
INTRODUCTION
Cancer stem cells (CSCs) are resistant to conventional radiotherapy and chemotherapy regimens, leading to high relapse rates and poor overall survival [1]. Therefore new therapeutic strategies specifically targeting these CSCs may overcome disease resistance and improve treatment efficacy.
CSCs are characterized by their ability to undergo self-renewal and are highly tumorigenic, with the capability to phenotypically recapitulate the primary tumor from which they have been derived [2]. In breast cancer, CSCs have been implicated in essential stages of tumor progression such as invasiveness and distant organ metastasis along with drug resistance [3, 4]. Identification of CSC-specific markers has aided in the isolation and characterization of the CSCs in many tumor models, for example, the CD44+CD24-/lowLin- population of human breast cancers [5]. High ALDH1 (aldehyde dehydrogenase 1) activity has also been used as a functional marker to define the CSCs [6]. The phenotype in mouse breast CSCs is different from that of human. Mouse breast CSCs derived from MMTV-PyMT mice that develop spontaneous breast adenocarcinomas in female mice as early as 5 weeks have high expression of CD24 and CD49f [7]. CD49f (integrin subunit α6) is a marker for breast cancer progenitor cells, whereas CD24 is highly expressed on both CSCs and tumor transit amplifying cells [8, 9]. A cell line derived from the breast tumors in MMTV-PyMT mice, FMMC 419II, is highly enriched for breast CSCs using the characterization markers (Lin-CD24highCD49fhigh) and a single-cell tumor-initiation activity [7].
Multiple developmental pathways regulate CSC function and maintenance including the Notch, Hedgehog, and Wnt pathways [10]. Many molecules within these signal transduction pathways have been targeted to disrupt the CSC population [11]. One such molecule, Bmi1, is a Polycomb group (PcG) transcriptional repressor protein that silences gene expression by inducing chromatin modifications, hindering transcription factors and RNA polymerase binding [12]. Microarray analysis-defined 11-gene expression signature of the Bmi1 regulated stemness pathway identifies cancer patients with poor prognosis and increased likelihood of death from cancer [12]. Bmi1 is implicated in CSC self-renewal through regulation of genes important for cell cycle control and stem cell fate decisions, as well as regulation of survival genes and inhibition of cellular senescence in multiple cancer models [13, 14]. Bmi1 controls important stages in cancer progression including invasion and metastasis by modulating epithelial-mesenchymal transition (EMT) [15, 16] and drug resistance [17]. There is a statistically significant correlation between Bmi1 expression levels and poor prognosis and survival [18] as well as aggressiveness [19] in human breast cancer patients. Inhibiting expression of the Bmi1 gene by stable transfection with shRNA to Bmi1 also inhibits proliferation of FMMC 419II cells in vitro and blocks tumor initiation [7].
In this study, we use two parallel and independent approaches to inhibit Bmi1 in FMMC 419II breast CSCs: 1) PTC 209, a Bmi1-specific small molecule inhibitor and 2) shRNA to Bmi1. PTC 209 was discovered by high throughput screening of compounds utilizing the gene expression modulation by small molecules (GEMS) technology and shown to downregulate Bmi1 expression in colorectal cancer initiating cells [20]. PTC 209 was tested in vitro against biliary tract cancer and acute myeloid leukemia by other investigators and our group [21–23].
This is the first study directly assessing the effect of Bmi1 inhibition, using both molecular and pharmacological approaches, in a highly enriched population of CSCs in vitro and after transplantation into syngeneic fully immunocompetent animals.
We demonstrate that both PTC 209 treatment and stable transfection with a Bmi1-specific shRNA plasmid decrease Bmi1 mRNA and protein expression in the mammary CSCs and inhibit proliferation in vitro and tumor growth in vivo at relatively lower doses after orthotopic implantation into syngeneic fully immunocompetent host. Furthermore, nano-targeted delivery of PT 209 encapsulated into anti-CD49f poly (lactic-co-glycolic acid)poly (ethylene glycol) nanoparticles (PLGA-PEG NPs) resulted in a significant suppression of implanted mouse CSC into immunocompetent mice as compared to free PTC 209 or to PTC 209 encapsulated into PLGA-PEG NPs and without anti-CD49.
The application of nanotechnology in medicine offers unprecedented opportunities for addressing many of the current challenges in cancer therapy [24–26], in particular targeting stem cells to treat various kinds of cancers. NPs that are biodegradable and biocompatible are especially desirable for ease of in vivo preclinical and clinical utility [26, 27]. PLGA-based controlled release polymer has been utilized clinically, and its clinical safety and feasibility is well established [26–28]. PEG-functionalized NPs are important to enhance pharmacokinetics of these drugs [24]. Biodegradable PLGA-PEG NPs can be targeted for delivery of drugs along with potentially more sensitive diagnostic imaging options. As a proof of the concept, we have demonstrated our expertise in the field of imaging and targeted drug delivery [24, 26–33]. For this study we hypothesized that incorporation of PTC 209 into anti-CD49f PLGA-PEG NPs for targeted delivery will not only increase the accumulation of Bmi1 inhibitor PTC 209 into implanted breast CSC tumor and hence anti-cancer efficacy through active targeting, but will also enable improvement of its safety by using lower doses.
