2011 |
|
Chen et al., 2008 |
|
Cervical |
Liu et al., 2014 * |
|
|
Colorectal |
Lundberg et al., 2016 |
|
|
Esophageal |
|
|
Gen et al., 2013 |
|
|
Bass et al., 2009 |
|
Ewing's sarcoma |
|
|
Ren et al., 2016 |
Gastric |
Tian et al., 2012 |
Tian et al., 2012 |
Hütz et al., 2013 |
|
Tian et al., 2014 |
Tian et al., 2014 |
|
|
|
Wang et al., 2015 |
|
Glioblastoma |
Jeon et al., 2011 |
Hagerstrand et al., 2011 |
Fang et al., 2011 |
|
Jeon et al., 2011 |
Cox et al., 2012 |
|
|
|
Alonso et al., 2011 |
|
|
|
Hagerstrand et al., 2011 |
|
|
|
Gangemi et al., 2009 |
|
Head and neck squamous cell carcinoma |
Lee et al., 2014 * |
Lee et al., 2014 |
|
Bourguignon et al., 2012 |
|
|
|
Lung adenocarcinoma |
Nakatsugawa et al., 2011 * |
|
|
Lung cancer, non-small cell |
Singh et al., 2012 |
Chou et al., 2013 |
Chou et al., 2013 |
Xiang et al., 2011 |
Singh et al., 2012 |
|
|
Lung cancer, small cell |
|
|
Rudin et al., 2012 |
Lung cancer, squamous cell |
Hussenet et al., 2010 |
|
Bass et al., 2009 |
|
|
Hussenet et al., 2010 |
|
Medulloblastoma |
Vanner et al., 2014 * |
|
Cox et al., 2012 |
Melanoma |
Santini et al., 2014 * |
|
Laga et al., 2010 |
Osteosarcoma |
Basu-Roy et al., 2015 |
|
Basu-Roy et al., 2012 |
Ovarian |
Ma et al., 2010 |
Ma et al., 2010 |
Wang et al., 2014 |
Yasuda et al., 2013 |
Yasuda et al., 2013 |
Yasuda et al., 2013 |
|
Bareiss et al., 2013 * |
Bareiss et al., 2013 |
|
|
Pancreas |
Herreros-Villanueva et al., 2013 |
Wuebben et al., 2016 |
Wuebben et al., 2016 |
Prostate |
Rybak et al., 2013 |
Li et al., 2014 |
Cox et al., 2012 |
|
Jia et al., 2011 |
Jia et al., 2011 |
|
Skin squamous-cell carcinoma |
Boumahdi et al., 2014 * |
|
|
* Limiting cell dilution assays performed in connection with SOX2
REGULATION OF SOX2 EXPRESSION
Transcriptional regulation of SOX2
The SOX2 gene in mammals, as well as birds, is located within a gene desert (a large genomic region largely devoid of other protein coding genes). Analysis of a 200 kb region of the chicken gene that surrounds the SOX2 single exon identified at least 27 distinct enhancers that are transcriptionally active for the regulation of SOX2 during neuro-sensory development in the chicken [130]. Eleven of the enhancers are distributed fairly evenly over a 97 kb region located upstream of the coding region of the SOX2 gene, and 16 enhancers are fairly evenly distributed over a 110 kb region downstream of the coding region of the SOX2 gene The large majority of the enhancers identified in the chicken genome are located in regions that are conserved in mammals [130]. Thus, it is likely that the mammalian SOX2 gene is also transcriptionally regulated by a large number of distinct distal enhancers during different stages of development. However, far more work will be needed to define the regulatory regions of mammalian SOX2 gene that are active in specific cell types. As discussed below, only three enhancers have been identified as functionally active in mammalian cells, one of which is located ~100 kb downstream of the Sox2 gene.
In mammalian cells, transcriptional regulation of the Sox2 gene, including enhancers that drive Sox2 expression, has been primarily studied in mouse ESC. In addition to the basal promoter of the Sox2 gene [131], early studies identified two enhancers, SRR1 and SRR2, which influence the activity of the Sox2 promoter [132]. SRR1 is located ~4kb upstream of the Sox2 transcription start site; whereas, SRR2 is located ~2.5 kb downstream of the 3’ end of the Sox2 coding region. Although SRR1 has been shown to be active in promoter/reporter gene constructs expressed in ESC, its impact on the expression of Sox2 in ESC is minimal when SRR1 is deleted from the endogenous Sox2 gene [133]. However, deleting a region -5.7 to -3.3 kb upstream of the Sox2 transcription start site, which contains SRR1, abolished expression of SOX2 in telencephalic neural stem cells and precursors during murine development [134, 135]. SRR2 is not only active in mouse ESC; it has been used to isolate human iPS cells [136]. For these studies, the SRR2 enhancer was multimerized (4 tandem repeats) and inserted into a lentiviral vectors which drives the expression of enhanced green fluorescence protein (EGFP) via a minimal promoter only when SRR2 is active. Subsequently, this lentiviral vector which drives EGFP was shown to be active in breast cancer cells [115]; and isolation of the subset of EGFP-expressing breast tumor cells were shown to exhibit enhanced tumorigenic potential, but, unexpectedly, only when NOD/SCID mice were engrafted with a large number of cells [137].
Several studies have examined the transcriptional machinery that regulates the activity of SRR2. The sequence of SRR2 contains adjacent HMG and POU motifs (referred to as an HMG/POU cassette) that have been shown to be essential for the activity of SRR2 in ESC and bind Sox2 and Oct4 in ESC [132, 138]. These studies led to the conclusion that Sox2 in combination with Oct4 contributes to the transcription of Sox2 in ESC. However, this may not be the only role of SRR2 in the transcription of Sox2. In fact, several recent studies lead us to suggest that SRR2 may also repress Sox2 transcription, especially during differentiation. First, as in the case of SRR1, deletion of SRR2 from the endogenous Sox2 gene did not significantly reduce Sox2 expression in ESC [133]. Even more suggestive of a repressive role for SRR2 is the finding that SRR2 is able to bind transcriptional repressors, such as p21, p27Kip1, and the p130/E2F4-SIN3A repressor complex, in neural stem cells and iPS cells undergoing differentiation [139, 140]. Consistent with these findings, Sox2 mRNA is elevated in Rb (p105) null and p130 (retinoblastoma family member) null MEFs and it is elevated in the pituitary tissue of Rb heterozygous mice [141]. Moreover, in pituitary tumors, loss of Rb or p130 has been linked to a defect in the repression of Sox2 expression [141]. Given the roles of p21, p27Kip1, and Rb proteins in the G1 cell cycle check point, Sox2 expression may be reduced in the G1 phase of the cell cycle. Thus, future studies should consider whether SOX2 expression is cell cycle regulated.
In ESC, a critical enhancer region (referred to as SCR - Sox2 control region) that is required for Sox2 transcription is located ~100 kb downstream of the Sox2 gene [133]. Previous studies predicted 10 enhancers surrounding the Sox2 gene, including two that overlapped SRR1 and SRR2. When tested in promoter/reporter gene constructs, three of the 10 putative enhancers, which are located 18, 107, and 111 kb downstream of the Sox2 gene, were found to drive the expression of the reporter gene more potently than SRR1 and SRR2 in ESC. More definitive results were obtained by generating deletions of these enhancers in one allele of the SOX2 gene using CRISPR based gene editing [133]. Deletion of SRR1, SRR2, or the enhancer located 18 kb downstream of SOX2 did not affect the expression of the targeted allele. In strong contrast, deletion of the SCR reduced expression of the targeted allele. (For these studies, expression of the targeted and non-targeted alleles was monitored separately by PCR in a heterozygous ESC line containing one allele from mouse strain Mus musculus and one allele from Mus castaneus.) Notably, targeting one Sox2 allele in ESC did not impact the maintenance of pluripotent ESC, due to upregulation of the non-targeted Sox2 allele. This finding and earlier studies involving Sox2 overexpression in ESC (see below) indicate that Sox2 influences its own expression in ESC by a feedback loop. Going forward, it will be important to determine whether the SCR, which is active in ESC, is also active in other SOX2-expressing cells, in particular SOX2-positive tumor cells. Thus far, only SRR2 has been reported to be active in SOX2-positive tumor cells.
Sox2 not only positively influences Sox2 expression in ESC when it is under expressed, it has the opposite effect when Sox2 is overexpressed in ESC. As mentioned earlier in this review, ESC engineered for inducible overexpression of Sox2 undergo differentiation when Sox2 is elevated by 2-fold or more. Interestingly, overexpression of exogenous Flag-tagged Sox2 in ESC reduces expression of endogenous Sox2 at the protein level [122] and at the transcriptional level [142]. Specifically, it was determined that elevation of exogenous Sox2 activated a negative feedback loop mediated at least in part by increased phosphorylation of AKT and one of its downstream targets, FoxO1, which regulates transcription of Sox2 [142]. When FoxO1 is phosphorylated, it translocates out of the nucleus, reducing Sox2 transcription. Thus in ESC, Sox2 can regulate its own expression at the transcriptional levels by both positive and negative feedback loops when Sox2 expression is too low and when Sox2 expression is too high, respectively. Importantly, overexpression of SOX2 in brain tumor cells and PDAC cells from an inducible promoter does not reduce expression of the endogenous SOX2 [104, 114], which suggests that the negative feedback loop is not active in at least some tumor cells. From the discussion in this section, it is evident that the transcriptional regulation of SOX2 has been extensively studied. However, there is far more to learn regarding how this gene is regulated at the transcriptional level.
MicroRNAs and SOX2 expression
A large body of data has implicated microRNAs (miRs) in the function of normal embryonic and adult cells, as well as diseased tissues, in particular cancer. More than 10 years ago, ChIP-Chip studies conducted by Boyer et al determined that SOX2 associates with the regulatory regions of many miR genes in human ESC [143]. This finding was extended by ChIP-seq analysis of Sox2 chromatin binding in mouse ESC [144]. More recently, Fang et al determined by ChIP-seq that SOX2 is bound to over 100 miR genes in a glioblastoma cell line [116]. Further study is expected to show that SOX2 regulates the transcription of a large number of miRs in a wide variety of SOX2-positive tumors. However, the specific miR genes regulated by SOX2 are expected to differ widely between tumor cell types due to differences in their transcriptional circuitries.
In addition to the regulation of miRs by SOX2, there is a growing list of miRs that are capable of regulating SOX2 at the post-transcriptional level. In the case of cancer, at least 18 miRs have been reported to regulate SOX2 expression in tumor cell lines (Figure 1A, Table 3). Of these, miR-145 has been implicated directly or indirectly in ESC and at least seven cancers, glioblastoma, prostate cancer, non-small cell lung carcinoma, Ewing sarcoma, hepatocellular carcinoma, PDAC, and urothelial carcinoma [145-152]. Interestingly, in glioblastoma, SOX2 and miR145 have been reported to form a negative feedback loop with one another (Figure 1A). In this tumor, SOX2 can associate with the gene regulatory regions of miR145, where it is believed to repress miR145 transcription; whereas miR145 reduces the expression of SOX2 by interfering with its translation [116]. In colorectal cancer, miR-200c and SOX2 also appear to regulate one another by a negative feedback loop [153].
