
INTRODUCTION
Stanley Cohen described on “The Journal of Biological Chemistry” a protein called Epidermal Growth Factor (EGF) able to induce epidermis proliferation and to elicit precocious eye disclosure and teeth eruption [1].Some years later it was discovered that radioactivity of labelled EGF may be localized in many tissues (e.g. epidermis, corneal epithelium, liver, mammary gland) so it was described an EGF pleyotropic action [2-5]; just 30 years ago on 1986 the Nobel Committee proclaimed Stanley Cohen and Rita Levi-Montalcini, the first scientist who discovered the Nerve Growth Factor, winners of the prize for Medicine and Physiology.
The interaction between epithelial and stromal cells is crucial for differentiation and epithelial growth in the course of morphogenesis, embryonal stage and in wound healing of various tissues [6, 7]. A part of growth factors, such as the Epidermal growth factor (EGF), Fibroblast growth factor (FGF), Transforming growth factor-alpha (TGF-α), Keratinocyte growth factor (KGF) and Insulin-like growth factor 2 (IGF2y) can induce, both morphogenesis in different organisms as well as, the development and growth of many tumor entities. Most oncogenic pathways mediated by peptide mitogens FGF, EGF, TGF-α and IGF2y, and Src, Ras, Ets, Integrin, Wnt/b-catenin and Notch signalings, can promote Epithelial Mesenchymal Transition (EMT) [8] (Figure 1A-1B). Cadherins genes activated in embryogenesis are also activated during carcinoma progression and metastasis, indicating that EMT involves a reactivation during tumor progression. Growth factors activity overlaps with the Cadherinsreported to be involved in EMT.The elucidation of the various processes that modulate Cadherins and Growth Factor Receptors (GFR) signal transduction, like receptor heterodimerization and endocytosis, has highlightened new therapeutic opportunities and focused the mechanisms contributing to the effectiveness of existing anticancer treatments [9]. The involvement of cadherins is recognized in the normal epithelial tissue organization and in cancer promoting as discussed previously [10]. The authors describe that metastatic phenotype and EMT are typically linked together with E-cadherin down-regulation [11], and that the interplay between Growth Factor Receptors and cadherins promotes tumorigenesis. The aim of this review is focused on the key role of growth factors signaling and its downstream effectors promoting various biological events. The authors purpose is to emphasize the governance of growth factor receptors in progression and cancer invasion.
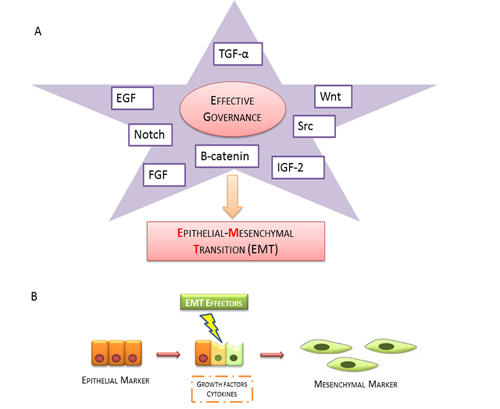
Figure 1: Epithelial mesenchymal transition scheme. A. The broad spectrum of growth factors and related activating downstream effectors (EGF, TGF-α, WNT, Notch, Src, β-catenin, IGF-2 and FGF) involved in induction of EMT B. Epithelial cells promote the expression of mesenchymal markers by the production of growth factors cytokines.
CROSS-TALK BETWEEN EPITHELIAL Growth Factor Receptors and Steroid Receptors in EMT
In humans, more than 30 ligands of the group of the EGF receptors (EGFR) are located, as a multi-layered signal-transduction network, at the beginning of a complex. The interaction between EGFR and its ligand leads to an EGFR monomer dimerization or with another member of erbB family, then these events mediates tyrosine kinase activation. Ligand dependent or independent activation of receptor tyrosine kinases (RTKs) can support EMT signaling in normal and epithelial cells, but finally that cell migration and invasion depend additionally upon i) which receptor heterodimers are assembled ii) which downstream targets are activated iii) how RTKs signaling influences cellular adhesion and matrix ECM interactions in tissue environment [12].
The EGFR has two domains, an extracellular domain that accept the ligands and an intracellular TK domain; these two domains are connected by a transmembrane hydrophobic peptide region. EGFR is a glycoprotein with a molecular weight of 170-kD. EGFR ligands are betacellulin, EGF, amphiregulin, TGF-α, heparin, and epiregulin [13].
EGFR is localized in its monomeric form in the cell membrane, upon ligand binding, EGFR dimerization leads to Tyrosine Kinase (TK) activation and autophosphorylation on tyrosine residues. These phosphorilations are crucial steps indownstream physiologic and pathogenic events [14]. The binding of EGFR-ligand can activate various signaling pathways, main pathways comprise PI-3K/Akt and MAPK ways. The Ras-Raf-MEK-MAPK signaling prevents apoptosis, induces cell proliferation and the release of angiogenetic factors and promote metastasis, while PI-3K/Akt pathway regulates cell surviving, metabolic effects, proliferation and apoptosis (Figure 2). The described signaling is “turned off” by endocytosis of EGFR/ligand. Upon activation, EGFR is translocated and transported to endosome and it is either reprocessed back to the plasma membrane or degraded in lysosomes (Figure 3) [15, 16].
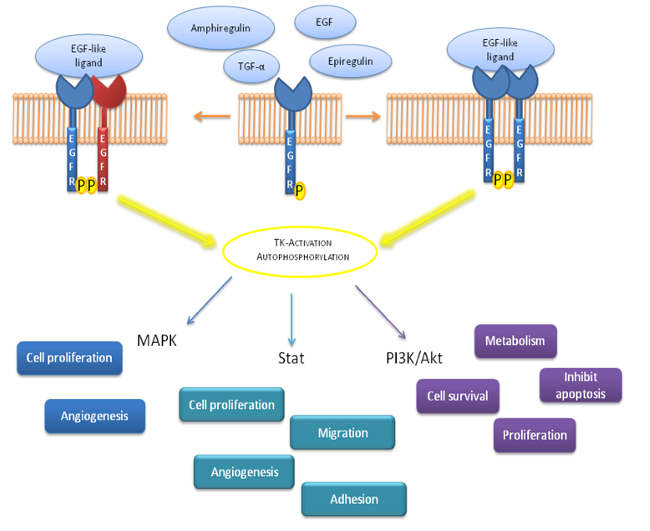
Figure 2: Schematic representation of signal transduction pathways involved after EGFR activation. EGFR ligands binding leads to a receptor dimerization followed by Tyrosine Kinase (TK) autophosphorylation; this mechanism induces a down-stream precise signaling correlated to different biological effects.
EGFR signaling activation is triggered by the phosphorylation of the intrinsic kinase receptor domain and this event contributes to the recruitment of different substrates [17]. This binding is allowed by Src homology 2 motifs (SH2) included on tyrosine kinase domains. On following disruption, the related adaptors and signaling cascades effectors, which consist of KRAS-BRAF-MEK-ERK, phosphoinositide 3-kinase (PI3K), phospholipase C gamma protein, the anti-apoptotic AKT kinase and the STAT , are activated and lead to angiogenesis, survival and cancer promoting [18-23]. Numerous mutations related to different genes implicated in described targets signaling are on the basis of this biological event.
Transcription factors such as ZEB1/TCF8, Snail, ZEB2, Snail2, E12/E47, FOXC2 are involved in growth factors signaling [21] (Figure 3) and it is triggered by the induction of anti-apoptotic signals via receptors such as EGFR, Fibroblast growth factor receptor (FGFR), Platelet derived growth factor receptor (PDGFR), Keratinocytes growth factor receptor (KGFR), cMET, Transforming growth factor β receptor (TGFβR), Human growth factor receptor (HGFR) and Akt, mTOR which are PI3K down-effectors. In order to demonstrate tumor motility, strong evidences in literature proposed the involvement of Snail, Snail2 and E2a transcription factors to explain the disruption of cell adhesion [8].