RESULTS
PTC 209 treatment or shRNA stable transfection decreases Bmi1 expression
Ma et al. have previously identified that the CD24+CD49f+ FMMC 419II cells isolated from the MMTV-PyMT tumors express Bmi1 at a significantly higher level than the other cell populations in these tumors that do not exhibit tumor initiation and other CSC-like properties [7]. Here, we observed that RT-PCR analysis of the CD24+CD49f+ FMMC 419II cells that were treated with various concentrations of the Bmi1 specific small molecule inhibitor PTC 209 show a significant decrease in Bmi1 expression (Figure 1A), as does the analysis of cells after transfection with Bmi1-specific shRNAs (Figure 1B). For the stable transfection with inhibitory RNA, the FMMC 419II cells were transfected with a Bmi1-specific shRNA plasmid construct and colonies 2, 4, and 5 were obtained. RT-PCR analysis of these three colonies confirms that knockdown of Bmi1 with shRNA transfection significantly decreases Bmi1 mRNA expression. The significant decrease in expression of Bmi1 protein is seen with western blot analysis (Figure 1C and 1D).
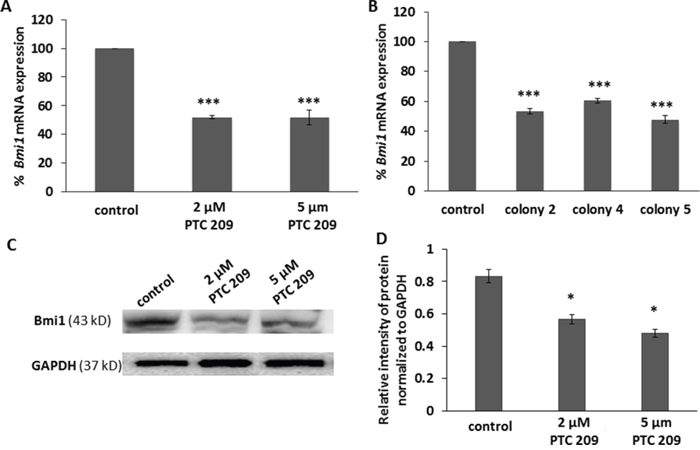
Figure 1: PTC 209 treatment and Bmi1 shRNA transfection decreases Bmi1 mRNA expression. (A) Cells treated with PTC 209 and FMMC 419II cells stably transfected with Bmi1 shRNA plasmid show a decrease in Bmi1 mRNA expression. (B) Purified mRNA from the cells was reverse transcribed into cDNA and then analyzed for Bmi1 mRNA expression with quantitative PCR using TaqMan gene expression assays. The fold difference in expression between control samples and the PTC 209 treated of the Bmi1 shRNA transfected samples was calculated using the average of the Ct (threshold cycle) per group, relative to the expression of the internal control gene GAPDH. (C) FMMC cells were treated with PTC 209 (2 μM and 5 μM) for 24 hours and the expression of Bmi1 protein was detected with western blot. (D) PTC 209 treatment decreased Bmi1 protein expression in FMMC cells. Results are represented as mean ± S.E.M., *P <0.01, ***P <0.005.
Inhibition of Bmi1 expression inhibits cell cycle progression and proliferation
Bmi1 inhibits expression of the Cdkn2a locus and therefore reduces the expression of the p16Ink4a and p19ARF proteins that negatively regulate cell cycle progression [13]. Flow cytometry shows inhibition of cell cycle progression in PTC 209 treated FMMC cells and in cells from colonies 2, 4, and 5. There is a marked decrease in the number of cells that are in the G2 phase of cell cycle in the treated cells (9.4%, 2 μM PTC 209) and (14.1%, 5 μM PTC 209) compared to the untreated cells (49.4%), with more cells arrested at the G0/G1 phase in the treated cells (2 μM PTC 209: 19.6% G0; 55.9% G1, 5 μM PTC 209: 16.3% G0 and 59.8% G1) than in the untreated cells (0.3% G0 and 29.6% G1) (Figure 2A-2D). Cells from colonies 2, 4, and 5 also display a similar cell cycle pattern with more cells arrested at the G1 stage, a phenotype associated with reduced Bmi1 expression (G1 phase: control, 32.6%; colony 2, 71.6%; colony 4, 56.9%; colony 5, 46.6%) (Figure 2E-2I and Supplementary Table 1).
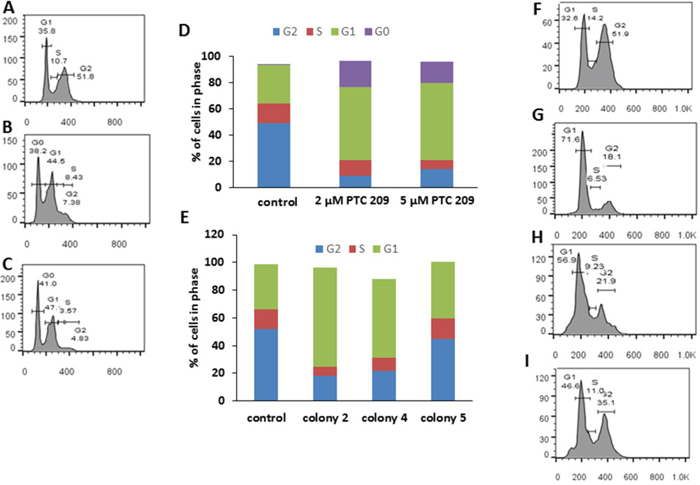
Figure 2: Bmi1 downregulation causes cell cycle arrest. (A) Untreated (control) FMMC 419II cells. (B) Cells treated with 2 μM PTC 209. (C) Cells treated with 5 μM PTC 209. (D) Results of PTC 209 treatment shown as bar graphs. There is a G0/G1 cell cycle arrest in FMMC 419II cells treated with PTC 209 when compared to untreated cells. Similarly, FMMC 419II cells that have been transfected with a Bmi1 shRNA show a G1 arrest. (E) Bar graphs of cell cycle profiles for FMMC 419II cells from control (F), colony 2 (G), colony 4 (H), and colony 5 (I). Cells stained with PI/RNAse staining buffer were run on a FACSAria flow cytometer and cell cycle progression was analyzed and quantified (D, E) using FlowJo.