Table 3: MicroRNAs regulating SOX2 in cancer
Tumor Type |
miR |
Effects Observed |
Reference |
Breast cancer |
miR-140 |
can target SOX2 3'UTR |
Zhang et al., 2012 |
altered expression alters SOX2 expression |
|||
miR-378 |
Enhances SOX2 expression indirctly |
Deng et al., 2013 |
|
Colorectal cancer |
miR-200c |
can target SOX2 3'UTR |
Lu et al., 2014 |
altered expression alters SOX2 expression |
|||
miR-638 |
can target SOX2 3'UTR |
Ma et al., 2014 |
|
altered expression alters SOX2 expression |
|||
miR-450b |
targets SOX2 directly |
Jin et al., 2016 |
|
miR-429 |
can target SOX2 3'UTR |
Li et al., 2013 |
|
altered expression alters SOX2 expression |
|||
Embryonal carcinoma |
miR-211 |
targets both SOX2 and SOX2OT |
Shafiee et al., 2016 |
Embryonic stem cells |
miR-145 |
targets SOX2 3'UTR upon differentiation |
Xu et al., 2009 |
Esophageal cancer |
miR-625 |
can target SOX2 3'UTR |
Wang et al., 2014 |
altered expression alters SOX2 expression |
|||
Ewing sarcoma |
miR-145 |
altered expression alters SOX2 expression |
Riggi et al., 2010 |
Gastric cancer |
miR-371 |
can target SOX2 3'UTR |
Li et al., 2016 |
altered expression alters SOX2 expression |
|||
miR-126 |
can target SOX2 3'UTR |
Otsubo et al., 2011 |
|
altered expression alters SOX2 expression |
|||
Glioblastoma |
miR-21 |
low miR-21/high SOX2 in one subgroup |
Sathyan et al., 2015 |
high miR-21/low SOX2 in different subgroup |
|||
miR-145 |
SOX2 and miR-145 regulate each other |
Fang et al., 2011 |
|
Glioma stem cells |
miR-218 |
elevated miR-218-5p reduced SOX2 |
Wu et al., 2016 |
miR-218-5p may not target SOX2 directly |
|||
miR-9* |
ID4 decreases miR-9* and increases SOX2 |
Jeon et al., 2011 |
|
SOX2 3'UTR activity elvated as ID4 increases |
|||
Hepatocellular carcinoma |
miR-126 |
can target SOX2 3'UTR |
Zhao et al., 2015 |
miR-145 |
|
Jia et al., 2012 |
|
Nasopharyngeal carcinoma |
miR-30a |
targets SOX2 3'UTR |
Qin et al., 2015 |
Neuroblastoma |
miR-340 |
can target SOX2 3'UTR |
Daa et al., 2013 |
miR-340 gene is methylated in this tumor |
|||
Non-small cell lung carcinoma |
miR-638 |
can target SOX2 3'UTR |
Xia et al., 2014 |
miR-145 |
altered expression alters SOX2 expression |
Campayo et al., 2013 |
|
Osteosarcoma |
miR-126 |
can target SOX2 3'UTR |
Yang et al., 2013 |
altered expression alters SOX2 expression |
|||
Pancreatic cancer |
miR-145 |
can target SOX2 |
Sureban et al., 2013 |
miR-1181 |
directly targets SOX2 |
Jiang et al., 2015 |
|
Prostate cancer |
miR-145 |
altered expression alters SOX2 expression |
Ozen et al., 2015 |
miR-34b |
unclear if it directly targets SOX2 |
Forno et al., 2015 |
|
Urothelial carcinoma |
miR-145 |
altered expression alters SOX2 expression |
Fujii et al., 2015 |
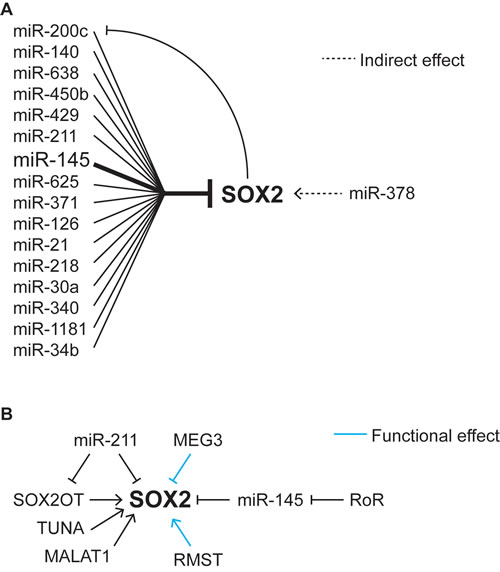
Figure 1: Regulation of SOX2 by miRNAs and lncRNAs. A. Effects of miRNAs on the expression of SOX2. B. Effects of long non-coding RNAs on SOX2 expression and function.
With one notable exception, SOX2-targeting miRs are associated with downregulation of SOX2. However, Deng et al reported that miR-378 increases SOX2 expression in breast cancer [154]. Although the types of breast cancer specimens examined were not described, they noted miR-378 was expressed at higher levels in breast tumor tissue than adjacent non-tumorigenic tissue. Rather than directly affecting SOX2, miR-378 targets vimentin, which influences SOX2 expression. The influence of vimentin was shown by overexpression of vimentin and the resulting downregulation of SOX2, but the mechanism by which vimentin regulates SOX2 was not determined. The association of SOX2 and miRs in specific cancers has been inferred predominately from the correlation between elevated SOX2 expression and low miR expression. In most studies, this association is supported by two additional lines of evidence, down regulation of SOX2 when the miR in question is ectopically elevated in tumor cell lines, and down regulation of a reporter gene construct, typically luciferase, containing a portion of the SOX2 3’UTR when the miR is ectopically expressed in tumor cell lines (Table 3).
For some cancers only a single miR has been implicated thus far in the regulation of SOX2. For example, miR-30a, when upregulated in nasopharyngeal carcinoma cells, appears to be capable of reducing SOX2 protein by targeting the 3’ UTR of SOX2 mRNA [155]. However, it is likely that SOX2 can be regulated by several miRs in the same cell type. In gastric carcinoma (see below), prostate cancer, and colorectal cancer, more than one miR has been implicated in the regulation of SOX2 (Table 3). In prostate cancer, SOX2 expression is associated with low expression of both miR-145 and miR-34b [147, 156]. However, unlike miR-145, which has been shown to target the 3’ UTR of the SOX2 transcript [145], it is unclear whether miR-34b targets SOX2 directly. As noted earlier, SOX2 is associated with a higher Gleason score in a subset of prostate tumors that express SOX2 [41]. In the case of colorectal cancer, miR-200c, miR-638, miR-450-5p, and miR-429 have been reported to regulate SOX2, but with different outcomes (Table 3). Lu et al reported that miR-200c, which is expressed at lower levels in colorectal specimens and highly metastatic colorectal cell lines, exhibits an inverse relationship with SOX2 [153]. Similarly, Ma et al has reported that miR-638, which is expressed at a lower level in colorectal tumors than adjacent non-tumorigenic tissue, is able to target SOX2 [157], and Jin et al reported that miR-450-5p, which is downregulated in recurrent colorectal cancer, is capable of downregulating SOX2 [158]. In contrast, Li et al reported that higher levels of miR-429 and lower levels of SOX2 mRNA in colorectal cancer are correlated with poor survival after surgery [159]. Interestingly, these investigators argued that high miR-429 expression exerts its anti-apoptotic function by downregulating SOX2. However, this is inconsistent with the apparent oncogenic role of SOX2 in a subgroup of colorectal cancer patients. In this regard, Lundberg et al reported that SOX2-positive colorectal cancer patients do not survive as long as SOX2-negative colorectal cancer patients, and this differential is larger for patients with BRAFV600E mutations who survive for substantially shorter periods than those who are SOX2-positive, but lack the BRAF mutation [160]. Going forward, it will be important to determine whether the levels of miR-429 are lower in colorectal cancer patients with BRAFV600E mutations. In view of our earlier proposal that SOX2 levels must be carefully titrated to maximize tumor growth, one of the mechanisms by which miR-429 promotes colorectal cancer may be to help maintain SOX2 within optimal levels for the BRAFV600E mutant subgroup of colorectal tumors.
More than one miR has also been reported to target SOX2 in gastric carcinoma. For miR-371-5p and miR-126, high miR expression is associated with low SOX2 expression. Li et al reported that miR-371-5p, which is elevated in gastric carcinoma compared to adjacent normal tissue, targets SOX2 [161]. In addition, these investigators reported that miR-371-5p downregulated a luciferase reporter gene construct containing a short sequence from the SOX2 3’UTR; whereas blocking expression of this miR in gastric tumor cell line increased SOX2 expression and cell proliferation in vitro. A similar conclusion was reached for miR-126. Otsubo et al reported that transiently elevating miR-126 in gastric cancer cell lines decreased SOX2 and increased cell proliferation in vitro [162]. They also demonstrated that miR-126 reduced the expression of a luciferase reporter gene containing regions taken from the SOX2 3’ UTR. Furthermore, these investigators reported low SOX2 expression and elevated miR-126 in some gastric tumor specimens, but the results reported do not show a clear pattern. Although, elevated miR-126 expression and low SOX2 expression was observed in several gastric cancer tumor specimens, low miR-126 expression was accompanied by low SOX2 expression in several other gastric tumor specimens. Thus, a larger number of tumor specimens will need to be evaluated to resolve the relationship between miR-126 and SOX2. In addition, the relationship between miR-126, SOX2, and patient survival remains to be determined. As noted earlier, high SOX2 in gastric cancer has been reported to be associated with longer patient survival [31, 42, 56]. Interestingly, there are reports that miR-126 can act as a tumor suppressor in other types of cancer. For example, Yang et al and Zhao et al reported that miR-126 behaves as a tumor suppressor in osteosarcoma and hepatocellular carcinoma, respectively, by targeting SOX2 [163, 164]. Additionally, Hamada et al reported that loss of miR-126 expression is observed in invasive PDAC [165].
Although miRs are recognized as important regulators of SOX2 expression, two important issues need to be considered. First, unless the cell of origin and its expression of miRs have been determined, it remains to be determined whether the miR in question has, in fact, been lost during tumor progression. Second, the full spectrum of SOX2-targeting miRs is likely to be far greater than those already identified.
Long non-coding RNAs and SOX2 expression
In addition to miRs, several long non-coding RNAs (lncRNAs) have been reported to influence the levels of SOX2 in tumor cells (Figure 1B). LncRNAs are a class of RNAs greater than 200 nucleotides that lack protein-coding sequences. They are transcribed by RNA polymerase II and they are spliced, 5’ capped, and 3’ polyadenylated. The human genome contains several thousand lncRNAs, and there is growing evidence that many play major roles in gene regulation by influencing chromatin structure, gene transcription, and processing of mRNA [166]. More recently, several lncRNAs have been implicated in the regulation of SOX2 expression and its transcriptional activity. The first direct link between SOX2 and lncRNAs was the discovery that the single exon SOX2 gene is embedded within an intron of a multi-exon lncRNA gene known as SOX2 overlapping transcript (SOX2OT, also known as non-protein-coding RNA 43) [167]. Like SOX2 itself, SOX2OT orthologues are expressed widely in other vertebrates, including in mouse, chicken, and zebrafish. SOX2 and SOX2OT are both transcribed in the same direction. SOX2OT is reported to possess at least 10 exons with up to four different transcription start sites. Through use of alternative transcription start sites and alternative splicing at least 8 splice variants of SOX2OT can be generated [168, 169].