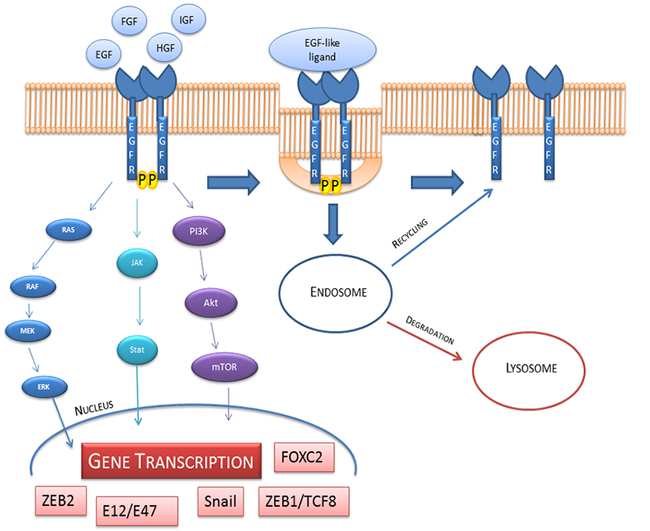
Figure 3: EGFR activation, internalization, recycling and degradation. The down-stream signaling involve Ras/Raf/Mek/Erk; Jak/Stat and PI3K/Akt/mTOR pathways. These pathways activate the transcription of ZEB1/TCF8, Snail, ZEB2, Snail2, E12/E47, FOXC2 that are able to influence the cell fate. On the right side of the figure we show the internalization and endosomal sorting of EGFR.
Modified transduction pathways, in which it was described a crosstalk between EGFRs and steroid receptors have been depicted in the progression of human tumors. An EGFR up-regulation has been represented in a large range of tumors, characterized more aggressive phenotype of these cancers compared to those with a low or normal expression [24-26]. The steroid ligand,17b-Oestradiol (E2)is able to transiently up-regulate EGFR mRNA and protein both in human breast cancer cells and in the rat uterus [27, 28]. The amount of appropriate levels of heterodimers between the two Estrogen Receptor (ER) isoforms mediates estrogenic effects through synergistic mechanism, these results are shown also by others authors in a different systems [29].
Zuo et al. using two human Basal phenotype breast cancers (BPBC) type with respect to Estrogen Receptor/Androgen Receptor ER/AR cell lines as model, have underlined a refractory hormone Progesterone Receptor pathway that is involved in EMT mechanisms through mPRα, Cav-1, EGFR, and PI3K/Akt mediators [30]. mPRα receptor is able to modulate on caveolar membrane of BPBC cells. It promotes EGFR signaling and related inhibition of PI3K pathway and EMT. Others authors shown that steroid receptors contributed to enhance EGF signaling, and Src dependent-EGFR tyrosine phosphorylation in mammary and prostate cancer cells [31]. The interplay between steroid receptors and EGF receptor results in the up-regulation of EGFR tyrosine phosphorylation by EGF in Cos cells transfected transiently with ERα and AR. This phosphorylation determined the assembly of the AR/ER/Src complex [31]. The EGFR signaling regulates the balance between proliferation, apoptosis, differentation and neoplastic development and progression of different human carcinomas. EGFR regulates focal stromal cells associated with invasion and migration in physiological and neoplastic epithelial cells [32] .
Salvatori et al.demonstrated a dose dependent activation of the EGFR gene expression by ligand-bound ERα [33].To explain this mode of action they described a model using the induction the EGFR gene expression by estrogens was dependent upon the formation of a ERα-Sp1. This model have shown a mechanism in which ERα enhances EGFR gene transcription through protein-protein interactions that substitutes DNA binding. The ERα target is considered to be the transcription factor Sp1, which contributes to the stabilization of an Sp1-dependent initiation complex on a TATA-free template, through enhancing the interaction of Sp1 to its site [34].
These authors have shown, in another report, a relevant dependent gene expression for ERα, that was revealed when ERβ transcriptional ability was tested on the minimal promoter. The sensitivity of target cells to estrogens and selective estradiol receptor modulators (SERMS) can be affected by the two different isoforms of the ER, depending on the formation of appropriate amount and ratio of heterodimers [34].
Keratinocyte growth factor and Fibroblast growth factor pathways
KGFR was described in CaCo-2 cells (intestinal epithelium) during spontaneous differentiation in vitro [35]. Many evidences demonstrated that functional KGFRs, in monolayer cells, have a plasma membranes distribution in a basolateral polarized manner and KGFRs are up-regulated during the beginning of differentiation induced by cell confluence. During differentiation, the KGFR expressing cells are not responsive to EGFR ligands [35, 36]. Another experimental study suggested that KGFs factors have a role of estromedin-like in mammary cancer progression. KGF-13 short peptides are suggested as possible therapeutic application for their biological features [37, 38]. FGFR is involved in various physiological processes during embryogenesis. Four related genes belong to the EGFRs gene family; FGFR1 and FGFR2 encoded during homeostasis of adult tissues [39]. Two splicing transcript variants are encoded by FGFR2. These variants were discovered in mesenchymal tissue, while KGFR was found in epithelial tissue [40]. D’Amici et al. proposed a clear model in which FGFR2 is modulated by cytokines and forms a paracrine loop together with KGFR. Inflammatory cytochines such as interleukin (IL 1β e IL6, Inf-6, Inf-γ and TNF-α) are involved in EMT [41, 42].
The described mechanism of action by D’Amici et al.have shown a way of TNF-α on both fibroblasts and keratinocytes at the site of inflammation. In this way, fibroblasts increase the expression of FGFR-IIIc on their surface and enhance the production of KGF followed by this ligand binding and activating its own receptor which is also stimulated by TNF-α. In keratinocytes TNF-α induces phosphorylation of pRb and therefore E2F interacts with FGFR2 promoter in order to induce KGFR biosynthesis(Figure 4) [3, 42-45].
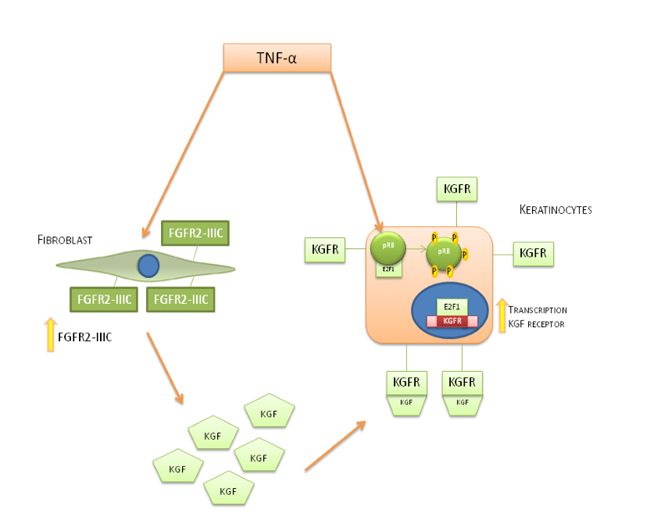
Figure 4: Mechanism of action of TNF-α on fibroblast cells and keratinocytes cells. TNF-α stimulation induces an increase of KGF biosynthesis in fibroblast cells and FGFR2-III expression and the expression of KGFR on keratynocites cells. The interplay between KGF and KGFR on keratinocytes leads to an hyper-phosphorylation of pRb, inducing the release of transcriptionally active E2F1 allowing KGFR biosynthesis.
Others pathways correlated to EMT
Notch pathway
Notch signaling pathway is involved both in physiological that in pathological EMT processes [46].
The activation of Notch signaling pathway is able to stimulate tumorigenesis via regulating EMT [47].
It is known, that in mammals, the Notch family consists of 4 receptors allocated on the membrane:Notch1-Notch4 and its 5 ligands: Jagged-1, Jagged-2, Delta-1, Delta-3, and Delta-4. Das I et al. proposed an interplay between EGF and Notch, in fact Notch receptors are characterized by an extracellular domain and an intracellular domains: RAM, Ankyrin repeats, and a C-terminal PEST [48].
Notch gene overexpression in immortalized endothelial cells leads to EMT by Snail activation and VE-cadherin repression [49, 50].
The complex of Notch ligands/Notch receptors induce the cleavage of intracellular domain of Notch (NIC) by c-secretase. In order to realize a complex with RBP-Jk/CBF1, Su(H), Lag-2 and mastermind-like, Notch intracellular domain is translocated into the nucleus [51-53].