We also observed changes in proliferation in the test cells in comparison to the control cells in a 48 hour MTT assay. Cells that are either treated with PTC 209 or transfected with Bmi1 shRNAs have a higher number of cells arrested at the G0/G1 stage than untreated cells (Supplementary Figure 1).
Decrease in Bmi1 reduces mammosphere formation
The potential to from tumorspheres, or mammospheres in the case of breast cancer, is indicative of self-renewal of CSCs [34]. The effect of Bmi1 downregulation on self-renewal was assessed by the ability of a single cell to form a mammosphere when cultured in non-adherent conditions in serum-free media. PTC 209 treated cells (Figure 3A, 3B) and cells from colonies 4 and 5 (Figure 3C, 3D) form fewer mammospheres than the control cells (Supplementary Table 2), and the formed mammospheres are much smaller. Thus blocking Bmi1 expression inhibits the self-renewal property of the FMMC 419II CSC-like population.
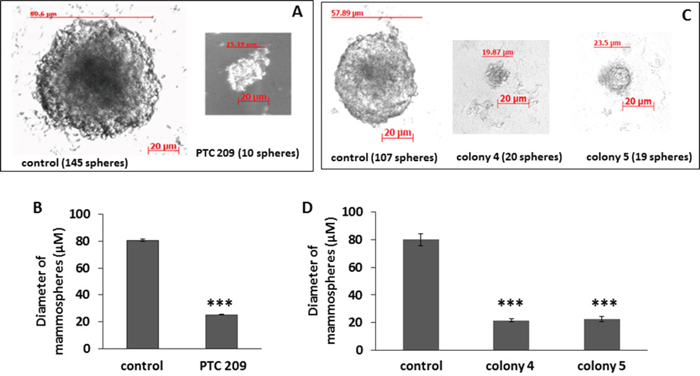
Figure 3: FMMC 419II cells treated with 2 μM PTC 209 or transfected with Bmi1 shRNA display significantly lower mammosphere formation potential. Treated cells were plated at a concentration of 1 cell/μl in an ultra-low attachment 96-well plate in serum free conditions. Phase contrast images at 20X magnification were taken of mammospheres (“spheres”) that were formed after a 2 week incubation (A, C). The graphs (B, D) represent the mean ± S.E.M. of the mammosphere diameters. ***P <0.005.
Flow cytometry analysis for expression of CD24 and CD49f
Flow cytometry analysis showed that on treatment with 2 μM PTC 209, the FMMC cells showed a decrease in cells with high CD49f expression (Figure 4A and 4B). Similarly, cells from the Bmi1 shRNA transfected colonies, colony 4 and colony 5, showed lower CD49f expression than un-transfected control cells (Figure 4A and 4B).
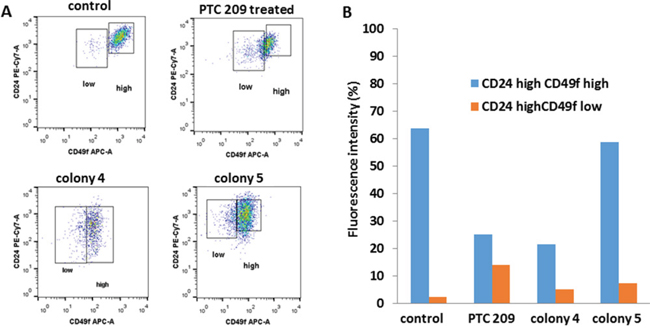
Figure 4: Flow cytometry analysis of CD49f and CD24 expression for FMCC 419II cells treated with 2 μM PTC 209 versus Bmi1 shRNA transfected colonies 4 and 5 were carried out (A), and the fluorescent intensities were quantitated (B).
Reduced Bmi1 expression inhibits tumorigenicity in vivo
To explore the effect of Bmi1 downregulation on breast CSC growth in vivo, we determined the effect of treating the FMMC 419II cells with PTC 209 when they were transplanted into female FVB animals. Tumors generated from mice that received cells mixed with PTC 209 are uniformly much smaller than control tumors and weigh significantly less than tumors generated from untreated cells (26±4 mg versus 176±33 mg, respectively, Figure 5A). Histological analysis of tumors derived from PTC 209 treated cells show fewer mitotic cells and more apoptotic cells than do transplanted tumors from untreated cells. The untreated control tumor sections also have areas that follow the normal tumor growth pattern with central coagulative necrosis, whereas the treated tumors have larger areas of apoptotic cells (Figure 5B).
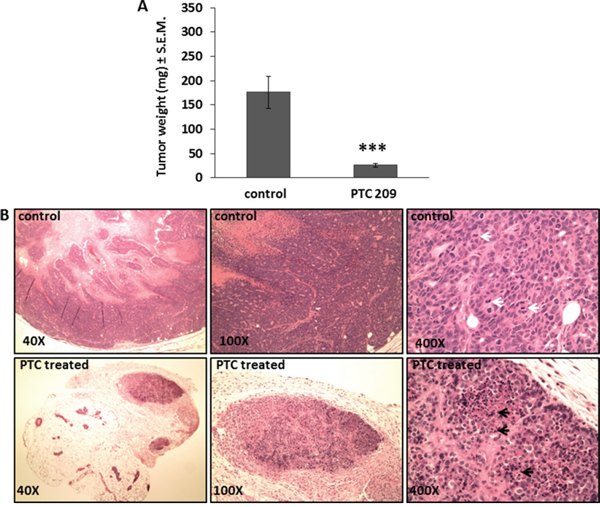
Figure 5: PTC 209 treatment results in decrease of tumor growth of FMMC 419II tumors. (A) Tumors resected from the right and left 4th mammary pad 4 weeks after implantation of 0.75x106 FMMC 419II cells mixed with 50 μg PTC 209 were weighed and results are represented as the average weight (mg) ± S.E.M., ***P <0.005, n=6. (B) H and E staining of 6 μM tumor tissue sections were imaged at 40X, 100X, and 400X magnifications. Representative images from untreated (control) and PTC 209 treated tumors are shown. White arrows denote mitosis, black arrows denote apoptosis.