SOX2 and SOX2OT have been shown to be co-expressed in ESC, as well as breast, lung, brain, and esophageal tumors [170-174]. In each of these cancers, more than one splice variant is expressed, and the splice variants expressed differ between different cancers. SOX2 and SOX2OT are also likely to be co-expressed in hepatocellular carcinoma. Separate studies have reported that expression of SOX2 and SOX2OT in hepatocellular carcinoma is each associated with poor prognosis [175, 176]. Although the mechanistic relationship between SOX2 expression and SOX2OT remains to be determined, several studies support the conclusion that SOX2OT lncRNA contributes to the expression of SOX2. Knockdown of SOX2OT by siRNA in the lung adenocarcinoma cell line A549 reduced the expression of SOX2 transcripts [173]. Conversely, forced overexpression of SOX2OT in the breast tumor cell line MDA-MB-231 increased the expression of SOX2 transcripts and protein [171]. Intriguingly, SOX2 and SOX2OT expression may both be related by at least one miR. miR-211 has been reported to target the same sequence in transcripts of SOX2OT and SOX2 and lead to their downregulation when miR-211 is overexpressed in the human embryonal carcinoma cell line NT-2 [177]. Thus far, only miR-211 has been reported to downregulate SOX2 and SOX2OT. Future studies should examine whether other miRs that have been shown to target SOX2 (Table 3) also target one or more of the SOX2OT splice variants.
In addition to SOX2OT, several other lncRNAs have been directly implicated in the expression of SOX2. The lncRNA TUNA (Tcl1 Upstream Neuron-Associated), which can form a complex with three RNA-binding proteins, has been shown by performing chromatin isolation via RNA purification (ChIRP) to associate with the Sox2 promoter in mouse ESC [178]. Furthermore, knockdown of TUNA by shRNA reduced the expression of Sox2 and led to the differentiation of mouse ESC. Interestingly, TUNA and Sox2 are also co-expressed in the brain [178]. Thus, it will be interesting to determine whether TUNA is expressed in glioblastoma and medulloblastoma and other SOX2-positive tumors, where it may also contribute to SOX2 expression. However, further study will be needed to determine how TUNA influences SOX2 expression.
The lncRNA MALAT1 (Metastasis-Associated Lung Adenocarcinoma Transcript 1) also appears to influence the expression of SOX2. MALAT1 has been shown to be expressed in the glioma tumor cell line SHG139S and in two pancreatic tumor cell lines, AsPC1 and CFPAC-1 [179, 180]. Knockdown of MALAT1 in each of these tumor cell lines reduced the expression of SOX2. However, it is unclear whether the effect of MALAT1 on SOX2 in these tumor cells is direct or indirect. Equally interesting is the report that lncRNA RoR supports SOX2 expression by functioning as a miRNA sponge. Specifically, RoR helps maintain SOX2 expression by serving as an RNA decoy that competes for miRs (e.g. miR145) that target SOX2 expression [181].
LncRNAs also appear to regulate the transcriptional activity of SOX2. The lncRNA RMST (RhabdoMyoSarcoma 2-associated Transcript) has been reported to coregulate SOX2 target genes during neurogenesis [182]. RMST interacts physically with SOX2 and it promotes the binding of SOX2 to the regulatory regions of neurogenic transcription factors. Impressively, knockdown of RMST reduces SOX2 association with approximately half of its chromatin binding sites [182]. Although RMST appears to enhance the transcriptional activity of SOX2, at least one lncRNA, MEG3 (Maternally Expressed Gene 3) that physically associates with SOX2, can interfere with its action. Knockdown of MEG3 has been reported to increase the association of SOX2 with the BMP4 gene, which is inhibited by SOX2, and decrease the transcription of this gene [183]. Thus far, the domains of SOX2 that associate with these lncRNA have not been determined, nor has it been determined how they influence the transcriptional activity of SOX2. Going forward, it will be interesting to determine whether RMST and MEG3 are commonly expressed in SOX2-positive tumors, including glioblastoma and medulloblastoma. Moreover, MEG3 has been shown to be expressed in PDAC cell lines, where its knockdown led to a reduction in cell number in vitro [184]. Thus, it will be interesting to determine whether knockdown of MEG3 alters the function of SOX2 in PDAC cells.
Post-translational modifications of SOX2
Another important mechanism used to regulate SOX2, including its transcriptional activity, nuclear localization, and stability, is post-translational modifications, which include phosphorylation, glycosylation, sumoylation, methylation, ubiquitination, and acetylation. Thus far, nearly all reports of SOX2 post-translational modifications have been conducted with mouse pluripotent stem cells and mostly with ESC. In the future, it will be important to characterize the post-translational modifications of SOX2 in tumor cells. Recently, one study described a SOX2 post-translational modification (phosphorylation) in human lung squamous cell carcinoma cells [185]. For the purposes of clarity, and to avoid confusion, the reader is reminded that human SOX2 and mouse Sox2 differ in length by 2 amino acids: 317 amino acids and 319 amino acids, respectively, due to a two amino acid insertion beginning at residue 23 in mouse Sox2 (Figure 2, Table 4).
Table 4: Post-translations modifications of mouse Sox2
Site |
Modification(s) |
Effects Observed |
Modifying enzyme |
Reference |
S39 |
Phosphorylation |
Reduces reprogramming |
Cdk2 |
Ouyang et al., 2015 |
K75 |
Acetylation |
Low level acetylation increases reprogramming. Nuclear export? |
likely p300/CBP |
Baltus et al., 2009 |
Sirt1 (deacetylate) |
||||
S83 |
Phosphorylation |
ND |
ND |
Malak et al., 2015 |
R113 |
Methylation |
Increased transcriptional activity, increased self-association |
CARM1 |
Zhao et al., 2011 |
T118 |
Phosphorylation |
Increased transcriptional activity, increased stability |
AKT |
Jeong et al., 2010 |
Blocks monomethylation at K119 |
ND |
Fang et al., 2014 |
||
K119 |
Methylation |
Increases Sox2 ubiquitination |
set7 |
Fang et al., 2014 |
K123 |
Ubiquitination |
Targets Sox2 for proteasomal degradation |
Ube2s |
Wang et al., 2016 |
K247 |
Sumoylation |
Block Sox2's transcription of Fgf2 and Nanog |
ND |
Tsuruzoe et al., 2006 |
S248 |
Phosphorylation |
Alter transcriptional activity |
ND |
Ouyang et al., 2015 |
O-GlcNAcylation |
Alter transcriptional activity |
ND |
Myers et al., 2011 |
|
Jang et al., 2012 |
||||
S251 |
Phosphorylation |
Regulate sumoylation |
ND |
Tsuruzoe et al., 2006 |
S252 |
Phosphorylation |
Regulate sumoylation, reduces reprogramming |
ND |
Tsuruzoe et al., 2006 |
S253 |
Phosphorylation |
Regulate sumoylation |
Cdk2 |
Tsuruzoe et al., 2006 |
T258 |
O-GlcNAcylation |
ND |
ND |
Myers et al., 2011 |
Jang et al., 2012 |
||||
T118* (H) |
Phosphorylation |
Increase SOX2 transcriptional activity |
PKCι |
Justilien et al., 2014 |
The sequence numbering shown refers to mouse Sox2 except for human SOX2 T118* (where indicated).
Not determined (ND).
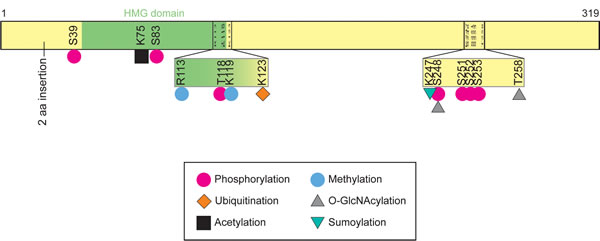
Figure 2: Mouse Sox2 structure and post-translational modification sites. Illustration of the 319 amino acid (aa) Mus musculus Sox2 and established post-translational modifications. Positions of comparable post-translational modifications in human SOX2 would differ due to the 2 aa inserted at residue 23 in mouse Sox2. Human SOX2 totals 317 aa; whereas mouse Sox2 totals 319 aa.
The most common and diverse post-translational modification reported for Sox2 is phosphorylation. Sox2 phosphorylation influences its transcriptional activity and its stability. Studies by several research teams have shown that Sox2 can be phosphorylated in vivo on at least 6 serine residues (mouse S39, S83, S248, S251, S252, S253) and two threonine resides (mouse Sox2 T118 and human SOX2 T118) (Figure 2) [186-188]. Sox2 has also been reported to be phosphorylated on tyrosine residues when ectopically expressed in 293T cells, which express little if any endogenous SOX2 [186]. It remains to be determined whether these tyrosine residues are phosphorylated in cells that endogenously express SOX2. The kinases responsible for serine phosphorylation of SOX2 have only begun to be determined. For example, Cdk2 can phosphorylate both S39 and S253 in vitro [186]. Modifying both serine residues by conversion to alanine (S39A, S253A) reduces the ability of mutant Sox2 to reprogram somatic cells into iPS cells. Surprisingly, even though S39 and S253 are phosphorylated in mouse ESC, and most highly phosphorylated during mitosis, a mutant form of Sox2 (S39A/S253A) is able to support the self-renewal of mouse ESC when endogenous Sox2 is depleted [186]. It is possible that at different levels of Sox2, phosphorylation of these serine residues is dispensable.
Serine 248 of mouse Sox2 has been reported to be phosphorylated in mouse ESC. Phosphorylation of this serine is likely to have a significant role in the function of SOX2, because this serine, along with T258 in mouse Sox2 (see below) can also be modified by O-GlcNAcylation [189, 190]. Studies conducted thus far suggest that modification of S248 in mouse Sox2 may alter the transcriptional activity of Sox2, but further work will be needed to properly dissect the impact of phosphorylation and O-GlcNAcylation of Sox2 S248. Similar to this serine residue, the serine triplet S249-S250-S251 in human SOX2 appears to regulate another post-translational modification, sumoylation. Human SOX2 has been shown to be sumoylated on K245 and K247 in mouse Sox2 [188, 191]. Importantly, sumoylation of K245 is abolished in the SOX2 mutant (S249A-S250A-S251A) [191]. Thus, phosphorylation of one or more serine residues in the triplet appears to serve as a priming step in the subsequent sumoylation of SOX2. Although the roles of SOX2 sumoylation remain to be fully characterized, sumoylation of mouse Sox2 has been reported to reduce the ability of Sox2 to increase the transcription of Fgf4 and Nanog [188]. In the future, it will be important to determine whether this reduction in transcriptional activity is restricted to a small number of genes or is true for most Sox2-regulated genes.