Understanding the molecular mechanism of Notch pathway suggests new rational approaches for cancer therapy [54].
The Notch signaling activation induce the expression of different targets involved in cellular proliferation, such as Cyclin D1 and surviving gamma-secretase inhibitors prevent oxaliplatin-stimulated activation of Notch-1 signaling contributing to the increase of chemo sensitivity in colon cancer cells [55].
Notch-1 overexpression was found in T-cell leukemias and Notch1 pathway is activated in different tumors such as lung adenocarcinoma [56].
Oskarsson and Joan Massague [57] have shown that pulmonary metastatic niches providethe survival of disseminated tumor cells (DTCs) in different tissues. The involved molecular components such as Notch-1 and WNT represent a novelty to develop target therapies to target DTCs. Fiorelli A et al.isolated Circulating cells from peripheral blood , in order to propose a validmarker in the diagnostic workup of lung lesions [58].
Notch is considered a key tumor suppressor gene and its genetic alterations lead to atypical pathway activation [59, 60].
Wnt/β catenin pathway
Wnt’s family includes 19 glycoproteins that have a governance position in the control of different biological events, including self-renewal, polarity, motility and cell fate in stem cells. Wnt/β catenin signaling are activated by interaction with Frizzled (FZD) family receptors. The pathway is induced by ligand-receptor complex, the stimulation from outside of the cell to the internal surface activate Disheveled (DVL) which can induce a multitude of downstream target proteins. This protein prevents β-catenin degradation. In addition Wnt regulates calcium dependent and small-GTPases, such as RAC, RhoA, and CD42 [61, 62].
Another class of FZD molecules, the Wnt receptors ROR1 and ROR 2 induce tumor progression [63]. During the EMT the signaling Wnt/β-catenin complex depends by SLUG activation which translocates into the nucleus for different functions after the interaction with β-catenin. Slug cooperates with TCF/LEF [64], and enhances the target gene transcription [65]. Others receptors such as PTK7, VANG and CELSR bind Wnt receptors to control planar cell polarity (PCP) pathways in vertebrates [66].
Wnt/ β-catenin is involved in regulation of physiological and pathological processes [67].
Wnt/β-catenin activation promotes EMT during gastrulation [68], controls EMT in zebrafish [69, 70] and when deregulated, leads to an early EMT in the mouse [50, 71]. Wnt/β-catenin induces EMT during progression of colorectal cancer metastasis and squamous cell carcinoma (SCC) progression (Figure 5) [72, 73].
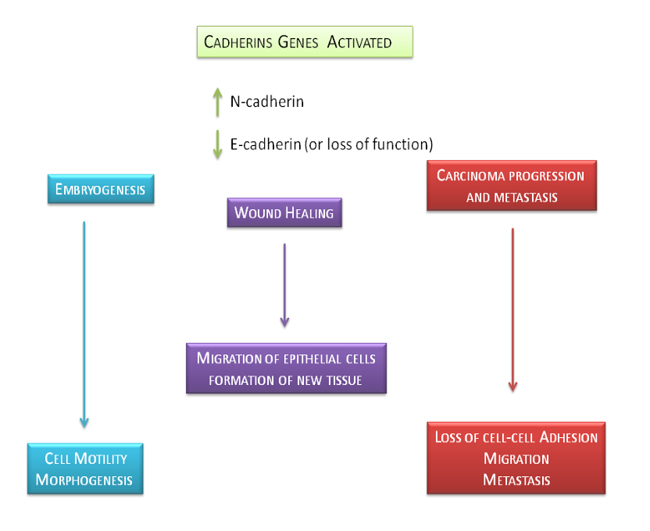
Figure 5: Physiological and pathological role of E-cadherin and N-cadherin genes. This figure shows the precise role of E-cadherin and N-cadherin in physiological and pathological biological effects such as embryogenesis, wound healing and carcinoma progression and metastasis.
Cadherins in epithelial mesenchymal transition
During embryogenesis, E-cadherin goes through spatial and temporal changes of the which allow the occurrence of cell movement and morphogenesis [74]. It induces a migration of epithelial cells on the surface of the wound with the formation of new tissue [75]. In tumors, the cell-cell adhesion is often weak, giving the cells the possibility to promote metastasis. E-cadherin controls cell motility as “invasion suppressor” [76, 77]. The expression of cadherins, in particular of E-cadherin, has been studied in different human tumors, such as cancers of the prostate, breast, pancreas, lung, cervix, liver, stomach, colon, bladder and squamous cell carcinoma [78]. The neoplastic cells show the E-cadherin loss or can maintain an high expression forming possible cell aggregates [79]. Poorly differentiated cancers show a large expression of E-cadherin without keeping its function, probably due to alterations in the complex of catenin [80, 81]or due to mutation of the catenin [82]. In general, however, the low expression of E-cadherin is described in aggressive phenotype cancers characterized by significant cellular infiltration [83-87]. These evidences are confirmed by in vitro assays in which the E-cadherin transfected cells prevent tumorigenesis, therefore deficit in E-cadherin is increasing cell adhesiveness and down-regulating EGFR [77, 88, 89]. Conversely, the knockdown on E-cadherin and β-catenin using siRNA induces an invasive phenotype in epithelial cells [90]. Expression of E-cadherin is correlated to an increased incidence of metastases in lymphatic tissue in different cancers [91]. The mouse ovary carcinoma cells, strongly expressing E-cadherin, show a low metastatic potential [92]. However, no correlation has been described between the presence of E-cadherin and gastric adenocarcinoma and colon metastasis [93].
The role of cadherins in the progression of cancer is much more complex than it was thought at the beginning. The function of E-cadherin is related to the interaction with catenins sites juxta-membrane to form functional units often called catenin-E-cadherin units [94]. The loss of the expression of surface E-cadherin was correlated with a loss of some molecules in lymph node metastases of breast cancer [95]. A previous study has shown that lymph node metastases of breast cancer have a re-expression of E-cadherin or catenins, suggesting that the loss may not be necessary for the metastasis [94].
The E-cadherin is also involved in the regulation of different proteases. Indeed, the E-cadherin transfection in cells prostate cancer E-cadherin-negative line caused a reducing of the production of metalloproteinase MMP-2 and a concomitant reduction of invasiveness [96]. In a similar manner, the cell-cell contacts mediated by E-cadherin involving in the down-regulation of the expression of MMP-9 in skin SCC [97].
In several carcinomas, inappropriate N-cadherins expression has been shown to contribute to a migratory phenotype [98, 99] and Katafiasz et al. have shown that in breast cancer cells the N-cadherin promotes motility independently from the expression of E-cadherin [100]. There is also increasing evidence of coordinate regulation or “cross-talk” between different families of adhesion molecules so that the sniff in the functional capacity of a class of receptors (eg. cadherins) has an impact on another class of receptors (integrins). Weaver et al and Fournier et al. have observed that variants of tumorigenic cell lines of breast cancer could be induced to re-express functional E-cadherin using antibodies blocking the β1 integrins [101, 102]. It was also noted that the cross-talk between integrins and E-cadherins complex and IGF-1 promote migration in colon cancer cells inducing adherence and signal transduction [103, 104]. Finally, increased circulating E-cadherins have been recognized in patients with gastric and hepatocellular cancer [105, 106] and also in several patients with skin cancer, suggesting a possible role as biomarker in certain malignancies [107]. In conclusion E-cadherin ability to maintain the epithelium stratified integrity is lost in more aggressive cancers, such as oral carcinomas [108, 109], because there is a reduction in E-cadherins expressions.