Similarly, transplanted tumors derived from cells stably transfected with Bmi1 are significantly smaller and weigh less (16±6 mg and 67±6 mg) than untreated (control) tumors (265±46 mg) (Figure 6A). Tumor sites transplanted with untreated cells display dense growth of undifferentiated tumor cells with many actively dividing mitotic cells. Sites from tumors implanted with transfected cells have collections of fibroblasts and giant cells consistent with granulomas, with very few tumor cells (Figure 6B).
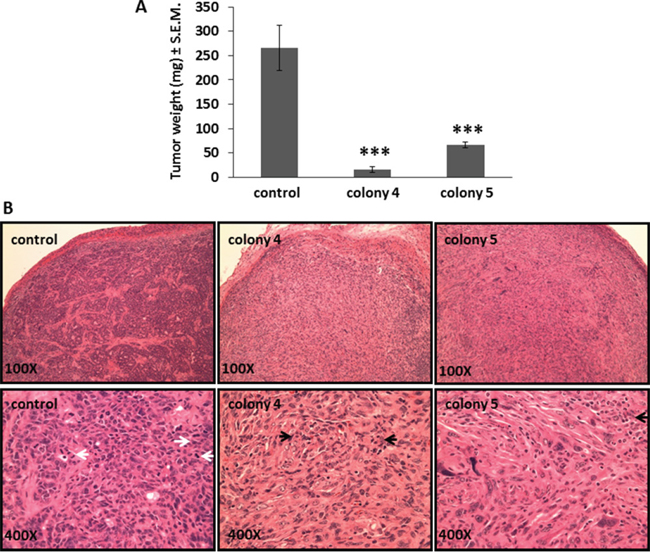
Figure 6: Bmi1 inhibition in FMMC 419II cells stably transfected with Bmi1 shRNA causes reduced tumor growth when transplanted in vivo. Tumors were collected 4 weeks after implantation from the 4th mammary pad of FVB animals transplanted with 0.75x106 control or transfected cells (colony 4 and colony 5) and weighed. (A) Tumor weights are represented as average (mg) ± S.E.M., ***P <0.005, n=5. (B) 6 μM tissue sections of tumor tissue were stained for H and E and imaged at 100X and 400X magnifications. Representative images of tumor derived from control, colony 4, and colony 5 cells are shown. White arrows denote mitosis, black arrows denote apoptosis.
Inhibition of Bmi1 reduces CD49f expression on the cell surface of breast CSCs
A previous study established that the subpopulation of FMMC 419II cells that initiated tumors had a high expression of the cell surface molecule CD49f [7]. CD49f is a cell surface integrin that is associated with self-renewal properties in breast CSCs [35], and its elevated expression correlates with reduced survival of breast cancer patients [36]. We therefore tested the expression levels of CD49f in the tumors that were resected from the control and experimental groups of animals. PLGA NPs coated with anti-CD49f and Cy7 were injected into the animals 24 hours before tumors were collected, and fluorescence levels were measured using an IVIS imager. As expected, tumors from control animals showed high expression of CD49f. In striking contrast, tumors derived from cells treated with PTC 209 showed little or no expression of CD49f (Figure 7). Similarly, the tumors obtained from colonies 4 and 5 showed significantly less CD49f expression. Analysis with flow cytometry of cells treated in vitro with PTC 209 also shows a 60.37% decrease in CD49f expression, and there is a 66.14% decrease in CD49f expression in colony 4 and a 7.68% decrease in colony 5 as compared to untreated 419II cells. Thus, inhibition of Bmi1 led to a decrease in CD49f expression and potentially reduced self-renewal capabilities.
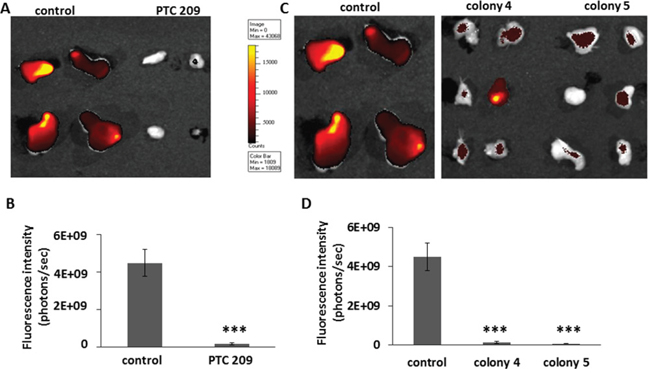
Figure 7: PTC 209 treated tumors with decreased Bmi1 expression show significantly lower expression of CSC marker CD49f. (A) Tumors derived from transplantation of FMMC 419II cells treated with PTC 209, or (C) from colony 4 and colony 5 were imaged for CD49f fluorescence. The probe was an empty PLGA nano shell coated with anti-mouse CD49f antibody and conjugated to a Cy7 fluorescent molecule that was injected into the animals 24 hours before tumors were extracted. (B, D) Fluorescence intensity was calculated as photons emitted per second and graphed as average ± S.E.M., ***P <0.005.