The kinases that phosphorylate threonine residues of mouse Sox2 T118 and humanT118 have been identified. These threonine residues are both located within a consensus nuclear localization sequence and the HMG domain of SOX2, which is responsible for DNA binding. Phosphorylation of human SOX2 on T118 is mediated by PKCβ [185]. Phosphorylation of this threonine is associated with an increase in the transcriptional activity of SOX2, which was shown using SOX2 mutants. The transcriptional activity observed with wild-type SOX2 was not observed with the SOX2 mutant (T118A), but exhibited by the SOX2 phospho-mimic mutant (SOX2-T118D). Interestingly, human SOX2-T118A does not appear to alter SOX2 stability. In stark contrast, the mouse mutant Sox2-T118A exhibits reduced stability. Mouse Sox2 can be phosphorylated on T118 by AKT in mouse ESC [192]. Phosphorylation of this serine not only increases SOX2 stability, it also increases its transcriptional activity. Remarkably, phosphorylation of mouse T118 blocks the monomethylation of Sox2 on the adjacent K119 by the methyltransferase set7 [193]. Methylation of K119 induces the ubiquitination of Sox2 by the E3 ligase WWP2 and the degradation of Sox2 [193]. Thus, the antagonistic phosphorylation-methylation switch mediated by T118-K119 alters the transcription activity and stability of Sox2, respectively. Importantly, we are not aware of any studies reporting that AKT inhibitors reduce the stability of SOX2 in tumor cells. This warrants attention given the use of AKT inhibitors in many cancer clinical trials. As discussed earlier, AKT has been implicated in a negative feedback loop that influences the transcription of the Sox2 gene in ESC [142].
Sox2 can also be ubiquitinated on lysine K123, which is located just beyond the C-terminal portion of the DNA binding domain of Sox2 (the HMG domain). The ubiquitin-conjugating enzyme E2S (Ube2s) mediates K11-linked polyubiquitination of Sox2 at this site [194]. When ubiquitinated on K123, Sox2 is targeted for proteasome-mediated degradation. The comparable lysine of human SOX2 is K121. Although SOX2-T118A does not appear to be less stable than wild-type SOX2 in lung squamous cell carcinoma cells, it is possible in some tumor cells that phosphorylation of T118 (human SOX2) may block ubiquitination of SOX2 at K121, as was discussed above for T118 (mouse Sox2), and its influence on the methylation of K119 and the subsequent degradation of Sox2.
In addition to Sox2 methylation and O-GlcNAcylation discussed above, SOX2 can also be methylated and O-GlcNAcylated on other amino acids. Sox2 T258 has been shown to be modified by O-GlcNAcylation in mouse ESC. Thus far, the function of T258 O-GlcNAcylation has only been studied in the context of double and triple mutants (T258A/S259A and S248A/T258A/S259A). The double mutant reduced the ability of Sox2 to reprogram somatic cells to iPS cells; whereas the triple mutant did not [190]. Additionally, Sox2 can be methylated on R113 by the arginine methyltransferase CARM1, which increases SOX2 self-association and increases the transcriptional activity of Sox2 [195]. However, further study will be needed to determine whether the increase in Sox2 transcriptional activity is linked to its self-association. Furthermore, it is possible that methylation of Sox2 R113 increases its association with other Sox family members [195, 196]. R113, which is located within the HMG domain of Sox2, is located within a second Sox2 nuclear localization sequences (NLS2). However, the Sox2-R113K mutant, which cannot be methylated, did not alter the subcellular location or the stability of Sox2 [195].
Finally, Sox2 has been shown to be acetylated within its DNA binding domain on K75 in vitro [197]. Although the acetyltransferase that acetylates Sox2 in vivo has not been determined definitively, p300/CBP is a likely candidate, especially since Sox2 can be acetylated by p300/CBP on K75 in vitro [197]. Moreover, Sox2 has been shown to recruit p300 to the Fgf4 enhancer in ESC [198]. Blocking acetylation of Sox2 in ESC, as shown with the Sox2-K75A mutant, led to retention of Sox2 in the nucleus and maintenance of its transcriptional activity; whereas, the acetyl-mimic Sox2-K75Q mutant, associates with the nuclear export machinery, specifically Crm1 [197]. Other studies indicate that Sox2 can be deacetylated by Sirt1, a member of the sirtuin family of NAD-dependent protein deacetylases [199, 200]. Acetylation of Sox2 not only affects its function in ESC, a low level of Sox2 acetylation enhances reprogramming of somatic cells to iPS cells [200].
It is clear from the discussion in this section that post-translational modifications of SOX2 dramatically alter its function, and undoubtedly play key roles in helping to adjust the function and levels of SOX2 needed to support cellular activity. However, many questions remain to be addressed. In addition to the enzymes responsible for creating the variety of SOX2 post-translation modifications, enzymes that remove some of these modifications of SOX2 have not been identified. Besides phosphatases, likely candidates include deubiquitinating enzymes (DUBs). Interestingly, proteomic analysis of the SOX2-interactome indicates that SOX2 associates with several DUBs that exert important roles in tumor cells, including USP9X, USP7, USP15, USP24, and USP34 [125, 128, 201]. In the future, defining the roles of each of the SOX2 modifications and the enzymes involved in tumor cells may provide valuable insights into possible strategies for targeting SOX2 in a large number of cancers (Tables 1-2). An equally important question that warrants careful attention is the extent to which any given SOX2 molecule is simultaneously modified by more than one post-translational modification. By analogy to the histone code, a “SOX2 code” of post-translational modifications is likely to play a key role in orchestrating the formation of the multitude of SOX2-protein complexes (SOX2-interactome) needed to properly control the level, transcriptional activity, subcellular localization, and stability of SOX2.
CONCLUSIONS AND FUTURE PERSPECTIVES
It is evident from work conducted over the past 15 years that SOX2 is far from monolithic. Life is not possible without expression of SOX2, in particular during embryogenesis, but SOX2 also has a dark side. SOX2 is expressed in at least 25 different cancers and in many of these cancers SOX2 expression has been directly implicated in increased tumor growth, metastasis, drug resistance, and poor survival. Thus, targeting SOX2 expression or its mode of action could improve the survival of patients with some of the most difficult to treat cancers.
For the many cancers where SOX2 expression poses a serious threat, much more work needs to be conducted to understand mechanistically how SOX2 contributes to the biology of the tumor. This is particularly evident for the transcriptional regulation of SOX2, which has been woefully understudied in cancer biology. Studies conducted primarily in mouse ESC have identified several essential distal enhancers that control SOX2 transcription. However, the roles of these enhancers in SOX2 expression in human tumor cells are poorly understood. In addition, ChIP-seq has only been used in two tumor studies to identify genome-wide binding of SOX2 [116, 202]. A similar situation exists regarding the roles of SOX2 post-translational modifications, which, thus far, have only been examined in one human tumor study. Interestingly, studies conducted in ESC indicate that there is significant cross-talk between different post-translation modifications, but this too has not been explored in human tumor cells.
In contrast to our lack of understanding of SOX2 transcriptional regulation and post-translational modification in human cancer, a significant amount of work has focused on miRs and SOX2 expression in many cancers. Similarly, several lncRNAs have been shown to influence the expression and function of SOX2. Particularly interesting is the ability of SOX2OT to regulate SOX2 expression. However, far more work needs to be performed before the impact of lncRNAs on SOX2 expression is understood in human cancer. Similarly, efforts are needed to determine whether SOX2 regulates the expression of lncRNAs. Given the large number of genes bound by SOX2, it would not be surprising to find that SOX2 influences the expression of many lncRNA, including some that regulate SOX2 expression and function.
Intriguingly, recent studies indicate that small changes in the levels of SOX2 can radically affect tumor cell behavior. Thus far, both small increases and small decreases in SOX2 expression have been shown to adversely influence tumor cell growth in at least five different types of cancer. In the future, it will be important to determine how small decreases as well as small increases in SOX2 expression adversely affect tumor cell proliferation. Finally, we discussed potential problems using cell lines engineered to stably overexpress SOX2. It is evident that the initial response of many, if not most, tumor cells is growth inhibition when SOX2 levels are elevated. Thus, we emphasize the benefit of using cell lines engineered for inducible overexpression of SOX2, both in vitro and in vivo. Similarly, we have pointed out that markers used in many studies to isolate cancer stem cells may lead to erroneous conclusions and over estimation of the size of the tumor-initiating population.
In conclusion, we believe the work performed thus far indicates that the expression and function of SOX2 in cancer clearly warrants further study. Although significant progress has been made during the past 10 years, far too many questions remain to be answered about SOX2 and this deadly disease. Addressing these questions is expected to lead to new insights into the functions of SOX2 in cancer, which will help design new strategies for more effectively treating cancer.
ACKNOWLEDGMENTS
Phillip Wilder is thanked for reading this review and providing helpful comments. Research in this laboratory was supported in part by grants from the Nebraska Department of Health and Human Services (2016-42 and Stem Cell 2015-01), the Pediatric Cancer Research Program, the Regenerative Medicine Program and the Fred & Pamela Buffett Cancer Center. Core facilities used by this laboratory were supported in part by a Cancer Center Support Grant from the National Cancer Institute (P30CA036727).
ConflictS of Interest
There is no conflict of interest.
References
1. Yuan H, Corbi N, Basilico C, Dailey L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes & development. 1995; 9: 2635-2645.
2. Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes & development. 2003; 17: 126-140.
3. Chew JL, Loh YH, Zhang W, Chen X, Tam WL, Yeap LS, Li P, Ang YS, Lim B, Robson P, Ng HH. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Molecular and cellular biology. 2005; 25: 6031-6046.
4. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126: 663-676.
5. Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clinical and translational medicine. 2014; 3: 19.
6. Rizzino A, Wuebben EL. Sox2/Oct4: A delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochimica et biophysica acta. 2016; 1859: 780-791.
7. Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell stem cell. 2013; 12: 15-30.
8. Alonso MM, Diez-Valle R, Manterola L, Rubio A, Liu D, Cortes-Santiago N, Urquiza L, Jauregi P, Lopez de Munain A, Sampron N, Aramburu A, Tejada-Solis S, Vicente C, et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PloS one. 2011; 6: e26740.
9. Belotte J, Fletcher NM, Alexis M, Morris RT, Munkarah AR, Diamond MP, Saed GM. Sox2 gene amplification significantly impacts overall survival in serous epithelial ovarian cancer. Reproductive sciences (Thousand Oaks, Calif.). 2015; 22: 38-46.
10. Gen Y, Yasui K, Zen Y, Zen K, Dohi O, Endo M, Tsuji K, Wakabayashi N, Itoh Y, Naito Y, Taniwaki M, Nakanuma Y, Okanoue T, et al. SOX2 identified as a target gene for the amplification at 3q26 that is frequently detected in esophageal squamous cell carcinoma. Cancer genetics and cytogenetics. 2010; 202: 82-93.
11. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature genetics. 2012; 44: 1111-1116.
12. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nature genetics. 2009; 41: 1238-1242.
13. Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D, Martinet N, Thibault C, Huelsken J, Brambilla E, du Manoir S. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PloS one. 2010; 5: e8960.
14. Annovazzi L, Mellai M, Caldera V, Valente G, Schiffer D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer genomics & proteomics. 2011; 8: 139-147.
15. Long KB, Hornick JL. SOX2 is highly expressed in squamous cell carcinomas of the gastrointestinal tract. Human pathology. 2009; 40: 1768-1773.