Cross-talk between Growth factor receptors and cadherins
Cross-talk between RTKs and cadherins [110, 111] is considered as a pivotal mechanism inducing biological cellular events in tumor environment. Kyriakakis et al. have demonstrated that an atypical glycosylphosphatidylinositol (GPI)-anchored T-cadherin (T-cad) is considered as a negative regulator of EGFR signaling in A431 cells [112, 113]. After EGF induction, T-cad was reallocated to cellular adhesion where it interacts with Tyr1068 phosphorylated EGFR. Cellular adhesion of T-cad relocalization induced by EGF is prevented by EGFR inhibitors such as gefitinib or lapatinib filipin, actin microfilament polymerization (cytochalasin D or cytochalasin B), p38MAPK (SB203580) or Rac1 (compound4). These data demonstrate that T-cad translocation is responsive of EGFR transactivation. EGFR and T-cad in concert represents a novelty in EGF activation pathway [113]. Another recent experimental study reports that the promoter methylation status of E-cadherin gene (CDH1) in Oral Squamous Cancer cells is correlated with E-cadherin protein expression in a large statistical evaluations, and that this finding correlated with patient outcome. In this contest, it was reported that EGFR participates to a bidirectional cross-talk with E-cadherin [114, 115]. It was described that EGFR phosphorylation enhances its internalization into endocytosis vesicles together with e-cadherin. Here, the EGF interacts with its membrane receptor and its activation induces co-localization of E-cadherin and EGF-R in the cytoplasm of cancer cells [109].
Epithelial Mesenchymal Transition and tumorigenesis
Many carcinomas show a different expression of cadherins, although this is often most evident in poorly differentiated tumors or in advancement of the lesions. Some authors have suggested that a loss or a reduction of E-cadherins determines an increase invasion and metastasis in SCC of the head and neck [116]. In this study the expression of E-cadherin appears well correlated with the differentiation of neoplasia, where the poorly differentiated tumors appear to be completely negative. Dayan and Vered have found a similar pattern of reduced expression in poorly differentiated tumors although this was not statistically significant [117]. Tsanget al. have found an up-regulation in the expression of P-cadherin in severe dysplasia and a decrese in E-cadherin expression and/or cadherins in situ carcinomas and infiltrating tumors [118]. They concluded that the destruction of the complex E-cadherin-catenin is a tardive result correlated with tumor infiltration. Some authors have found that the tumors shown a heterogeneous distribution of E-cadherin and that the loss of E-cadherin shows a statistically significant correlation with the degree of oral cancer or with lesions in the advanced stage and nodal metastasis [119, 120]. In contrast, others authors did not show a link between tumor differentiation and E-cadherin expression [121, 122]. In oral squamous cell carcinoma was described a correlation between tumor stage and E-cadherin and integrin expression, instead P-cadherin expression is mutable [123]. Their results concluded that in all SCC, there could be the precence of cell adhesion molecules and that the loss of integrins and E-cadherin are strictly associated. Harada et al., have observed a desmosomial cadherin in oral cells carcinoma and its low expression in undifferentiated tumors, while more recently they correlated the overexpressed desmosomalcadherins to the loss of invasiveness in SCC cells. [124, 125]. An oral SCC cell lines in vitro study has shown a reduced cadherins expression during tumorigenesis [126]. Schipper et al. shown that differentiated tumor lymph node metastatic sites were E-cadherin negative [127]. In opposition with these results, Bowie et al. have shown the same pattern of staining between lymph node metastatic tumors and primary tumors. Sorscher et al. and Hirao et al. obtained similar results [91, 128, 129].
The expression of E-cadherin is considered as tumor predictor biomarker. Yamada et al. have connected the E-cadherin reduction to tumor advanced stages and poor survival [130]. Mattijssen et al. have underlined the correlation between E-cadherin expression and patient survival while Bowie et al. not found this correlation [131].
Islam et al. recognized an inadequate expression of N-cadherin in a fibroblastic-like phenotype SCC cell line [132]. In this cell line there is a E-and P-cadherin down-regulation. Transfection of this cell line with an antisense N-cadherin led to the reversion toward apparently normal squamous epithelial cells with increased expression of E-and P-cadherin. In a similar manner, the transfection of a squamous epithelial cell, apparently normal, with the N-cadherin resulted in a fibroblastic phenotype and a down-regulation of E-and P-cadherin. In this review it was discussed about a distint epigenetic modification of DNA, which promotes methylation-induced gene silencing, has been described in a large number of tumors, and includes an increasing number of tumor suppressors. Infact aberrant methylation of genomic DNA are partly responsible for transcriptional silencing during carcinogenesis. Recent evidence indicates that the loss of the protein E-cadherin (wich has a key role in adhesion and tissue formation) due to epigenetic aberrations, including the hypermethylation of the promoter of the gene coding for itself, has a crucial role in the onset of progression triggering cancer invasion and metastasis in various human cancers [133]. The gene coding mutations for the E-cadherin is the gene CDH1 (cadherin 1, type 1) located on chromosome 16q22.1 [134], have already been described with regard to carcinomas of the head and neck [135, 136]; even though most of the studies were limited to small sample sizes, in short follow-up, or incomplete information of the treatment of the patient for which it was not possible to formulate a definitive statement about carcinogenesis(Figure 6).
Recently Kabashima-Niibe et al. have demonstrated that also mesenchymal stem cells are implicatedin tumor-promoting cancer stroma development and in the regulation of epithelial-mesenchymal transition in pancreatic tumor cells [137]. Mesenchymal stem cells are multipotent adult stem cells, often used in numerous clinical trials. MSCs are isolated from a wide of tissue and organs such as adipose tissue [138-140], it’s generally acknowledged that their plasticity properties allow them, in response to specific stimuli, to transdifferentiate into mesodermal lineagescells which include smooth, skeletal and cardiac muscle and to be committed towards a cardiac-like phenotype. [141-144].
Battula et al. have demonstrated that EMT-derived cells and MSCs are characterized by an MSCs antigenic profile; moreover MSCs express Twist, Snail (EMT-associated genes) and mesenchyme forkhead 1 (FOXC2). In addition, functional analyses have revealed that EMT-derived cells can differentiate, as MSCs, into chondrocytes, osteoblasts and adipocytes. These authors shown that EMT-derived cells are able to invade MDA-MB-231 breast cancer cells comparable to MSCs [145].
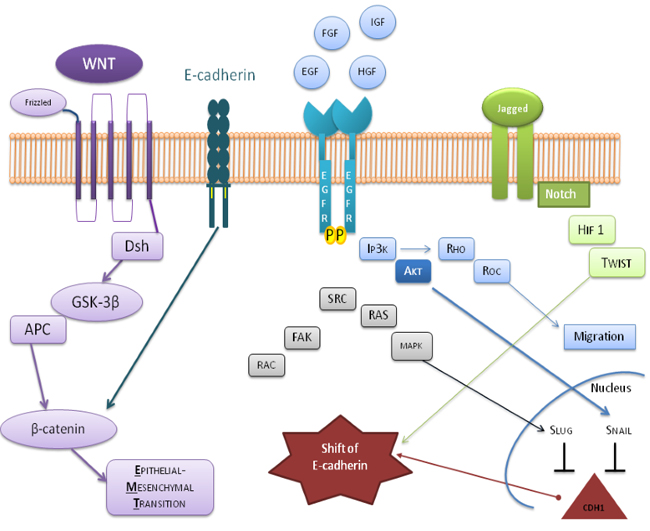
Figure 6: Summary diagram of positive or negative feedback circuits that regulate key processes during EMT. The critical crosstalk between important oncogenic factors, and tumor suppressive proteins are represented by the involved pathways which consist of the activation of WNT by β-catenin, the involvement of PI3K and MAPK in response to EGF ligands and HGF, describes the expression of Jagged-1 (JAG1), induced by Notch ligand in the regulation of Snail/Slug and Twist at transcriptional, translational, and posttranslational levels. All these effectors converge on the shift of E-cadherin.
CONCLUSIONS
The understanding of biological mechanisms associated with cancer could contribute to isolate new targets for novel therapeutic interventions.
It’s known that EMT is induced by growth factors and cytokines signals released from the neoplastic microenvironment including [146-148]. Cells undergoing EMT lose their epithelial morphology, reorganize their membrane ruffling and gain motility. The up- and down-regulation of several molecules including RTKs, steroid receptors, cadherins and their correlate pathways can represent useful target for cancer treatment [149].
Aknowledgments
We thank Prof. Luigi Frati for his pioneering studies of EGFR, above all “Selective binding of the epidermal growth factor and its specific effects on the epithelial cells of the cornea”his innovative works were source of inspiration for this review.
We thank Prof. Gerhard Unteregger, Prof. Marina Di Domenico german mentor, for his editorial support to this review.