Anti-cancer effects of systemic treatment with PTC 209
Treatment of syngeneic mice bearing transplanted FMMC 419II tumors with anti-CD49f coated NPs containing PTC 209 resulted in reduction in tumor weight and size not observed in mice receiving other NP formulations. Tumor weights in each group (n=5) are shown in Figure 8. Nano-targeted delivery of PTC 209 (PLGA-PEG conjugated with anti-CD49f and encapsulating PTC 209) administered daily at 1 mg/Kg, subcutaneously for 2 weeks, demonstrated distinct suppression of breast tumor growth (**P <0.01) as compared to free PTC 209 or PLGA-PEG NPs encapsulated with PTC209 (Figure 8).
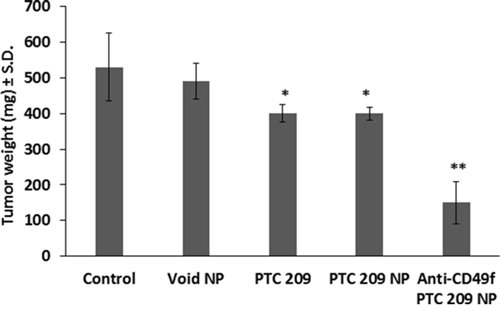
Figure 8: Effect of daily systemic treatment of PTC 209 in different forms on tumor growth. Syngeneic mice were implanted in the 4th mammary pad of FVB animals with mouse CSC, FMMC 419II at 0.75x106 cells per implant. Animals were treated daily at 1 mg/Kg, subcutaneously, with free PTC 209, PLGA-PEG encapsulating PTC 209 (PTC 209 NP), or PLGA-PEG conjugated with anti-CD49f and encapsulating PTC 209 (Anti-CD49f PTC 209 NP) as compared to vehicle control or void PLGA-PEG NPs. Data represent mean ± S.D., n = 5/group, *P <0.05 versus control or void NPs, ** P <0.01 versus all arms.
DISCUSSION
Our results indicate that Bmi1 plays a functional role in regulation of CSCs in breast cancer. To downregulate Bmi1 we used two parallel and independent strategies: a small molecule Bmi1 inhibitor and a transfection of Bmi1-specific shRNA plasmid. Results of these experiments documented significant inhibition of the Bmi1 mRNA and protein expression in CSCs and showed that inhibition of Bmi1 expression in these cells abrogates stem cell-specific properties and significantly reduces tumor growth in animals.
Bmi1 downregulation led to a G0/G1 arrest during cell cycle progression and a growth inhibition of CSCs. Colonies 2 and 4 were more effective in affecting the cell cycle as compared to colony 5, while all colonies exhibited comparable suppression of Bmi1 expression; the exact reason for the differences is not clear. However, PTC 209 showed effective suppression of Bmi1 expression and cell cycle effects. These results are highly consistent with the previously reported mechanisms of biological activity of the Bmi1 protein. Bmi1 acts as a positive regulator of cell cycle progression by elimination of the P16/p14 cell cycle check points [37]. Bmi1 causes the active repression of the cyclin kinase inhibitors (CKI) p16INK4a and p19ARF proteins [38]. The decreased expression of Bmi1 in the tumor stem cells relieves this repression, thus causing increased expression of the negative regulators of cell cycle progression. Bmi1 has also been implicated in regulation of the PI3K/AKT pathways involved in cancer cell progression [16].
It is generally accepted that mammosphere formation assays help identify and characterize breast CSCs. Dontu et al. first showed that mammary epithelial cells cultivated and propagated in non-adherent and serum free conditions consist of a core population of undifferentiated cells that recapitulate the properties of stem cells [39]. Mammosphere formation was later employed to identify breast CSCs in a number of studies [40]. We show that Bmi1 is necessary for mammosphere formation and thereby is essential for self-renewal of the mammary CSCs. PTC 209 treated cells and cells derived from the stable transfection colonies show a decrease in both the percentage of mammospheres formed as well as the diameter of the spheres formed. Thus inhibition of Bmi1 expression in these CSCs causes lower potential of self-renewal and decreases clonogenecity compared to untreated (control) cells.
Kreso et al. first characterized PTC 209 as a pharmacological inhibitor of Bmi1 in a colorectal cancer model and described the loss of stemness and growth inhibition in vitro of human colorectal CSCs upon PTC 209 treatment, as well as inhibition of tumor growth in vivo when nude mice with tumor cell xenografts were treated with the Bmi1 inhibitor [20]. We show that implantation of mammary CSCs mixed with a one-time dose of the inhibitor causes a significant decrease in tumor growth, which is likely caused by the inhibition of tumor initiation. Histological analysis of the tumor mass and surrounding tissue indicates that the treatment with the Bmi1 inhibitor results in increased apoptosis and inhibition of mitoses. We note the areas in control tumor masses that are clear of cancer cells show a pattern of coagulative necrosis, which is the typical growth to death pattern of cancer cells as they grow away from the blood supply. Tumors generated by cells from the stable transfection colonies are significantly smaller than the control tumors. The large areas of granuloma present in the tumor masses might indicate the early clearance of the weakly tumorigenic cancer cells and subsequent wound healing. It should be noted that these experiments were short-term with the tumors being resected out of the animals within 4 weeks of implantation. We plan to follow tumor growth for a longer time to better elucidate the effect of Bmi1 on growth and progression of the mammary tumors. This study proves however, that inhibition of just Bmi1 alone is sufficient to lower the tumor initiation capabilities of the mouse mammary CSCs.
CD49f is a cell surface integrin that was initially used as a biomarker to identify CSCs from the MMTV-PyMT primary tumors. CD49f has been implicated in self-renewal and tumorsphere formation in breast cancer cells [41], and high expression of CD49f positive breast cancer cells show higher tumorigenicity [42], higher metastatic potential [43], and reduced survival [36] compared to CD49f negative cells. The reduced CD49f expression seen both in vitro and in vivo as a result of Bmi1 inhibition suggests that Bmi1 may regulate CD49f expression and function in CSCs. In our mouse mammary stem cell model, CD49f therefore has a dual role: as a CSC identifying moiety to specifically target CSCs and as a functional predictor of the relative strength of the stemness potential in these cells. Bmi1 is a transcriptional repressor and regulates multiple downstream targets both directly and indirectly [44]. Whereas the transcriptional link between Bmi1 and CD49f has not been elucidated, there is evidence that Bmi1-mediated upregulation of Sox2 [45] may maintain expression of CD49f and thereby, self-renewal and tumorigenicity [46].