16. Wilbertz T, Wagner P, Petersen K, Stiedl AC, Scheble VJ, Maier S, Reischl M, Mikut R, Altorki NK, Moch H, Fend F, Staebler A, Bass AJ, et al. SOX2 gene amplification and protein overexpression are associated with better outcome in squamous cell lung cancer. Modern pathology. 2011; 24: 944-953.
17. Freier K, Knoepfle K, Flechtenmacher C, Pungs S, Devens F, Toedt G, Hofele C, Joos S, Lichter P, Radlwimmer B. Recurrent copy number gain of transcription factor SOX2 and corresponding high protein expression in oral squamous cell carcinoma. Genes, chromosomes & cancer. 2010; 49: 9-16.
18. Sattler HP, Lensch R, Rohde V, Zimmer E, Meese E, Bonkhoff H, Retz M, Zwergel T, Bex A, Stoeckle M, Wullich B. Novel amplification unit at chromosome 3q25-q27 in human prostate cancer. The Prostate. 2000; 45: 207-215.
19. Schrock A, Goke F, Wagner P, Bode M, Franzen A, Braun M, Huss S, Agaimy A, Ihrler S, Menon R, Kirsten R, Kristiansen G, Bootz F, et al. Sex determining region Y-box 2 (SOX2) amplification is an independent indicator of disease recurrence in sinonasal cancer. PloS one. 2013; 8: e59201.
20. Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, Rodriguez-Gil Y, Martinez MA, Hernandez L, Hardisson D, Reis-Filho JS, Palacios J. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Modern pathology. 2007; 20: 474-481.
21. Schmitz M, Temme A, Senner V, Ebner R, Schwind S, Stevanovic S, Wehner R, Schackert G, Schackert HK, Fussel M, Bachmann M, Rieber EP, Weigle B. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. British journal of cancer. 2007; 96: 1293-1301.
22. Phi JH, Park SH, Kim SK, Paek SH, Kim JH, Lee YJ, Cho BK, Park CK, Lee DH, Wang KC. Sox2 expression in brain tumors: a reflection of the neuroglial differentiation pathway. The American Journal of Surgical Pathology. 2008; 32: 103-112.
23. Sholl LM, Long KB, Hornick JL. Sox2 expression in pulmonary non-small cell and neuroendocrine carcinomas. Applied Immunohistochemistry & Molecular Morphology. 2010; 18: 55-61.
24. Yuan P, Kadara H, Behrens C, Tang X, Woods D, Solis LM, Huang J, Spinola M, Dong W, Yin G, Fujimoto J, Kim E, Xie Y, et al. Sex determining region Y-Box 2 (SOX2) is a potential cell-lineage gene highly expressed in the pathogenesis of squamous cell carcinomas of the lung. PloS one. 2010; 5: e9112.
25. Laga AC, Zhan Q, Weishaupt C, Ma J, Frank MH, Murphy GF. SOX2 and nestin expression in human melanoma: an immunohistochemical and experimental study. Experimental dermatology. 2011; 20: 339-345.
26. Ye F, Li Y, Hu Y, Zhou C, Hu Y, Chen H. Expression of Sox2 in human ovarian epithelial carcinoma. Journal of cancer research and clinical oncology. 2011; 137: 131-137.
27. Zhang J, Chang DY, Mercado-Uribe I, Liu J. Sex-determining region Y-box 2 expression predicts poor prognosis in human ovarian carcinoma. Human pathology. 2012; 43: 1405-1412.
28. Sanada Y, Yoshida K, Ohara M, Oeda M, Konishi K, Tsutani Y. Histopathologic evaluation of stepwise progression of pancreatic carcinoma with immunohistochemical analysis of gastric epithelial transcription factor SOX2: comparison of expression patterns between invasive components and cancerous or nonneoplastic intraductal components. Pancreas. 2006; 32: 164-170.
29. Jia X, Li X, Xu Y, Zhang S, Mou W, Liu Y, Liu Y, Lv D, Liu CH, Tan X, Xiang R, Li N. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. Journal of molecular cell biology. 2011; 3: 230-238.
30. Li XL, Eishi Y, Bai YQ, Sakai H, Akiyama Y, Tani M, Takizawa T, Koike M, Yuasa Y. Expression of the SRY-related HMG box protein SOX2 in human gastric carcinoma. International journal of oncology. 2004; 24: 257-263.
31. Chen Y, Huang Y, Zhu L, Chen M, Huang Y, Zhang J, He S, Li A, Chen R, Zhou J. SOX2 inhibits metastasis in gastric cancer. Journal of cancer research and clinical oncology. 2016; 142: 1221-1230.
32. Ren C, Ren T, Yang K, Wang S, Bao X, Zhang F, Guo W. Inhibition of SOX2 induces cell apoptosis and G1/S arrest in Ewing’s sarcoma through the PI3K/Akt pathway. J Exp Clin Cancer Res. 2016; 35:44.
33. Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, Shang Y. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. The Journal of biological chemistry. 2008; 283: 17969-17978.
34. Tian Y, Jia X, Wang S, Li Y, Zhao P, Cai D, Zhou Z, Wang J, Luo Y, Dong M. SOX2 oncogenes amplified and operate to activate AKT signaling in gastric cancer and predict immunotherapy responsiveness. Journal of cancer research and clinical oncology. 2014; 140: 1117-1124.
35. Bourguignon LY, Wong G, Earle C, Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. The Journal of biological chemistry. 2012; 287: 32800-32824.
36. Gure AO, Stockert E, Scanlan MJ, Keresztes RS, Jager D, Altorki NK, Old LJ, Chen YT. Serological identification of embryonic neural proteins as highly immunogenic tumor antigens in small cell lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97: 4198-4203.
37. Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, Kushida M, Head R, Morrissy S, Zhu X, Aviv T, Voisin V, Clarke ID, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer cell. 2014; 26: 33-47.
38. Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, Barredo I, Lopez-Ruiz JA, Zabalza I, Kypta R, Vivanco M. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO molecular medicine. 2014; 6: 66-79.
39. Ma YH, Mentlein R, Knerlich F, Kruse ML, Mehdorn HM, Held-Feindt J. Expression of stem cell markers in human astrocytomas of different WHO grades. Journal of neuro-oncology. 2008; 86: 31-45.
40. Wang Q, He W, Lu C, Wang Z, Wang J, Giercksky KE, Nesland JM, Suo Z. Oct3/4 and Sox2 are significantly associated with an unfavorable clinical outcome in human esophageal squamous cell carcinoma. Anticancer Research. 2009; 29: 1233-1241.
41. Kregel S, Kiriluk KJ, Rosen AM, Cai Y, Reyes EE, Otto KB, Tom W, Paner GP, Szmulewitz RZ, Vander Griend DJ. Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PloS one. 2013; 8: e53701.
42. Wang S, Tie J, Wang R, Hu F, Gao L, Wang W, Wang L, Li Z, Hu S, Tang S, Li M, Wang X, Nie Y, et al. SOX2, a predictor of survival in gastric cancer, inhibits cell proliferation and metastasis by regulating PTEN. Cancer letters. 2015; 358: 210-219.
43. Otsubo T, Akiyama Y, Yanagihara K, Yuasa Y. SOX2 is frequently downregulated in gastric cancers and inhibits cell growth through cell-cycle arrest and apoptosis. British journal of cancer. 2008; 98: 824-831.
44. Tsukamoto T, Mizoshita T, Mihara M, Tanaka H, Takenaka Y, Yamamura Y, Nakamura S, Ushijima T, Tatematsu M. Sox2 expression in human stomach adenocarcinomas with gastric and gastric-and-intestinal-mixed phenotypes. Histopathology. 2005; 46: 649-658.
45. Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Ioue Y, Miki C, Kusunoki M. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Annals of surgical oncology. 2009; 16: 3488-3498.
46. Wang X, Liang Y, Chen Q, Xu HM, Ge N, Luo RZ, Shao JY, He Z, Zeng YX, Kang T, Yun JP, Xie F. Prognostic significance of SOX2 expression in nasopharyngeal carcinoma. Cancer investigation. 2012; 30: 79-85.
47. Sholl LM, Barletta JA, Yeap BY, Chirieac LR, Hornick JL. Sox2 protein expression is an independent poor prognostic indicator in stage I lung adenocarcinoma. The American Journal of Surgical Pathology. 2010; 34: 1193-1198.
48. Chou YT, Lee CC, Hsiao SH, Lin SE, Lin SC, Chung CH, Chung CH, Kao YR, Wang YH, Chen CT, Wei YH, Wu CW. The emerging role of SOX2 in cell proliferation and survival and its crosstalk with oncogenic signaling in lung cancer. Stem cells (Dayton, Ohio). 2013; 31: 2607-2619.
49. Lee SH, Oh SY, Do SI, Lee HJ, Kang HJ, Rho YS, Bae WJ, Lim YC. SOX2 regulates self-renewal and tumorigenicity of stem-like cells of head and neck squamous cell carcinoma. British journal of cancer. 2014; 111: 2122-2130.
50. Sun C, Sun L, Li Y, Kang X, Zhang S, Liu Y. Sox2 expression predicts poor survival of hepatocellular carcinoma patients and it promotes liver cancer cell invasion by activating Slug. Medical oncology. 2013; 30: 503.
51. Du L, Yang Y, Xiao X, Wang C, Zhang X, Wang L, Zhang X, Li W, Zheng G, Wang S, Dong Z. Sox2 nuclear expression is closely associated with poor prognosis in patients with histologically node-negative oral tongue squamous cell carcinoma. Oral oncology. 2011; 47: 709-713.
52. Wang X, Ji X, Chen J, Yan D, Zhang Z, Wang Q, Xi X, Feng Y. SOX2 enhances the migration and invasion of ovarian cancer cells via Src kinase. PloS one. 2014; 9: e99594.
53. Neumann J, Bahr F, Horst D, Kriegl L, Engel J, Luque RM, Gerhard M, Kirchner T, Jung A. SOX2 expression correlates with lymph-node metastases and distant spread in right-sided colon cancer. BMC cancer. 2011; 11:518.
54. Han X, Fang X, Lou X, Hua D, Ding W, Foltz G, Hood L, Yuan Y, Lin B. Silencing SOX2 induced mesenchymal-epithelial transition and its expression predicts liver and lymph node metastasis of CRC patients. PloS one. 2012; 7: e41335.
55. Saigusa S, Mohri Y, Ohi M, Toiyama Y, Ishino Y, Okugawa Y, Tanaka K, Inoue Y, Kusunoki M. Podoplanin and SOX2 expression in esophageal squamous cell carcinoma after neoadjuvant chemo-radiotherapy. Oncology reports. 2011; 26: 1069-1074.
56. Zhang X, Yu H, Yang Y, Zhu R, Bai J, Peng Z, He Y, Chen L, Chen W, Fang D, Bian X, Wang R. SOX2 in gastric carcinoma, but not Hath1, is related to patients’ clinicopathological features and prognosis. Journal of gastrointestinal surgery. 2010; 14: 1220-1226.
57. Gao H, Teng C, Huang W, Peng J, Wang C. SOX2 Promotes the Epithelial to Mesenchymal Transition of Esophageal Squamous Cells by Modulating Slug Expression through the Activation of STAT3/HIF-alpha Signaling. International journal of molecular sciences. 2015; 16: 21643-21657.