CONFLICTS OF INTEREST
The authors declare no conflict of interests.
GRANT SUPPORT
No funding was requested for this study.
References
1. Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem. 1962; 237:1555–62.
2. Covelli I, Rossi R, Mozzi R, Frati L. Synthesis of bioactive 131 I-labeled epidermal growth factor and its distribution in rat tissues. Eur J Biochem. 1972; 27:225–30.
3. Frati L, Daniele S, Delogu A, Covelli I. Selective binding of the epidermal growth factor and its specific effects on the epithelial cells of the cornea. Exp Eye Res. 1972; 14:135–41.
4. Covelli I, Mozzi R, Rossi R, Frati L. The mechanism of action of the epidermal growth factor. 3. Stimulation of the uptake of labeled precursors into RNA, DNA and proteins induced by EGF in isolated tumor cells. Hormones. 1972; 3:183–91.
5. Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem. 1990; 265:7709–12.
6. Donjacour AA, Cunha GR. Stromal regulation of epithelial function. Cancer Treat Res. 1991; 53:335–64.
7. Kvidera A, Mackenzie IC. Rates of clearance of the epithelial surfaces of mouse oral mucosa and skin. Epithelial Cell Biol. 1994; 3:175–80.
8. Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007; 101:816–29.
9. Mellman I, Yarden Y. Endocytosis and cancer. Cold Spring Harb Perspect Biol. 2013; 5:a016949.
10. Choi Y, Lee HJ, Jang MH, Gwak JM, Lee KS, Kim EJ, Kim HJ, Lee HE, Park SY. Epithelial-mesenchymal transition increases during the progression of in situ to invasive basal-like breast cancer. Hum Pathol. 2013; 44:2581–89.
11. Baranwal S, Alahari SK. Molecular mechanisms controlling E-cadherin expression in breast cancer. Biochem Biophys Res Commun. 2009; 384:6–11.
12. Feigin ME, Muthuswamy SK. ErbB receptors and cell polarity: new pathways and paradigms for understanding cell migration and invasion. Exp Cell Res. 2009; 315:707–16.
13. Wells A. Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res. 2000; 78:31–101.
14. Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol. 2005; 25:7734-42.
15. Roepstorff K, Grandal MV, Henriksen L, Knudsen SL, Lerdrup M, Grøvdal L, Willumsen BM, van Deurs B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic. 2009; 10:1115-27.
16. Roepstorff K, Grøvdal L, Grandal M, Lerdrup M, van Deurs B. Endocytic downregulation of ErbB receptors: mechanisms and relevance in cancer. Histochem Cell Biol. 2008; 129:563-78.
17. Normanno N, Gullick WJ. Epidermal growth factor receptor tyrosine kinase inhibitors and bone metastases: different mechanisms of action for a novel therapeutic application? Endocr Relat Cancer. 2006; 13:3–6.
18. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996; 16:5276-87.
19. Di Zazzo E, Feola A, Zuchegna C, Romano A, Donini CF, Bartollino S, Costagliola C, Frunzio R, Laccetti P, Di Domenico M, Porcellini A. The p85 regulatory subunit of PI3K mediates cAMP-PKA and insulin biological effects on MCF-7 cell growth and motility. ScientificWorldJournal. 2014; 2014:565839.
20. Donini CF, Di Zazzo E, Zuchegna C, Di Domenico M, D’Inzeo S, Nicolussi A, Avvedimento EV, Coppa A, Porcellini A. The p85α regulatory subunit of PI3K mediates cAMP-PKA and retinoic acid biological effects on MCF7 cell growth and migration. Int J Oncol. 2012; 40:1627–35.
21. Santoro A, Pannone G, Carosi MA, Francesconi A, Pescarmona E, Russo GM, Feola A, Losito S, Franco R, Nappi L, Aquino G, De Rosa G, Di Domenico M, et al. BRAF mutation and RASSF1A expression in thyroid carcinoma of southern Italy. J Cell Biochem. 2013; 114:1174–82.
22. Baselga J, Albanell J. Epithelial growth factor receptor interacting agents. Hematol Oncol Clin North Am. 2002; 16:1041–63.
23. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012; 16:15–31.
24. Fiorelli A, Ricciardi C, Pannone G, Santoro A, Bufo P, Santini M, Serpico R, Rullo R, Pierantoni GM, Di Domenico M. Interplay between steroid receptors and neoplastic progression in sarcoma tumors. J Cell Physiol. 2011; 226:2997–3003.
25. Neal DE, Marsh C, Bennett MK, Abel PD, Hall RR, Sainsbury JR, Harris AL. Epidermal-growth-factor receptors in human bladder cancer: comparison of invasive and superficial tumours. Lancet. 1985; 1:366–68.
26. Yasui W, Sumiyoshi H, Hata J, Kameda T, Ochiai A, Ito H, Tahara E. Expression of epidermal growth factor receptor in human gastric and colonic carcinomas. Cancer Res. 1988; 48:137–41.
27. Lingham RB, Stancel GM, Loose-Mitchell DS. Estrogen regulation of epidermal growth factor receptor messenger ribonucleic acid. Mol Endocrinol. 1988; 2:230–35.
28. Yarden RI, Lauber AH, El-Ashry D, Chrysogelos SA. Bimodal regulation of epidermal growth factor receptor by estrogen in breast cancer cells. Endocrinology. 1996; 137:2739–47.
29. van den Wijngaard A, Mulder WR, Dijkema R, Boersma CJ, Mosselman S, van Zoelen EJ, Olijve W. Antiestrogens specifically up-regulate bone morphogenetic protein-4 promoter activity in human osteoblastic cells. Mol Endocrinol. 2000; 14:623–33.
30. Zuo L, Li W, You S. Progesterone reverses the mesenchymal phenotypes of basal phenotype breast cancer cells via a membrane progesterone receptor mediated pathway. Breast Cancer Res. 2010; 12:R34.
31. Migliaccio A, Di Domenico M, Castoria G, Nanayakkara M, Lombardi M, de Falco A, Bilancio A, Varricchio L, Ciociola A, Auricchio F. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res. 2005; 65:10585–93.
32. Al Moustafa AE, Achkhar A, Yasmeen A. EGF-receptor signaling and epithelial-mesenchymal transition in human carcinomas. Front Biosci (Schol Ed). 2012; 4:671–84.
33. Salvatori L, Caporuscio F, Coroniti G, Starace G, Frati L, Russo MA, Petrangeli E. Down-regulation of epidermal growth factor receptor induced by estrogens and phytoestrogens promotes the differentiation of U2OS human osteosarcoma cells. J Cell Physiol. 2009; 220:35–44.
34. Salvatori L, Pallante P, Ravenna L, Chinzari P, Frati L, Russo MA, Petrangeli E. Oestrogens and selective oestrogen receptor (ER) modulators regulate EGF receptor gene expression through human ER alpha and beta subtypes via an Sp1 site. Oncogene. 2003; 22:4875–81.
35. Visco V, Bava FA, d’Alessandro F, Cavallini M, Ziparo V, Torrisi MR. Human colon fibroblasts induce differentiation and proliferation of intestinal epithelial cells through the direct paracrine action of keratinocyte growth factor. J Cell Physiol. 2009; 220:204–13.
36. Di Domenico M, Castoria G, Bilancio A, Migliaccio A, Auricchio F. Estradiol activation of human colon carcinoma-derived Caco-2 cell growth. Cancer Res. 1996; 56:4516–21.
37. Chang HL, Sugimoto Y, Liu S, Ye W, Wang LS, Huang YW, Lin YC. Keratinocyte growth factor (KGF) induces tamoxifen (Tam) resistance in human breast cancer MCF-7 cells. Anticancer Res. 2006; 26:1773–84.
38. Al Saleh S, Sharaf LH, Luqmani YA. Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition (Review). Int J Oncol. 2011; 38:1197–217.
39. Ho HK, Yeo AH, Kang TS, Chua BT. Current strategies for inhibiting FGFR activities in clinical applications: opportunities, challenges and toxicological considerations. Drug Discov Today. 2014; 19:51–62.