An important advantage of using FMMC 419II cells is the ability of these cells to grow in an immunocompetent mouse without the risk of rejection. The FVB strain of mice is syngeneic to the MMTV-PyMT mice from which the FMMC 419II cells were originally derived and therefore offers an in vivo model system that is physiologically more relevant than tumor transplantation into immunodeficient mice.
Many studies have focused on Bmi1 expression and function in primary breast cancer cells from patients, or human breast cancer cell lines, and determined that loss of Bmi1 leads to loss of characteristics associated with CSCs. PTC 209 reduced Bmi1 mRNA in vitro at 2-5 μM by ~ 50%, reflecting its potency, but this does not suggest its effect to be direct or indirect. For proof of the concept, we have used molecular approaches to illustrate the impact of Bmi1 downregulation on breast CSC in a novel model of mouse immunocompetent breast CSC-driven breast tumor generation. Additionally, we tested a nano-targeting strategy using anti-CD49f conjugated to PLGA-PEG NPs encapsulated with PTC 209 to illustrate an effective strategy in suppressing breast CSC derived tumor growth.
The present contribution is the first study to directly assess the effect of Bmi1 inhibition in a highly enriched population of CSCs in vitro and after transplantation in vivo into syngeneic fully immunocompetent animals. We believe this is critically important given the central role of the Bmi1 gene in the maintenance of stemness state in both normal and malignant stem cells. We have shown through both pharmacological inhibition and by genetic silencing that Bmi1 is implicated in growth and maintenance of stemness of these mammary CSCs. Unfortunately, systemic administration of PTC 209 after tumor implantation resulted in early mortality of the animals, even when doses were reduced to 15-fold less than what was used in the study by Kreso et al. [20] (data not shown). The reasons for this unexpected toxicity in vivo of the PTC 209 remain unknown. Therefore, we used anti-CD49f conjugated NPs containing PTC 209 to deliver the Bmi1 inhibitor directly to CSCs at lower doses of PTC 209 where free PTC 209 was safe but ineffective as compared to the anti-CD49f conjugated NPs containing PTC 209.
In our study, PTC 209 administered systemically illustrated major toxicity in mice, which was not discussed in the previous report by Kreso et al. [20]. For initial proof of the concept for the role of Bmi1, we treated the mouse breast CSC line FMMC 419II with PTC 209 and implanted in mice versus implanting FMMC 419II stably transfected with Bmi1 plasmid. Data showed comparable efficacy in all in vitro and in vivo assay and model systems used. We did not do a clearance study for PTC 209 from the tumor but rather compared local treatment of FMMC 419II cells with PTC 209 as compared to FMMC 419II stably transfected with Bmi1 plasmid where comparable data were shown. Intra-tumoral implant of FMMC 419II stably transfected with Bmi1 plasmid in Matrigel versus FMMC 419II cells treated with PTC 209 demonstrated comparable efficacy. Further confirmations were provided by the systemic nano-targeted delivery of PTC 209 (1 mg/Kg, subcutaneously, daily for 2 weeks) where the anti-cancer efficacy versus the same dose of free PTC 209 was significantly (**P <0.01) improved.
MATERIALS AND METHODS
A more detailed Materials and Methods is provided in the Supplementary Materials and includes quantitative PCR, flow cytometry, cell cycle analysis, cell proliferation and viability assay, mammosphere formation assay, NPs’ synthesis and characterization, in vitro (IVIS) imaging, and histology.
Cells and cell culture
Cell culture reagents such as Dubelco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), trypsin-EDTA, penicillin/streptomycin and glutamate were purchased from Sigma Aldrich (St. Louis, MO). The CSC-enriched FMMC 419II mouse mammary tumor cells were isolated from primary tumors in MMTV-PyMT transgenic mice and characterized with flow cytometry and sorting in the lab of S.S. Cells were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin (1%) and grown to sub-confluency at 37°C in a humidified atmosphere of 5% CO2/95% air, then sub-cultivated and expanded for experimental use. For treatment with the Bmi1 small molecule inhibitor PTC 209 (Xcess Biosciences, San Diego, CA), cells were trypsinized, washed, and seeded into 6-well plates at around 30% confluency. Twenty-four hours later, appropriate concentrations of PTC 209 were added to the cells and incubated for 48 hours. After treatment, cells were trypsinized, washed, and equal number of viable cells from the PTC 209 treated and DMSO control wells were used for further experiments.
shRNA transfection
Mouse Bmi1 shRNA plasmid (Santa Cruz Biotechnology, Dallas, TX) consists of 3 target-specific lentivirus vector plasmids to knock down Bmi1 expression along with a puromycin selection gene. FMMC cells were transfected according to the manufacturer’s protocol using transfection support reagents (Santa Cruz Biotechnology). Briefly, cells were grown in 6-well culture plates until they were 60% confluent. They were then transfected with the Bmi1 shRNA plasmid in the transfection medium provided in the absence of serum or antibiotics and incubated at 37°C. Five to 7 hours later, DMEM supplemented with 20% FBS and 2 μg/ml puromycin was added and cells were further incubated per the manufacturer’s protocol. Cells were then cultured and maintained in DMEM supplemented with 10% FBS and 1 μg/ml puromycin until the formation of transfected colonies appeared. The colonies were spot trypsinized using small spherical discs of sterile filter paper, and the discs were transferred to new culture plates for expansion of the transfected colonies. Three colonies were identified and named as colony 2, colony 4, and colony 5. Knockdown of Bmi1 expression was confirmed from these expanded colonies with PCR and western blot analysis.