58. Yang N, Hui L, Wang Y, Yang H, Jiang X. Overexpression of SOX2 promotes migration, invasion, and epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in laryngeal cancer Hep-2 cells. Tumour biology. 2014; 35: 7965-7973.
59. Singh SK, Chen NM, Hessmann E, Siveke J, Lahmann M, Singh G, Voelker N, Vogt S, Esposito I, Schmidt A, Brendel C, Stiewe T, Gaedcke J, et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. The EMBO journal. 2015; 34: 517-530.
60. Xia Y, Wu Y, Liu B, Wang P, Chen Y. Downregulation of miR-638 promotes invasion and proliferation by regulating SOX2 and induces EMT in NSCLC. FEBS letters. 2014; 588: 2238-2245.
61. Guo J, Wang B, Fu Z, Wei J, Lu W. Hypoxic Microenvironment Induces EMT and Upgrades Stem-Like Properties of Gastric Cancer Cells. Technology in cancer research & treatment. 2016; 15: 60-68.
62. Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T, Liu Y, Li X, Xiang R, Li N. SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/beta-catenin signal network. Cancer letters. 2013; 336: 379-389.
63. Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, Sarkar FH. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PloS one. 2010; 5: e12445.
64. Lu Y, Futtner C, Rock JR, Xu X, Whitworth W, Hogan BL, Onaitis MW. Evidence that SOX2 overexpression is oncogenic in the lung. PloS one. 2010; 5: e11022.
65. Bayo P, Jou A, Stenzinger A, Shao C, Gross M, Jensen A, Grabe N, Mende CH, Rados PV, Debus J, Weichert W, Plinkert PK, Lichter P, et al. Loss of SOX2 expression induces cell motility via vimentin up-regulation and is an unfavorable risk factor for survival of head and neck squamous cell carcinoma. Molecular oncology. 2015; 9: 1704-1719.
66. Hepburn AC, Veeratterapillay R, Williamson SC, El-Sherif A, Sahay N, Thomas HD, Mantilla A, Pickard RS, Robson CN, Heer R. Side population in human non-muscle invasive bladder cancer enriches for cancer stem cells that are maintained by MAPK signalling. PloS one. 2012; 7: e50690.
67. Tian T, Zhang Y, Wang S, Zhou J, Xu S. Sox2 enhances the tumorigenicity and chemoresistance of cancer stem-like cells derived from gastric cancer. Journal of biomedical research. 2012; 26: 336-345.
68. Xiang R, Liao D, Cheng T, Zhou H, Shi Q, Chuang TS, Markowitz D, Reisfeld RA, Luo Y. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. British journal of cancer. 2011; 104: 1410-1417.
69. Yasuda K, Torigoe T, Morita R, Kuroda T, Takahashi A, Matsuzaki J, Kochin V, Asanuma H, Hasegawa T, Saito T, Hirohashi Y, Sato N. Ovarian cancer stem cells are enriched in side population and aldehyde dehydrogenase bright overlapping population. PloS one. 2013; 8: e68187.
70. Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA, Gomez TS, Simeone DM, Bujanda L, Billadeau DD. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2013; 2: e61.
71. Lee YJ, Wu CC, Li JW, Ou CC, Hsu SC, Tseng HH, Kao MC, Liu JY. A rational approach for cancer stem-like cell isolation and characterization using CD44 and prominin-1(CD133) as selection markers. Oncotarget. 2016; 7: 78499-78515. doi: 10.18632/oncotarget.12100.
72. Li Z. CD133: a stem cell biomarker and beyond. Exp Hematol Oncol. 2013; 2:17.
73. Simoes BM, Piva M, Iriondo O, Comaills V, Lopez-Ruiz JA, Zabalza I, Mieza JA, Acinas O, Vivanco MD. Effects of estrogen on the proportion of stem cells in the breast. Breast cancer research and treatment. 2011; 129: 23-35.
74. Lundberg IV, Edin S, Eklof V, Oberg A, Palmqvist R, Wikberg ML. SOX2 expression is associated with a cancer stem cell state and down-regulation of CDX2 in colorectal cancer. BMC Cancer. 2016; 16:471.
75. Jeon HM, Sohn YW, Oh SY, Kim SH, Beck S, Kim S, Kim H. ID4 imparts chemoresistance and cancer stemness to glioma cells by derepressing miR-9*-mediated suppression of SOX2. Cancer research. 2011; 71: 3410-3421.
76. Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, Chellappan SP. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer. 2012;11:73.
77. Rybak AP, Tang D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cellular signalling. 2013; 25: 2734-2742.
78. Santini R, Pietrobono S, Pandolfi S, Montagnani V, D’Amico M, Penachioni JY, Vinci MC, Borgognoni L, Stecca B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014; 33: 4697-4708.
79. Nakatsugawa M, Takahashi A, Hirohashi Y, Torigoe T, Inoda S, Murase M, Asanuma H, Tamura Y, Morita R, Michifuri Y, Kondo T, Hasegawa T, Takahashi H, et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Laboratory investigation. 2011; 91: 1796-1804.
80. Bareiss PM, Paczulla A, Wang H, Schairer R, Wiehr S, Kohlhofer U, Rothfuss OC, Fischer A, Perner S, Staebler A, Wallwiener D, Fend F, Fehm T, et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer research. 2013; 73: 5544-5555.
81. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015; 526: 131-135.
82. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, Brohee S, Salmon I, Dubois C, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014; 511: 246-250.
83. Liu XF, Yang WT, Xu R, Liu JT, Zheng PS. Cervical cancer cells with positive Sox2 expression exhibit the properties of cancer stem cells. PloS one. 2014; 9: e87092.
84. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, Chan KS. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015; 517: 209-213.
85. Jung K, Wang P, Gupta N, Gopal K, Wu F, Ye X, Alshareef A, Bigras G, McMullen TP, Abdulkarim BS, Lai R. Profiling gene promoter occupancy of Sox2 in two phenotypically distinct breast cancer cell subsets using chromatin immunoprecipitation and genome-wide promoter microarrays. Breast Cancer Res. 2014; 16:470.
86. Holmberg Olausson K, Maire CL, Haidar S, Ling J, Learner E, Nister M, Ligon KL. Prominin-1 (CD133) defines both stem and non-stem cell populations in CNS development and gliomas. PloS one. 2014; 9: e106694.
87. Iacopino F, Angelucci C, Piacentini R, Biamonte F, Mangiola A, Maira G, Grassi C, Sica G. Isolation of cancer stem cells from three human glioblastoma cell lines: characterization of two selected clones. PloS one. 2014; 9: e105166.
88. Wu CP, Du HD, Gong HL, Li DW, Tao L, Tian J, Zhou L. Hypoxia promotes stem-like properties of laryngeal cancer cell lines by increasing the CD133+ stem cell fraction. International journal of oncology. 2014; 44: 1652-1660.
89. Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin(+) CD133(-) novel stem-like hepatocellular carcinoma cells expressing EMT markers. International journal of cancer. 2015; 137: 491-496.
90. Zhang X, Hua R, Wang X, Huang M, Gan L, Wu Z, Zhang J, Wang H, Cheng Y, Li J, Guo W. Identification of stem-like cells and clinical significance of candidate stem cell markers in gastric cancer. Oncotarget. 2016; 7: 9815-9831. doi: 10.18632/oncotarget.6890.
91. Lai YJ, Tsai JC, Tseng YT, Wu MS, Liu WS, Lam HI, Yu JH, Nozell SE, Benveniste EN. Small G protein Rac GTPases regulate the maintenance of glioblastoma stem-like cells in vitro and in vivo. Oncotarget. 2017; 8:18031-18049. doi: 10.18632/oncotarget.14949.
92. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer research. 2003; 63: 5821-5828.
93. Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004; 23: 7267-7273.
94. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell stem cell. 2007; 1: 313-323.
95. Maeda S, Shinchi H, Kurahara H, Mataki Y, Maemura K, Sato M, Natsugoe S, Aikou T, Takao S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. British journal of cancer. 2008; 98: 1389-1397.
96. Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. Journal of cell science. 2004; 117: 3539-3545.
97. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell death and differentiation. 2008; 15: 504-514.
98. Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochemical and biophysical research communications. 2006; 351: 820-824.
99. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007; 445: 106-110.
100. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer research. 2007; 67: 4010-4015.
101. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013; 501: 328-337.
102. Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133- cells, and the CD133+ sector is enlarged by hypoxia. International journal of radiation oncology, biology, physics. 2007; 67: 1-5.
103. Annabi B, Doumit J, Plouffe K, Laflamme C, Lord-Dufour S, Beliveau R. Members of the low-density lipoprotein receptor-related proteins provide a differential molecular signature between parental and CD133+ DAOY medulloblastoma cells. Molecular carcinogenesis. 2010; 49: 710-717.
104. Cox JL, Wilder PJ, Desler M, Rizzino A. Elevating SOX2 levels deleteriously affects the growth of medulloblastoma and glioblastoma cells. PloS one. 2012; 7: e44087.
105. Dong L, Qi N, Ge RM, Cao CL, Lan F, Shen L. Overexpression of CD133 promotes the phosphorylation of Erk in U87MG human glioblastoma cells. Neuroscience letters. 2010; 484: 210-214.
106. Boivin D, Labbe D, Fontaine N, Lamy S, Beaulieu E, Gingras D, Beliveau R. The stem cell marker CD133 (prominin-1) is phosphorylated on cytoplasmic tyrosine-828 and tyrosine-852 by Src and Fyn tyrosine kinases. Biochemistry. 2009; 48: 3998-4007.
107. Sgambato A, Puglisi MA, Errico F, Rafanelli F, Boninsegna A, Rettino A, Genovese G, Coco C, Gasbarrini A, Cittadini A. Post-translational modulation of CD133 expression during sodium butyrate-induced differentiation of HT29 human colon cancer cells: implications for its detection. Journal of cellular physiology. 2010; 224: 234-241.
108. Singh PK, Behrens ME, Eggers JP, Cerny RL, Bailey JM, Shanmugam K, Gendler SJ, Bennett EP, Hollingsworth MA. Phosphorylation of MUC1 by Met modulates interaction with p53 and MMP1 expression. The Journal of biological chemistry. 2008; 283: 26985-26995.
109. Singh PK, Wen Y, Swanson BJ, Shanmugam K, Kazlauskas A, Cerny RL, Gendler SJ, Hollingsworth MA. Platelet-derived growth factor receptor beta-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer research. 2007; 67: 5201-5210.
110. Behrens ME, Grandgenett PM, Bailey JM, Singh PK, Yi CH, Yu F, Hollingsworth MA. The reactive tumor microenvironment: MUC1 signaling directly reprograms transcription of CTGF. Oncogene. 2010; 29: 5667-5677.
111. Hagerstrand D, He X, Bradic Lindh M, Hoefs S, Hesselager G, Ostman A, Nister M. Identification of a SOX2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro-oncology. 2011; 13: 1178-1191.
112. Ma L, Lai D, Liu T, Cheng W, Guo L. Cancer stem-like cells can be isolated with drug selection in human ovarian cancer cell line SKOV3. Acta biochimica et biophysica Sinica. 2010; 42: 593-602.