40. Xu R, Rudd TR, Hughes AJ, Siligardi G, Fernig DG, Yates EA. Analysis of the fibroblast growth factor receptor (FGFR) signalling network with heparin as coreceptor: evidence for the expansion of the core FGFR signalling network. FEBS J. 2013; 280:2260–70.
41. Rizzo A, Di Domenico M, Carratelli CR, Mazzola N, Paolillo R. Induction of proinflammatory cytokines in human osteoblastic cells by Chlamydia pneumoniae. Cytokine. 2011; 56:450–57.
42. D’Amici S, Ceccarelli S, Vescarelli E, Romano F, Frati L, Marchese C, Angeloni A. TNFα modulates Fibroblast Growth Factor Receptor 2 gene expression through the pRB/E2F1 pathway: identification of a non-canonical E2F binding motif. PLoS One. 2013; 8:e61491.
43. Cito L, Indovina P, Forte IM, Pentimalli F, Di Marzo D, Somma P, Barone D, Penon A, Penon D, Ceccherini E, Micheli P, Saragoni L, Di Domenico M, et al. pRb2/p130 localizes to the cytoplasm in diffuse gastric cancer. J Cell Physiol. 2015; 230:802–05.
44. Cito L, Pentimalli F, Forte I, Mattioli E, Giordano A. Rb family proteins in gastric cancer (review). Oncol Rep. 2010; 24:1411–18.
45. Frati L, Cenci G, Sbaraglia G, Teti DV, Covelli I. Levels of epidermal growth factor in mice: tissues measured by a specific radioreceptor assay. Life Sci. 1976; 18:905–11.
46. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006; 7:678–89.
47. Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. 2010; 11:745–51.
48. Das I, Craig C, Funahashi Y, Jung KM, Kim TW, Byers R, Weng AP, Kutok JL, Aster JC, Kitajewski J. Notch oncoproteins depend on gamma-secretase/presenilin activity for processing and function. J Biol Chem. 2004; 279:30771–80.
49. Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, Aranda S, Palomo S, McCormick F, Izpisúa-Belmonte JC, de la Pompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004; 18:99–115.
50. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene. 2005; 24:7443–54.
51. Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995; 377:355–58.
52. Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr Biol. 1995; 5:1416–23.
53. Du R, Sun W, Xia L, Zhao A, Yu Y, Zhao L, Wang H, Huang C, Sun S. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS One. 2012; 7:e30771.
54. Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000; 406:536–40.
55. Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, Schwartz GK. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009; 69:573-82.
56. Allen TD, Rodriguez EM, Jones KD, Bishop JM. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 2011; 71:6010–8.
57. Oskarsson T, Massagué J. Extracellular matrix players in metastatic niches. EMBO J. 2012; 31:254–6.
58. Fiorelli A, Accardo M, Carelli E, Angioletti D, Santini M, Di Domenico M. Circulating Tumor Cells in Diagnosing Lung Cancer: Clinical and Morphologic Analysis. Ann Thorac Surg. 2015; 99:1899–905.
59. Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006; 1:e116.
60. Kulshreshtha R, Ferracin M, Negrini M, Calin GA, Davuluri RV, Ivan M. Regulation of microRNA expression: the hypoxic component. Cell Cycle. 2007; 6:1426–31.
61. Schlessinger K, McManus EJ, Hall A. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J Cell Biol. 2007; 178:355–61.
62. Lai SL, Chien AJ, Moon RT. Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res. 2009; 19:532–45.
63. Zhang S, Chen L, Wang-Rodriguez J, Zhang L, Cui B, Frankel W, Wu R, Kipps TJ. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am J Pathol. 2012; 181:1903–10.
64. Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze’ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003; 163:847-57.
65. Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005; 280:11740–48.
66. Wansleeben C, Meijlink F. The planar cell polarity pathway in vertebrate development. Dev Dyn. 2011; 240:616–26.
67. Horvath A, Mathyakina L, Vong Q, Baxendale V, Pang AL, Chan WY, Stratakis CA. Serial analysis of gene expression in adrenocortical hyperplasia caused by a germline PRKAR1A mutation. J Clin Endocrinol Metab. 2006; 91:584–96.
68. Logan CY, Miller JR, Ferkowicz MJ, McClay DR. Nuclear beta-catenin is required to specify vegetal cell fates in the sea urchin embryo. Development. 1999; 126:345–57.
69. Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003; 425:633–37.
70. Malafoglia V, Colasanti M, Raffaeli W, Balciunas D, Giordano A, Bellipanni G. Extreme thermal noxious stimuli induce pain responses in zebrafish larvae. J Cell Physiol. 2014; 229:300-8.
71. Kemler R, Hierholzer A, Kanzler B, Kuppig S, Hansen K, Taketo MM, de Vries WN, Knowles BB, Solter D. Stabilization of beta-catenin in the mouse zygote leads to premature epithelial-mesenchymal transition in the epiblast. Development. 2004; 131:5817–24.
72. Taki M, Kamata N, Yokoyama K, Fujimoto R, Tsutsumi S, Nagayama M. Down-regulation of Wnt-4 and up-regulation of Wnt-5a expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Cancer Sci. 2003; 94:593–97.
73. Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, Kirchner T. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005; 179:56–65.
74. Sotomayor M, Weihofen WA, Gaudet R, Corey DP. Structure of a force-conveying cadherin bond essential for inner-ear mechanotransduction. Nature. 2012; 492:128–32.
75. Doan A, Wang ZD, Spencer J. E-cadherin expression in intestinal epithelium. J Clin Pathol. 1995; 48:143–6.
76. Vleminckx K, Kemler R, Hecht A. The C-terminal transactivation domain of beta-catenin is necessary and sufficient for signaling by the LEF-1/beta-catenin complex in Xenopus laevis. Mech Dev. 1999; 81:65–74.
77. Vleminckx K, Vakaet L Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991; 66:107–19.
78. Kuphal F, Behrens J. E-cadherin modulates Wnt-dependent transcription in colorectal cancer cells but does not alter Wnt-independent gene expression in fibroblasts. Exp Cell Res. 2006; 312:457–67.
79. Ceteci F, Ceteci S, Zanucco E, Thakur C, Becker M, El-Nikhely N, Fink L, Seeger W, Savai R, Rapp UR. E-cadherin controls bronchiolar progenitor cells and onset of preneoplastic lesions in mice. Neoplasia. 2012; 14:1164–77.
80. Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993; 120:757–66.
81. Oesterreich S, Deng W, Jiang S, Cui X, Ivanova M, Schiff R, Kang K, Hadsell DL, Behrens J, Lee AV. Estrogen-mediated down-regulation of E-cadherin in breast cancer cells. Cancer Res. 2003; 63:5203–08.
82. Muta H, Noguchi M, Kanai Y, Ochiai A, Nawata H, Hirohashi S. E-cadherin gene mutations in signet ring cell carcinoma of the stomach. Jpn J Cancer Res. 1996; 87:843–48.
83. Schipper JH, Unger A, Jahnke K. E-cadherin as a functional marker of the differentiation and invasiveness of squamous cell carcinoma of the head and neck. Clin Otolaryngol Allied Sci. 1994; 19:381–84.
84. Pignatelli M, Ansari TW, Gunter P, Liu D, Hirano S, Takeichi M, Klöppel G, Lemoine NR. Loss of membranous E-cadherin expression in pancreatic cancer: correlation with lymph node metastasis, high grade, and advanced stage. J Pathol. 1994; 174:243–48.
85. Tamura S, Shiozaki H, Miyata M, Kadowaki T, Inoue M, Matsui S, Iwazawa T, Takayama T, Takeichi M, Monden M. Decreased E-cadherin expression is associated with haematogenous recurrence and poor prognosis in patients with squamous cell carcinoma of the oesophagus. Br J Surg. 1996; 83:1608–14.
86. Tamura G, Sakata K, Nishizuka S, Maesawa C, Suzuki Y, Iwaya T, Terashima M, Saito K, Satodate R. Inactivation of the E-cadherin gene in primary gastric carcinomas and gastric carcinoma cell lines. Jpn J Cancer Res. 1996; 87:1153–59.
87. Tucker E, Buda A, Janghra B, Cpoad J, Moorghan M, Havler M, Dettmar P, Pignatelli M. Abnormalities of the cadherin-catenin complex in chemically-induced colo-rectal carcinogenesis. Proc Nutr Soc. 2003; 62:229–36.