Flow cytometry analysis for expression of CD24 and CD49f
FMMC cells were counted, plated at 1 x 106 cells/plate and treated with 2 μM PTC 209 for 48 hours. Cells were then washed, trypsinized, and incubated with fluorescent antibodies against CD24 and CD49f. Expression of these proteins was then determined with flow cytometry.
Nanoparticles synthesis and characterization
PLGA-PEG NPs encapsulating PTC 209 were synthesized by a solvent diffusion method, previously described by our group [26–28]. Details are included in the Supplementary.
In vivo animal studies
Six to 8 week old female wild type FVB mice (Harlan Laboratories, Indianapolis, IN) were used for the in vivo experiments. FVB mice are syngeneic to MMTV-PyMT transgenic mice, from which the FMMC 419II tumor cells were derived. Mice were maintained at the animal facility of the Albany Stratton Veterans Affairs Medical Center (VAMC), Albany, NY, under an animal protocol approved by the VAMC Institutional Animal Care and Use Committee. Mice were allowed to acclimate for 5 days after arrival at the animal facility prior to experimental treatments and were housed under conditions of a 12 hour light/dark cycle, 20-24°C, and 60-70% humidity. Food and water were provided ad libitum. We mixed 0.75x106 FMMC cells with Matrigel alone or Matrigel and 50 μg PTC 209 and implanted the cells into the left and right 4th mammary fat pad. Four weeks after implantation, tumors were excised and weighed and analyzed histologically. The CD49f+ CSCs in the tumor were identified by using a PLGA nano shell coated with anti-CD49f antibody and tagged with fluorescent Cy7. The tagged NPs were injected subcutaneously into the animals 13 days after cell implantation Twenty-four hours later, tumors were collected and the fluorescence intensity was measured using an in vivo imaging system (IVIS®, Xenogen, Toronto, ON, Canada) (Supplementary Figure 4). Photographic and fluorescent images were taken at constant exposure time. Signal intensity in regions of interest was analyzed using the Xenogen IVIS® Living Image software version 3.2.
Systemic treatment daily for 2 weeks
Systemic treatment was with PLGA-PEG NPs conjugated with anti-CD49f and encapsulating PTC 209 versus free PTC 209, or PLGA-PEG NPs encapsulating PTC 209 and as compared to control and void PLGA-PEG NPs. In all arms having PTC 209, it was dosed at 1 mg/Kg, subcutaneously, daily for 2 weeks.
Statistics
Results are presented as mean ± standard error means (S.E.M.) or ± standard deviation (S.D.) as given in Figure legends. Comparisons between control and experimental groups were statistically analyzed using students t-test and ANOVA using SigmaPlot 10.0 software. Differences between control and experimental end points were considered statistically significant if P <0.05.
Abbreviations
ALDH1, aldehyde dehydrogenase 1; CSC, cancer stem cell; NP, nanoparticle; PLGA-PEG NPs, poly (lactic-co-glycolic acid)poly (ethylene glycol) nanoparticles.
Author contributions
M. S. conceived of and designed experiments, collected and assembled data, did data analysis and interpretation and wrote the first drafts of the manuscript. D. J. B. synthesized and analyzed the nanoparticles. T. S. and M. K. collected and analyzed data. I. G. provided study materials and collected and assembled data. S. S. provided study materials, and did data analysis and interpretation. G. G. conceived of and designed experiments. S. A. M. conceived of and designed experiments, did data analysis and interpretation, and edited the manuscript. All authors reviewed the manuscript during editing and approved the final version.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011; 17:313-319.
2. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105-111.
3. Lindeman GJ, Visvader JE. Insights into the cell of origin in breast cancer and breast cancer stem cells. Asia Pac J Clin Oncol. 2010; 6:89-97.
4. Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. 2010; 28:4006-4012.
5. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100:3983-3988.
6. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555-567.
7. Ma J, Lanza DG, Guest I, Uk-Lim C, Glinskii A, Glinsky G, Sell S. Characterization of mammary cancer stem cells in the MMTV-PyMT mouse model. Tumour Biol. 2012; 33:1983-1996.
8. Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006; 439:993-997.
9. Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007; 7:791-799.
10. Malhotra GK, Zhao X, Band H, Band V. Shared signaling pathways in normal and breast cancer stem cells. J Carcinog. 2011; 10:38.
11. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011; 8:97-106.
12. Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005; 115:1503-1521.
13. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012; 30:372-378.
14. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004; 113:175-179.
15. Proctor E, Waghray M, Lee CJ, Heidt DG, Yalamanchili M, Li C, Bednar F, Simeone DM. Bmi1 enhances tumorigenicity and cancer stem cell function in pancreatic adenocarcinoma. PLoS One. 2013; 8:e55820.
16. Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, Kong QL, Xu LH, Zhang X, Liu WL, Li MZ, Zhang L, Kang TB, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009; 119:3626-3636.
17. Zhu Y, Yu F, Jiao Y, Feng J, Tang W, Yao H, Gong C, Chen J, Su F, Zhang Y, Song E. Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res. 2011; 17:7105-7115.
18. Silva J, Garcia V, Garcia JM, Pena C, Dominguez G, Diaz R, Lorenzo Y, Hurtado A, Sanchez A, Bonilla F. Circulating Bmi-1 mRNA as a possible prognostic factor for advanced breast cancer patients. Breast Cancer Res. 2007; 9:R55.