113. Li D, Zhao LN, Zheng XL, Lin P, Lin F, Li Y, Zou HF, Cui RJ, Chen H, Yu XG. Sox2 is involved in paclitaxel resistance of the prostate cancer cell line PC-3 via the PI3K/Akt pathway. Molecular medicine reports. 2014; 10: 3169-3176.
114. Wuebben EL, Wilder PJ, Cox JL, Grunkemeyer JA, Caffrey T, Hollingsworth MA, Rizzino A. SOX2 functions as a molecular rheostat to control the growth, tumorigenicity and drug responses of pancreatic ductal adenocarcinoma cells. Oncotarget. 2016; 7: 34890-34906. doi: 10.18632/oncotarget.8994.
115. Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012; 31: 1354-1365.
116. Fang X, Yoon JG, Li L, Yu W, Shao J, Hua D, Zheng S, Hood L, Goodlett DR, Foltz G, Lin B. The SOX2 response program in glioblastoma multiforme: an integrated ChIP-seq, expression microarray, and microRNA analysis. BMC genomics. 2011; 12: 11.
117. Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem cells (Dayton, Ohio). 2009; 27: 40-48.
118. Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB, Perry JA, Orkin SH, Mansukhani A, Basilico C. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. 2012; 31: 2270-2282.
119. Liu H, Du L, Wen Z, Yang Y, Li J, Dong Z, Zheng G, Wang L, Zhang X, Wang C. Sex determining region Y-box 2 inhibits the proliferation of colorectal adenocarcinoma cells through the mTOR signaling pathway. International journal of molecular medicine. 2013; 32: 59-66.
120. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, Long HW, Xu B, Brown M, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (New York, N.Y.). 2017; 355: 78-83.
121. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, Shah N, Adams EJ, Abida W, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (New York, N.Y.). 2017; 355: 84-88.
122. Kopp JL, Ormsbee BD, Desler M, Rizzino A. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem cells (Dayton, Ohio). 2008; 26: 903-911.
123. Rizzino A. Concise review: The Sox2-Oct4 connection: critical players in a much larger interdependent network integrated at multiple levels. Stem cells (Dayton, Ohio). 2013; 31: 1033-1039.
124. Gao Z, Cox JL, Gilmore JM, Ormsbee BD, Mallanna SK, Washburn MP, Rizzino A. Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. The Journal of biological chemistry. 2012; 287: 11384-11397.
125. Mallanna SK, Ormsbee BD, Iacovino M, Gilmore JM, Cox JL, Kyba M, Washburn MP, Rizzino A. Proteomic analysis of Sox2-associated proteins during early stages of mouse embryonic stem cell differentiation identifies Sox21 as a novel regulator of stem cell fate. Stem cells (Dayton, Ohio). 2010; 28: 1715-1727.
126. Wuebben EL, Mallanna SK, Cox JL, Rizzino A. Musashi2 is required for the self-renewal and pluripotency of embryonic stem cells. PloS one. 2012; 7: e34827.
127. Cox JL, Mallanna SK, Ormsbee BD, Desler M, Wiebe MS, Rizzino A. Banf1 is required to maintain the self-renewal of both mouse and human embryonic stem cells. Journal of cell science. 2011; 124: 2654-2665.
128. Cox JL, Wilder PJ, Gilmore JM, Wuebben EL, Washburn MP, Rizzino A. The SOX2-interactome in brain cancer cells identifies the requirement of MSI2 and USP9X for the growth of brain tumor cells. PloS one. 2013; 8: e62857.
129. Fang X, Yoon JG, Li L, Tsai YS, Zheng S, Hood L, Goodlett DR, Foltz G, Lin B. Landscape of the SOX2 protein-protein interactome. Proteomics. 2011; 11: 921-934.
130. Okamoto R, Uchikawa M, Kondoh H. Sixteen additional enhancers associated with the chicken Sox2 locus outside the central 50-kb region. Development, growth & differentiation. 2015; 57: 24-39.
131. Wiebe MS, Wilder PJ, Kelly D, Rizzino A. Isolation, characterization, and differential expression of the murine Sox-2 promoter. Gene. 2000; 246: 383-393.
132. Tomioka M, Nishimoto M, Miyagi S, Katayanagi T, Fukui N, Niwa H, Muramatsu M, Okuda A. Identification of Sox-2 regulatory region which is under the control of Oct-3/4-Sox-2 complex. Nucleic acids research. 2002; 30: 3202-3213.
133. Zhou HY, Katsman Y, Dhaliwal NK, Davidson S, Macpherson NN, Sakthidevi M, Collura F, Mitchell JA. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes & development. 2014; 28: 2699-2711.
134. Zappone MV, Galli R, Catena R, Meani N, De Biasi S, Mattei E, Tiveron C, Vescovi AL, Lovell-Badge R, Ottolenghi S, Nicolis SK. Sox2 regulatory sequences direct expression of a (beta)-geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development (Cambridge, England). 2000; 127: 2367-2382.
135. Miyagi S, Nishimoto M, Saito T, Ninomiya M, Sawamoto K, Okano H, Muramatsu M, Oguro H, Iwama A, Okuda A. The Sox2 regulatory region 2 functions as a neural stem cell-specific enhancer in the telencephalon. The Journal of biological chemistry. 2006; 281: 13374-13381.
136. Hotta A, Cheung AY, Farra N, Vijayaragavan K, Seguin CA, Draper JS, Pasceri P, Maksakova IA, Mager DL, Rossant J, Bhatia M, Ellis J. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nature methods. 2009; 6: 370-376.
137. Iglesias JM, Leis O, Perez Ruiz E, Gumuzio Barrie J, Garcia-Garcia F, Aduriz A, Beloqui I, Hernandez-Garcia S, Lopez-Mato MP, Dopazo J, Pandiella A, Menendez JA, Martin AG. The Activation of the Sox2 RR2 Pluripotency Transcriptional Reporter in Human Breast Cancer Cell Lines is Dynamic and Labels Cells with Higher Tumorigenic Potential. Frontiers in oncology. 2014; 4: 308.
138. Chakravarthy H, Boer B, Desler M, Mallanna SK, McKeithan TW, Rizzino A. Identification of DPPA4 and other genes as putative Sox2:Oct-3/4 target genes using a combination of in silico analysis and transcription-based assays. Journal of cellular physiology. 2008; 216: 651-662.
139. Marques-Torrejon MA, Porlan E, Banito A, Gomez-Ibarlucea E, Lopez-Contreras AJ, Fernandez-Capetillo O, Vidal A, Gil J, Torres J, Farinas I. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell stem cell. 2013; 12: 88-100.
140. Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Canamero M, Rizzoti K, Carneiro C, Martinez G, Vidal A, Lovell-Badge R, Serrano M. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell stem cell. 2012; 11: 845-852.
141. Vilas JM, Ferreiros A, Carneiro C, Morey L, Da Silva-Alvarez S, Fernandes T, Abad M, Di Croce L, Garcia-Caballero T, Serrano M, Rivas C, Vidal A, Collado M. Transcriptional regulation of Sox2 by the retinoblastoma family of pocket proteins. Oncotarget. 2015; 6: 2992-3002. doi: 10.18632/oncotarget.2996.
142. Ormsbee Golden BD, Wuebben EL, Rizzino A. Sox2 expression is regulated by a negative feedback loop in embryonic stem cells that involves AKT signaling and FoxO1. PloS one. 2013; 8: e76345.
143. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005; 122: 947-956.
144. Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008; 133: 1106-1117.
145. Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009; 137: 647-658.
146. Lee SJ, Kim SJ, Seo HH, Shin SP, Kim D, Park CS, Kim KT, Kim YH, Jeong JS, Kim IH. Over-expression of miR-145 enhances the effectiveness of HSVtk gene therapy for malignant glioma. Cancer letters. 2012; 320: 72-80.
147. Ozen M, Karatas OF, Gulluoglu S, Bayrak OF, Sevli S, Guzel E, Ekici ID, Caskurlu T, Solak M, Creighton CJ, Ittmann M. Overexpression of miR-145-5p inhibits proliferation of prostate cancer cells and reduces SOX2 expression. Cancer investigation. 2015; 33: 251-258.
148. Campayo M, Navarro A, Vinolas N, Diaz T, Tejero R, Gimferrer JM, Molins L, Cabanas ML, Ramirez J, Monzo M, Marrades R. Low miR-145 and high miR-367 are associated with unfavourable prognosis in resected nonsmall cell lung cancer. The European respiratory journal. 2013; 41: 1172-1178.
149. Riggi N, Suva ML, De Vito C, Provero P, Stehle JC, Baumer K, Cironi L, Janiszewska M, Petricevic T, Suva D, Tercier S, Joseph JM, Guillou L, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes & development. 2010; 24: 916-932.
150. Jia Y, Liu H, Zhuang Q, Xu S, Yang Z, Li J, Lou J, Zhang W. Tumorigenicity of cancer stem-like cells derived from hepatocarcinoma is regulated by microRNA-145. Oncology reports. 2012; 27: 1865-1872.
151. Sureban SM, May R, Qu D, Weygant N, Chandrakesan P, Ali N, Lightfoot SA, Pantazis P, Rao CV, Postier RG, Houchen CW. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PloS one. 2013; 8: e73940.
152. Fujii T, Shimada K, Tatsumi Y, Hatakeyama K, Obayashi C, Fujimoto K, Konishi N. microRNA-145 promotes differentiation in human urothelial carcinoma through down-regulation of syndecan-1. BMC cancer. 2015; 15: 818-015-1846-0.
153. Lu YX, Yuan L, Xue XL, Zhou M, Liu Y, Zhang C, Li JP, Zheng L, Hong M, Li XN. Regulation of colorectal carcinoma stemness, growth, and metastasis by an miR-200c-Sox2-negative feedback loop mechanism. Clinical cancer research. 2014; 20: 2631-2642.
154. Deng Z, Du WW, Fang L, Shan SW, Qian J, Lin J, Qian W, Ma J, Rutnam ZJ, Yang BB. The intermediate filament vimentin mediates microRNA miR-378 function in cellular self-renewal by regulating the expression of the Sox2 transcription factor. The Journal of biological chemistry. 2013; 288: 319-331.
155. Qin J, Ji J, Deng R, Tang J, Yang F, Feng GK, Chen WD, Wu XQ, Qian XJ, Ding K, Zhu XF. DC120, a novel AKT inhibitor, preferentially suppresses nasopharyngeal carcinoma cancer stem-like cells by downregulating Sox2. Oncotarget. 2015; 6: 6944-6958. doi: 10.18632/oncotarget.3128.
156. Forno I, Ferrero S, Russo MV, Gazzano G, Giangiobbe S, Montanari E, Del Nero A, Rocco B, Albo G, Languino LR, Altieri DC, Vaira V, Bosari S. Deregulation of MiR-34b/Sox2 Predicts Prostate Cancer Progression. PloS one. 2015; 10: e0130060.
157. Ma K, Pan X, Fan P, He Y, Gu J, Wang W, Zhang T, Li Z, Luo X. Loss of miR-638 in vitro promotes cell invasion and a mesenchymal-like transition by influencing SOX2 expression in colorectal carcinoma cells. Molecular cancer. 2014; 13: 118-4598-13-118.