88. Miyaki M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Konishi M, Takeichi M. Increased cell-substratum adhesion, and decreased gelatinase secretion and cell growth, induced by E-cadherin transfection of human colon carcinoma cells. Oncogene. 1995; 11:2547–52.
89. Wilding J, Vousden KH, Soutter WP, McCrea PD, Del Buono R, Pignatelli M. E-cadherin transfection down-regulates the epidermal growth factor receptor and reverses the invasive phenotype of human papilloma virus-transfected keratinocytes. Cancer Res. 1996; 56:5285–92.
90. Kaur J, Sawhney M, DattaGupta S, Shukla NK, Srivastava A, Walfish PG, Ralhan R. Clinical significance of altered expression of β-catenin and E-cadherin in oral dysplasia and cancer: potential link with ALCAM expression. PLoS One. 2013; 8:e67361.
91. Hirao T, Nanba D, Tanaka M, Ishiguro H, Kinugasa Y, Doki Y, Yano M, Matsuura N, Monden M, Higashiyama S. Overexpression of ADAM9 enhances growth factor-mediated recycling of E-cadherin in human colon cancer cell line HT29 cells. Exp Cell Res. 2006; 312:331–39.
92. Nakashima H, Hashimoto N, Aoyama D, Kohnoh T, Sakamoto K, Kusunose M, Imaizumi K, Takeyama Y, Sato M, Kawabe T, Hasegawa Y. Involvement of the transcription factor twist in phenotype alteration through epithelial-mesenchymal transition in lung cancer cells. Mol Carcinog. 2012; 51:400–10.
93. Dorudi S, Sheffield JP, Poulsom R, Northover JM, Hart IR. E-cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. Am J Pathol. 1993;142:981-6.
94. Ilyas M. Adhesion molecule expression in breast cancer: the phoenix in tumour metastasis? J Pathol. 2000; 190:3–5.
95. Bukholm IK, Nesland JM, Kåresen R, Jacobsen U, Børresen-Dale AL. E-cadherin and alpha-, beta-, and gamma-catenin protein expression in relation to metastasis in human breast carcinoma. J Pathol. 1998; 185:262–66.
96. Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999; 59:3552–56.
97. Llorens A, Rodrigo I, López-Barcons L, Gonzalez-Garrigues M, Lozano E, Vinyals A, Quintanilla M, Cano A, Fabra A. Down-regulation of E-cadherin in mouse skin carcinoma cells enhances a migratory and invasive phenotype linked to matrix metalloproteinase-9 gelatinase expression. Lab Invest. 1998; 78:1131–42.
98. Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999; 147:631-44.
99. DI Domenico M, Pierantoni GM, Feola A, Esposito F, Laino L, DE Rosa A, Rullo R, Mazzotta M, Martano M, Sanguedolce F, Perillo L, D’Angelo L, Papagerakis S, et al. Prognostic significance of N-Cadherin expression in oral squamous cell carcinoma. Anticancer Res. 2011; 31:4211–18.
100. Katafiasz BJ, Nieman MT, Wheelock MJ, Johnson KR. Characterization of cadherin-24, a novel alternatively spliced type II cadherin. J Biol Chem. 2003; 278:27513–19.
101. Fournier AK, Campbell LE, Castagnino P, Liu WF, Chung BM, Weaver VM, Chen CS, Assoian RK. Rac-dependent cyclin D1 gene expression regulated by cadherin- and integrin-mediated adhesion. J Cell Sci. 2008; 121:226–33.
102. Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997; 137:231–45.
103. André F, Rigot V, Thimonier J, Montixi C, Parat F, Pommier G, Marvaldi J, Luis J. Integrins and E-cadherin cooperate with IGF-I to induce migration of epithelial colonic cells. Int J Cancer. 1999; 83:497–505.
104. de Freitas Junior JC, Silva BR, de Souza WF, de Araújo WM, Abdelhay ES, Morgado-Díaz JA. Inhibition of N-linked glycosylation by tunicamycin induces E-cadherin-mediated cell-cell adhesion and inhibits cell proliferation in undifferentiated human colon cancer cells. Cancer Chemother Pharmacol. 2011; 68:227–38.
105. Katayama M, Hirai S, Yasumoto M, Nishikawa K, Nagata S, Otsuka M, Kamihagi K, Kato I. Soluble fragments of e-cadherin cell-adhesion molecule increase in urinary-excretion of cancer-patients, potentially indicating its shedding from epithelial tumor-cells. Int J Oncol. 1994; 5:1049–57.
106. Romano M, De Francesco F, Pirozzi G, Gringeri E, Boetto R, Di Domenico M, Zavan B, Ferraro GA, Cillo U. Expression of cancer stem cell biomarkers as a tool for a correct therapeutic approach to hepatocellular carcinoma. Oncoscience. 2015; 2:443–56. doi: 10.18632/oncoscience.163.
107. Furukawa F, Takigawa M, Matsuyoshi N, Shirahama S, Wakita H, Fujita M, Horiguchi Y, Imamura S. Cadherins in cutaneous biology. J Dermatol. 1994; 21:802–13.
108. Santoro A, Bufo P, Russo G, Cagiano S, Papagerakis S, Bucci P, Aquino G, Longo F, Feola A, Giordano A, Di Carlo A, Di Domenico M, Pannone G. Expression and clinical implication of cyclooxygenase-2 and e-cadherin in oral squamous cell carcinomas. Cancer Biol Ther. 2015; 0:0.
109. Pannone G, Santoro A, Feola A, Bufo P, Papagerakis P, Lo Muzio L, Staibano S, Ionna F, Longo F, Franco R, Aquino G, Contaldo M, De Maria S, et al. The Role Of E-Cadherin Down-Regulation In Oral Cancer: Cdh1 Gene Expression And Epigenetic Blockage. Curr Cancer Drug Targets. 2014; 14:115–27.
110. Fiorelli A, Morgillo F, Fasano M, Vicidomini G, Di Crescenzo VG, Di Domenico M, Accardo M, Santini M. The value of matrix metalloproteinase-9 and vascular endothelial growth factor receptor 1 pathway in diagnosing indeterminate pleural effusion. Interact Cardiovasc Thorac Surg. 2013; 16:263–9.
111. Di Domenico M, D’apuzzo F, Feola A, Cito L, Monsurrò A, Pierantoni GM, Berrino L, De Rosa A, Polimeni A, Perillo L. Cytokines and VEGF induction in orthodontic movement in animal models. J Biomed Biotechnol. 2012; 2012:201689.
112. Franco R, Nicoletti G, Lombardi A, Di Domenico M, Botti G, Zito Marino F, Caraglia M. Current treatment of cutaneous squamous cancer and molecular strategies for its sensitization to new target-based drugs. Expert Opin Biol Ther. 2013; 13:51–66.
113. Kyriakakis E, Maslova K, Frachet A, Ferri N, Contini A, Pfaff D, Erne P, Resink TJ, Philippova M. Cross-talk between EGFR and T-cadherin: EGFR activation promotes T-cadherin localization to intercellular contacts. Cell Signal. 2013; 25:1044–53.
114. Diniz-Freitas M, Garcia-Caballero T, Antunez-Lopez J, Gandara-Rey JM, Garcia-Garcia A. Reduced E-cadherin expression is an indicator of unfavourable prognosis in oral squamous cell carcinoma. Oral Oncol. 2006; 42:190–200.
115. Aquino G, Pannone G, Santoro A, Liguori G, Franco R, Serpico R, Florio G, De Rosa A, Mattoni M, Cozza V, Botti G, Losito S, Longo F, et al. pEGFR-Tyr 845 expression as prognostic factors in oral squamous cell carcinoma: a tissue-microarray study with clinic-pathological correlations. Cancer Biol Ther. 2012; 13:967–77.
116. Brana I, Siu LL. Locally advanced head and neck squamous cell cancer: treatment choice based on risk factors and optimizing drug prescription. Ann Oncol. 2012; 23 Suppl 10:x178-85.
117. Dayan D, Vered M. “Is immuno-expression of E-cadherin really a prognostic factor in head and neck cancer?”. Oral Oncol. 2013; 49:e5.