19. Wang Y, Zhe H, Ding Z, Gao P, Zhang N, Li G. Cancer stem cell marker Bmi-1 expression is associated with basal-like phenotype and poor survival in breast cancer. World J Surg. 2012; 36:1189-1194.
20. Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W, Sydorenko N, Moon YC, Gibson L, Wang Y, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014; 20:29-36.
21. Bolomsky A, Schlangen K, Schreiner W, Zojer N, Ludwig H. Targeting of BMI-1 with PTC-209 shows potent anti-myeloma activity and impairs the tumour microenvironment. J Hematol Oncol. 2016; 9:17.
22. Darwish NH, Sudha T, Godugu K, Elbaz O, Abdelghaffar HA, Hassan EE, Mousa SA. Acute myeloid leukemia stem cell markers in prognosis and targeted therapy: potential impact of BMI-1, TIM-3 and CLL-1. Oncotarget. 2016; 7:57811-57820. doi: 10.18632/oncotarget.11063.
23. Mayr C, Wagner A, Loeffelberger M, Bruckner D, Jakab M, Berr F, Di Fazio P, Ocker M, Neureiter D, Pichler M, Kiesslich T. The BMI1 inhibitor PTC-209 is a potential compound to halt cellular growth in biliary tract cancer cells. Oncotarget. 2016; 7:745-758. doi: 10.18632/oncotarget.6378.
24. Bharali DJ, Mousa SA. Emerging nanomedicines for early cancer detection and improved treatment: current perspective and future promise. Pharmacol Ther. 2010; 128:324-335.
25. Langer R. Drug delivery and targeting. Nature. 1998; 392:5-10.
26. Bharali DJ, Sudha T, Cui H, Mian BM, Mousa SA. Anti-CD24 nano-targeted delivery of docetaxel for the treatment of prostate cancer. Nanomedicine. 2017; 13:263-273.
27. Bharali DJ, Yalcin M, Davis PJ, Mousa SA. Tetraiodothyroacetic acid-conjugated PLGA nanoparticles: a nanomedicine approach to treat drug-resistant breast cancer. Nanomedicine (Lond). 2013; 8:1943-1954.
28. Hariri W, Sudha T, Bharali DJ, Cui H, Mousa SA. Nano-targeted delivery of toremifene, an estrogen receptor-alpha blocker in prostate cancer. Pharm Res. 2015; 32:2764-2774.
29. Bharali DJ, Klejbor I, Stachowiak EK, Dutta P, Roy I, Kaur N, Bergey EJ, Prasad PN, Stachowiak MK. Organically modified silica nanoparticles: a nonviral vector for in vivo gene delivery and expression in the brain. Proc Natl Acad Sci U S A. 2005; 102:11539-11544.
30. Bharali DJ, Lucey DW, Jayakumar H, Pudavar HE, Prasad PN. Folate-receptor-mediated delivery of InP quantum dots for bioimaging using confocal and two-photon microscopy. J Am Chem Soc. 2005; 127:11364-11371.
31. Bharali DJ, Pradhan V, Elkin G, Qi W, Hutson A, Mousa SA, Thanavala Y. Novel nanoparticles for the delivery of recombinant hepatitis B vaccine. Nanomedicine. 2008; 4:311-317.
32. Siddiqui IA, Adhami VM, Bharali DJ, Hafeez BB, Asim M, Khwaja SI, Ahmad N, Cui H, Mousa SA, Mukhtar H. Introducing nanochemoprevention as a novel approach for cancer control: proof of principle with green tea polyphenol epigallocatechin-3-gallate. Cancer Res. 2009; 69:1712-1716.
33. Yalcin M, Bharali DJ, Dyskin E, Dier E, Lansing L, Mousa SS, Davis FB, Davis PJ, Mousa SA. Tetraiodothyroacetic acid and tetraiodothyroacetic acid nanoparticle effectively inhibit the growth of human follicular thyroid cell carcinoma. Thyroid. 2010; 20:281-286.
34. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006; 66:6063-6071.
35. Bergmann S, Royer-Pokora B, Fietze E, Jurchott K, Hildebrandt B, Trost D, Leenders F, Claude JC, Theuring F, Bargou R, Dietel M, Royer HD. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005; 65:4078-4087.
36. Friedrichs K, Ruiz P, Franke F, Gille I, Terpe HJ, Imhof BA. High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res. 1995; 55:901-906.
37. Raaphorst FM. Deregulated expression of Polycomb-group oncogenes in human malignant lymphomas and epithelial tumors. Hum Mol Genet. 2005; 14 Spec No 1:R93-R100.
38. Dimri GP, Martinez JL, Jacobs JJ, Keblusek P, Itahana K, Van Lohuizen M, Campisi J, Wazer DE, Band V. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002; 62:4736-4745.
39. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003; 17:1253-1270.
40. Cho JH, Dimri M, Dimri GP. A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J Biol Chem. 2013; 288:3406-3418.
41. To K, Fotovati A, Reipas KM, Law JH, Hu K, Wang J, Astanehe A, Davies AH, Lee L, Stratford AL, Raouf A, Johnson P, Berquin IM, et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010; 70:2840-2851.
42. Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010; 70:4624-4633.
43. Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. 2014; 33:5477-5482.
44. Wicha MS. Targeting self-renewal, an Achilles' heel of cancer stem cells. Nat Med. 2014; 20:14-15.
45. Seo E, Basu-Roy U, Zavadil J, Basilico C, Mansukhani A. Distinct functions of Sox2 control self-renewal and differentiation in the osteoblast lineage. Mol Cell Biol. 2011; 31:4593-4608.
46. Yu KR, Yang SR, Jung JW, Kim H, Ko K, Han DW, Park SB, Choi SW, Kang SK, Scholer H, Kang KS. CD49f enhances multipotency and maintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells. 2012; 30:876-887.