158. Jin Y, Jiang Z, Guan X, Chen Y, Tang Q, Wang G, Wang X. miR-450b-5p Suppresses Stemness and the Development of Chemoresistance by Targeting SOX2 in Colorectal Cancer. DNA and cell biology. 2016; 35: 249-256.
159. Li J, Du L, Yang Y, Wang C, Liu H, Wang L, Zhang X, Li W, Zheng G, Dong Z. MiR-429 is an independent prognostic factor in colorectal cancer and exerts its anti-apoptotic function by targeting SOX2. Cancer letters. 2013; 329: 84-90.
160. Lundberg IV, Lofgren Burstrom A, Edin S, Eklof V, Oberg A, Stenling R, Palmqvist R, Wikberg ML. SOX2 expression is regulated by BRAF and contributes to poor patient prognosis in colorectal cancer. PloS one. 2014; 9: e101957.
161. Li YJ, Dong M, Kong FM, Zhou JP, Liang D, Xue HZ. MicroRNA-371-5p targets SOX2 in gastric cancer. Oncotarget. 2016; 7:31993-2005. doi: 10.18632/oncotarget.8289.
162. Otsubo T, Akiyama Y, Hashimoto Y, Shimada S, Goto K, Yuasa Y. MicroRNA-126 inhibits SOX2 expression and contributes to gastric carcinogenesis. PloS one. 2011; 6: e16617.
163. Yang C, Hou C, Zhang H, Wang D, Ma Y, Zhang Y, Xu X, Bi Z, Geng S. miR-126 functions as a tumor suppressor in osteosarcoma by targeting Sox2. International journal of molecular sciences. 2013; 15: 423-437.
164. Zhao C, Li Y, Zhang M, Yang Y, Chang L. miR-126 inhibits cell proliferation and induces cell apoptosis of hepatocellular carcinoma cells partially by targeting Sox2. Human cell. 2015; 28: 91-99.
165. Hamada S, Satoh K, Fujibuchi W, Hirota M, Kanno A, Unno J, Masamune A, Kikuta K, Kume K, Shimosegawa T. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Molecular cancer research. 2012; 10: 3-10.
166. Rutenberg-Schoenberg M, Sexton AN, Simon MD. The Properties of Long Noncoding RNAs that Regulate Chromatin. Annual review of genomics and human genetics. 2016; 17:69-94.
167. Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nature genetics. 2003; 33: 461-463.
168. Shahryari A, Jazi MS, Samaei NM, Mowla SJ. Long non-coding RNA SOX2OT: expression signature, splicing patterns, and emerging roles in pluripotency and tumorigenesis. Frontiers in genetics. 2015; 6: 196.
169. Saghaeian Jazi M, Samaei NM, Ghanei M, Shadmehr MB, Mowla SJ. Identification of new SOX2OT transcript variants highly expressed in human cancer cell lines and down regulated in stem cell differentiation. Molecular biology reports. 2016; 43: 65-72.
170. Amaral PP, Neyt C, Wilkins SJ, Askarian-Amiri ME, Sunkin SM, Perkins AC, Mattick JS. Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA (New York, N.Y.). 2009; 15: 2013-2027.
171. Askarian-Amiri ME, Seyfoddin V, Smart CE, Wang J, Kim JE, Hansji H, Baguley BC, Finlay GJ, Leung EY. Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in breast cancer. PloS one. 2014; 9: e102140.
172. Hou Z, Zhao W, Zhou J, Shen L, Zhan P, Xu C, Chang C, Bi H, Zou J, Yao X, Huang R, Yu L, Yan J. A long noncoding RNA Sox2ot regulates lung cancer cell proliferation and is a prognostic indicator of poor survival. The international journal of biochemistry & cell biology. 2014; 53: 380-388.
173. Saghaeian Jazi M, Samaei NM, Ghanei M, Shadmehr MB, Mowla SJ. Overexpression of the non-coding SOX2OT variants 4 and 7 in lung tumors suggests an oncogenic role in lung cancer. Tumour biology. 2016; 37:10329-38.
174. Shahryari A, Rafiee MR, Fouani Y, Oliae NA, Samaei NM, Shafiee M, Semnani S, Vasei M, Mowla SJ. Two novel splice variants of SOX2OT, SOX2OT-S1, and SOX2OT-S2 are coupregulated with SOX2 and OCT4 in esophageal squamous cell carcinoma. Stem cells (Dayton, Ohio). 2014; 32: 126-134.
175. Lin S, Qi W, Han K, Gan Z, Yao Y, Miu D. Prognostic value of SOX2 in digestive tumors: a meta-analysis. Hepato-gastroenterology. 2014; 61: 1274-1278.
176. Shi XM, Teng F. Up-regulation of long non-coding RNA Sox2ot promotes hepatocellular carcinoma cell metastasis and correlates with poor prognosis. International journal of clinical and experimental pathology. 2015; 8: 4008-4014.
177. Shafiee M, Aleyasin SA, Vasei M, Semnani SS, Mowla SJ. Down-Regulatory Effects of miR-211 on Long Non-Coding RNA SOX2OT and SOX2 Genes in Esophageal Squamous Cell Carcinoma. Cell journal. 2016; 17: 593-600.
178. Lin N, Chang KY, Li Z, Gates K, Rana ZA, Dang J, Zhang D, Han T, Yang CS, Cunningham TJ, Head SR, Duester G, Dong PD, et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Molecular cell. 2014; 53: 1005-1019.
179. Han Y, Zhou L, Wu T, Huang Y, Cheng Z, Li X, Sun T, Zhou Y, Du Z. Downregulation of lncRNA-MALAT1 Affects Proliferation and the Expression of Stemness Markers in Glioma Stem Cell Line SHG139S. Cellular and molecular neurobiology. 2016; 36: 1097-1107.
180. Jiao F, Hu H, Han T, Yuan C, Wang L, Jin Z, Guo Z, Wang L. Long noncoding RNA MALAT-1 enhances stem cell-like phenotypes in pancreatic cancer cells. International journal of molecular sciences. 2015; 16: 6677-6693.
181. Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, Wu M, Xiong J, Guo X, Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Developmental cell. 2013; 25: 69-80.
182. Ng SY, Bogu GK, Soh BS, Stanton LW. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Molecular cell. 2013; 51: 349-359.
183. Zhuang W, Ge X, Yang S, Huang M, Zhuang W, Chen P, Zhang X, Fu J, Qu J, Li B. Upregulation of lncRNA MEG3 Promotes Osteogenic Differentiation of Mesenchymal Stem Cells From Multiple Myeloma Patients By Targeting BMP4 Transcription. Stem cells (Dayton, Ohio). 2015; 33: 1985-1997.
184. Hu D, Su C, Jiang M, Shen Y, Shi A, Zhao F, Chen R, Shen Z, Bao J, Tang W. Fenofibrate inhibited pancreatic cancer cells proliferation via activation of p53 mediated by upregulation of LncRNA MEG3. Biochemical and biophysical research communications. 2016; 471: 290-295.
185. Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer cell. 2014; 25: 139-151.
186. Ouyang J, Yu W, Liu J, Zhang N, Florens L, Chen J, Liu H, Washburn M, Pei D, Xie T. Cyclin-dependent kinase-mediated Sox2 phosphorylation enhances the ability of Sox2 to establish the pluripotent state. The Journal of biological chemistry. 2015; 290: 22782-22794.
187. Malak PN, Dannenmann B, Hirth A, Rothfuss OC, Schulze-Osthoff K. Novel AKT phosphorylation sites identified in the pluripotency factors OCT4, SOX2 and KLF4. Cell cycle (Georgetown, Tex.). 2015; 14: 3748-3754.
188. Tsuruzoe S, Ishihara K, Uchimura Y, Watanabe S, Sekita Y, Aoto T, Saitoh H, Yuasa Y, Niwa H, Kawasuji M, Baba H, Nakao M. Inhibition of DNA binding of Sox2 by the SUMO conjugation. Biochemical and biophysical research communications. 2006; 351: 920-926.
189. Myers SA, Panning B, Burlingame AL. Polycomb repressive complex 2 is necessary for the normal site-specific O-GlcNAc distribution in mouse embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108: 9490-9495.
190. Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, Kwon YW, Cho EJ, Youn HD. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell stem cell. 2012; 11: 62-74.
191. Van Hoof D, Munoz J, Braam SR, Pinkse MW, Linding R, Heck AJ, Mummery CL, Krijgsveld J. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell stem cell. 2009; 5: 214-226.
192. Jeong CH, Cho YY, Kim MO, Kim SH, Cho EJ, Lee SY, Jeon YJ, Lee KY, Yao K, Keum YS, Bode AM, Dong Z. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem cells (Dayton, Ohio). 2010; 28: 2141-2150.
193. Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y, Li J, Du JX, Wong J. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Molecular cell. 2014; 55: 537-551.
194. Wang J, Zhang Y, Hou J, Qian X, Zhang H, Zhang Z, Li M, Wang R, Liao K, Wang Y, Li Z, Zhong D, Wan P, et al. Ube2s regulates Sox2 stability and mouse ES cell maintenance. Cell death and differentiation. 2016; 23: 393-404.
195. Zhao HY, Zhang YJ, Dai H, Zhang Y, Shen YF. CARM1 mediates modulation of Sox2. PloS one. 2011; 6: e27026.
196. Cox JL, Mallanna SK, Luo X, Rizzino A. Sox2 uses multiple domains to associate with proteins present in Sox2-protein complexes. PloS one. 2010; 5: e15486.
197. Baltus GA, Kowalski MP, Zhai H, Tutter AV, Quinn D, Wall D, Kadam S. Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem cells (Dayton, Ohio). 2009; 27: 2175-2184.
198. Nowling T, Bernadt C, Johnson L, Desler M, Rizzino A. The co-activator p300 associates physically with and can mediate the action of the distal enhancer of the FGF-4 gene. The Journal of biological chemistry. 2003; 278: 13696-13705.
199. Yoon DS, Choi Y, Jang Y, Lee M, Choi WJ, Kim SH, Lee JW. SIRT1 directly regulates SOX2 to maintain self-renewal and multipotency in bone marrow-derived mesenchymal stem cells. Stem cells (Dayton, Ohio). 2014; 32: 3219-3231.
200. Mu WL, Wang YJ, Xu P, Hao DL, Liu XZ, Wang TT, Chen F, Chen HZ, Lv X, Liu DP. Sox2 Deacetylation by Sirt1 Is Involved in Mouse Somatic Reprogramming. Stem cells (Dayton, Ohio). 2015; 33: 2135-2147.
201. Cox JL, Wilder PJ, Wuebben EL, Ouellette MM, Hollingsworth MA, Rizzino A. Context-dependent function of the deubiquitinating enzyme USP9X in pancreatic ductal adenocarcinoma. Cancer biology & therapy. 2014; 15: 1042-1052.
202. Fang X, Yu W, Li L, Shao J, Zhao N, Chen Q, Ye Z, Lin SC, Zheng S, Lin B. ChIP-seq and functional analysis of the SOX2 gene in colorectal cancers. Omics. 2010; 14: 369-384.