118. Tsang JY, Au SK, Ni YB, Shao MM, Siu WM, Hui SW, Chan SK, Chan KW, Kwok YK, Chan KF, Tse GM. P-cadherin and vimentin are useful basal markers in breast cancers. Hum Pathol. 2013; 44:2782–91.
119. Ge MH, Ling ZQ, Tan Z, Chen C, Xu JJ, Yu JL. [Expression and significance of E-cadherin in adenoid cystic carcinoma of salivary glands]. [Article in Chinese]. Zhonghua Yi Xue Za Zhi. 2012; 92:106–09.
120. Contaldo M, Di Napoli A, Pannone G, Franco R, Ionna F, Feola A, De Rosa A, Santoro A, Sbordone C, Longo F, Pasquali D, Loreto C, Ricciardiello F, et al. Prognostic implications of node metastatic features in OSCC: a retrospective study on 121 neck dissections. Oncol Rep. 2013; 30:2697–704.
121. Mattijssen V, Peters HM, Schalkwijk L, Manni JJ, van ’t Hof-Grootenboer B, de Mulder PH, Ruiter DJ. E-cadherin expression in head and neck squamous-cell carcinoma is associated with clinical outcome. Int J Cancer. 1993; 55:580–85.
122. Pirinen RT, Hirvikoski P, Johansson RT, Hollmén S, Kosma VM. Reduced expression of alpha-catenin, beta-catenin, and gamma-catenin is associated with high cell proliferative activity and poor differentiation in non-small cell lung cancer. J Clin Pathol. 2001; 54:391–5.
123. Bagutti C, Speight PM, Watt FM. Comparison of integrin, cadherin, and catenin expression in squamous cell carcinomas of the oral cavity. J Pathol. 1998; 186:8–16.
124. Harada T, Shinohara M, Nakamura S, Shimada M, Oka M. Immunohistochemical detection of desmosomes in oral squamous cell carcinomas: correlation with differentiation, mode of invasion, and metastatic potential. Int J Oral Maxillofac Surg. 1992; 21:346–49.
125. De Bruin A, Müller E, Wurm S, Caldelari R, Wyder M, Wheelock MJ, Suter MM. Loss of invasiveness in squamous cell carcinoma cells overexpressing desmosomal cadherins. Cell Adhes Commun. 1999; 7:13–28.
126. Huber MA, Maier HJ, Alacakaptan M, Wiedemann E, Braunger J, Boehmelt G, Madwed JB, Young ER, Marshall DR, Pehamberger H, Wirth T, Kraut N, Beug H. BI 5700, a Selective Chemical Inhibitor of IκB Kinase 2, Specifically Suppresses Epithelial-Mesenchymal Transition and Metastasis in Mouse Models of Tumor Progression. GenesCancer. 2010; 1:101–14. doi: 10.1177/1947601910361749.
127. Schipper JH, Frixen UH, Behrens J, Unger A, Jahnke K, Birchmeier W. E-cadherin expression in squamous cell carcinomas of head and neck: inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991; 51:6328–37.
128. Bowie GL, Caslin AW, Roland NJ, Field JK, Jones AS, Kinsella AR. Expression of the cell-cell adhesion molecule E-cadherin in squamous cell carcinoma of the head and neck. Clin Otolaryngol Allied Sci. 1993; 18:196–201.
129. Sorscher SM, Russack V, Graziano S, Cagle M, Feramisco JR, Green MR. Immunohistochemical evaluation of E-cadherin and epidermal growth factor receptor in non-small cell lung cancer. Mod Pathol. 1995; 8:450–55.
130. Yamada K, Jordan R, Mori M, Speight PM. The relationship between E-cadherin expression, clinical stage and tumour differentiation in oral squamous cell carcinoma. Oral Dis. 1997; 3:82–85.
131. Yamada Y, Wang XD, Yokoyama S, Fukuda N, Takakura N. Cardiac progenitor cells in brown adipose tissue repaired damaged myocardium. Biochem Biophys Res Commun. 2006; 342:662–70.
132. Islam S, Carey TE, Wolf GT, Wheelock MJ, Johnson KR. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J Cell Biol. 1996; 135:1643–54.
133. Kanai Y. Alterations of DNA methylation and clinicopathological diversity of human cancers. Pathol Int. 2008; 58:544–58.
134. Mansouri A, Spurr N, Goodfellow PN, Kemler R. Characterization and chromosomal localization of the gene encoding the human cell adhesion molecule uvomorulin. Differentiation. 1988; 38:67–71.
135. Sogabe Y, Suzuki H, Toyota M, Ogi K, Imai T, Nojima M, Sasaki Y, Hiratsuka H, Tokino T. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int J Oncol. 2008; 32:1253–61.
136. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, Kelsey KT. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005; 36:768–76.
137. Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y, Funakoshi S, Adachi M, Hamamoto Y, Kawachi S, Aiura K, Kitagawa Y, Sakamoto M, et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. 2013; 104:157–64.
138. Siciliano C, Ibrahim M, Scafetta G, Napoletano C, Mangino G, Pierelli L, Frati G, De Falco E. Optimization of the isolation and expansion method of human mediastinal-adipose tissue derived mesenchymal stem cells with virally inactivated GMP-grade platelet lysate. Cytotechnology. 2015; 67:165-74.
139. Tran KV, Gealekman O, Frontini A, Zingaretti MC, Morroni M, Giordano A, Smorlesi A, Perugini J, De Matteis R, Sbarbati A, Corvera S, Cinti S. The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metab. 2012; 15:222-9.
140. Feola A, Ricci S, Kouidhi S, Rizzo A, Penon A, Formisano P, Giordano A, Di Carlo A, Di Domenico M. Multifaceted Breast Cancer: The Molecular Connection With Obesity. J Cell Physiol. 2017; 232:69–77.
141. Siciliano C, Chimenti I, Ibrahim M, Napoletano C, Mangino G, Scafetta G, Zoccai GB, Rendina EA, Calogero A, Frati G, De Falco E. Cardiosphere conditioned media influence the plasticity of human mediastinal adipose tissue-derived mesenchymal stem cells. Cell Transplant. 2015; 24:2307–22.
142. Siciliano C, Chimenti I, Bordin A, Ponti D, Iudicone P, Peruzzi M, Rendina EA, Calogero A, Pierelli L, Ibrahim M, De Falco E. The potential of GMP-compliant platelet lysate to induce a permissive state for cardiovascular transdifferentiation in human mediastinal adipose tissue-derived mesenchymal stem cells. Biomed Res Int. 2015; 2015:162439.
143. Song L, Tuan RS. Transdifferentiation potential of human mesenchymal stem cells derived from bone marrow. FASEB J. 2004; 18:980–82.
144. Murdoch CE, Broadway-Stringer SA, Bartkeviciute M, Siciliano C, Altobelli R, Falco ED. Stem Cell Therapies for Ischemic Cardiovascular Diseases. Recent Patents on Regenerative Medicine. Bentham Science Publishers; 2014. pp. 149–67.
145. Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M, Mani SA. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010; 28:1435-45.
146. Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B, Giordano A, Tin S, Wu Q, Garza RJ, Cristofanilli M, Mani SA, Croix DA, et al. Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PLoS One. 2015; 10:e0132710.
147. Romano M, De Francesco F, Gringeri E, Giordano A, Ferraro GA, Di Domenico M, Cillo U. Tumor Microenvironment Versus Cancer Stem Cells in Cholangiocarcinoma: synergistic Effects? J Cell Physiol. 2016; 231:768–76.
148. Vergara D, Merlot B, Lucot JP, Collinet P, Vinatier D, Fournier I, Salzet M. Epithelial-mesenchymal transition in ovarian cancer. Cancer Lett. 2010; 291:59–66.
149. Di Domenico M, Santoro A, Ricciardi C, Iaccarino M, Iaccarino S, Freda M, Feola A, Sanguedolce F, Losito S, Pasquali D, Di Spiezio Sardo A, Bifulco G, Nappi C, et al. Epigenetic fingerprint in endometrial carcinogenesis: the hypothesis of a uterine field cancerization. Cancer Biol Ther. 2011; 12:447–57.