
INTRODUCTION
Worldwide, hepatocellular carcinoma (HCC) is among the most common types of cancer and the second leading cause of cancer-related deaths. Chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) accounts for up to 80% of HCC cases [1].
HBV infection causes the immune-mediated destruction of infected hepatocytes and subsequent liver regeneration, which are associated with liver fibrosis, cirrhosis, and HCC [2, 3]. The precise mechanism linking HBV infection to HCC is not entirely understood, especially as HBV is considered to be only weakly carcinogenic [4, 5]. Continual immune-mediated HBV clearance and increases in inflammatory cytokine levels have been suggested to activate cell proliferation and survival signals [6]. HBV X protein (HBx), a 154-amino-acid protein with a molecular mass of 17 kDa, is the major contributor to the long-term disease process of chronic active hepatitis. Rather than directly binding to DNA, HBx protein indirectly activates transcription factors such as NF-κB, activator protein-1 (AP-1), ATF/CREB, proto-oncogenes (c-myc and c-jun), and HIF-1 [7–12]. HBx also promotes cell proliferation by activating the RAS–RAF–MAPK pathway via associations with RAS, Shc, Grb2, and SOS, thereby stimulating RAS–GDP loading [13, 14]. Although many reports have described HBx activities, the molecular mechanism linking HBx activity to HCC remains poorly understood.
Nuclear factor-kappa B (NF-κB), a dimeric transcription factor, comprises a family of proteins, including NF-κB1 (p105 and p50), NF-κB2 (p100 and p52), RelA (p65), c-Rel, and RelB [15, 16]. NF-κB normally localizes to the cytoplasm and binds to members of the inhibitory IκB family (IκBα, IκBβ, p105, and p100), blocking the nuclear translocation of NF-κB [6]. To activate NF-κB, HBx forms complexes with IκBα in the cytoplasm or reduces the levels of cytoplasmic p105 proteins, leading to the nuclear accumulation of NF-κB and activation of the NF-κB pathway [17, 18]. NF-κB acts dual pro-inflammatory and anti-apoptotic roles in combating hepatocellular injury [6].
SHP2, encoded by Ptpn11, is a Src homology 2 domain-containing non-receptor tyrosine phosphatase and a proto-oncogene. Autosomal-dominant mutations in Ptpn11, which have been detected in approximately half of the patients with Noonan syndrome, predispose affected individuals to juvenile myelomonocytic leukemia and several other neoplasms that exhibit increased SHP2 phosphatase activity [19–21]. SHP2 overexpression has been reported in leukemia and breast cancers [22, 23]. Earlier studies have shown that SHP2 promotes EGFR–RAS–RAF–MEK–ERK pathway activity by dephosphorylating the RAS-GTPase-activating protein (RAS–GAP) binding site on the docking protein Grb2-associated binder 1 (Gab1), leading to prolonged RAS activation [24, 25].
A recent study reported that hepatocyte-specific SHP2 deletion promotes hepatic inflammation and necrosis, leading to regenerative hyperplasia, fibrosis, and HCC development [26]. SHP2 knockout was found to greatly increase signaling strength in the inflammatory IL-6–signal transducer and activator of transcription 3 (STAT3) pathway. In contrast, STAT3 ablation completely alleviates the effect of SHP2 depletion on HCC development. Therefore, SHP2 is a tumor suppressor that may counteract STAT3 activity in the liver. The contrasting roles of SHP2 in leukemogenesis and liver cancer suggest that SHP2 activity must be finely maintained [27]. Here we report for the first time the existence of an HBx–NF-κB–SHP2 pathway in HBV-infected livers. We found that HBx increased SHP2 expression through NF-κB, leading to prolonged EGFR signaling. Moreover, SHP2, NF-κB, and phospho-STAT3 levels were simultaneously higher in non-neoplastic background tissues than in corresponding HCC tissues. In an immunohistochemistry (IHC) analysis of 162 HCC tissues, SHP2 expression was decreased or absent in tumor tissues from a significant fraction of patients with HCC. By contrast, higher SHP2 expression levels were observed in the background liver tissues of patients with advanced fibrosis. In addition, SHP2 expression gradually increased from normal liver to HBV-associated cirrhotic liver, but decreased in dysplastic nodules. We also present evidence that SHP2 links HBx–NF-κB with EGFR signaling-mediated hepatocyte proliferation and that the HBx–NF-κB–SHP2 and IL-6–JAK–STAT3 pathways were active in parallel at an early stage of HCC development; accordingly, losses of activity in these pathways could accelerate HCC development.
RESULTS
HBx increases SHP2 mRNA and protein levels and stimulates growth signaling
Because SHP2 can act as both a proto-oncogene and tumor suppressor [27], we first questioned whether SHP2 is activated by oncogenic HBx in HCC cells. HBx and SHP2 transcript levels were measured in Huh7 and stably HBx expressing Huh7-X cells. Both the HBx and SHP2 mRNA levels were markedly higher in Huh7-X cells than in Huh7 cells (Figure 1A, 1B). SHP2 protein levels were also higher in Huh7-X cells than in Huh7 cells (Figure 1C). The expression of Gab1, an adaptor protein that recruits SHP2 to the EGFR signaling cascade, was slightly higher in cells with increased SHP2 levels. To examine the relationship between HBx and SHP2 overexpression, we transfected non-hepatic human embryonic kidney (HEK) 293 cells with increasing amounts of the HA–HBx expression vector. As shown in Figure 1D, the levels of SHP2 and its downstream effector, phospho-ERK, were dose-dependently increased with the amount of transfected HA–HBx. These data suggest that HBx activates SHP2 expression.
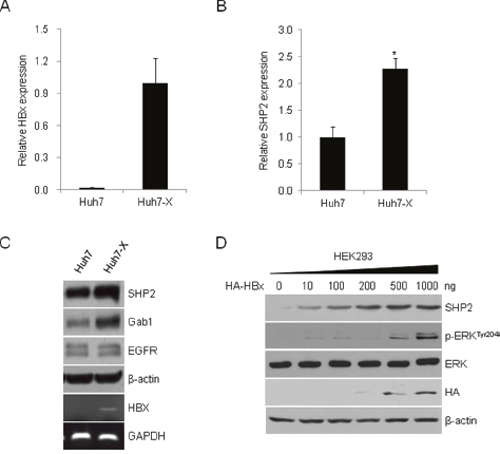
Figure 1: HBx induces SHP2 expression and activates ERK signaling. HBx (A) and SHP2 (B) transcript levels in Huh7 and Huh7-X (HBx-overexpressing) cells were analyzed by qRT-PCR. Data indicate the mRNA levels of HBx and SHP2 relative to that of the control gene, GAPDH. Significance vs. control, *, P < 0.05 (n = 3). (C) SHP2, Gab1, and EGFR protein levels were analyzed in Huh7 and Huh7-X cells by Western blotting with the indicated antibodies. HBx expression in Huh7-X cells was determined by RT-PCR. GAPDH was used as a control. (D) HEK293 cells were transfected with increasing amounts of HA-tagged HBx expression vectors. After 48 h, Western blotting was performed using the indicated antibodies.
HBx induces SHP2 expression via NF-κB–p65
Because HBx stimulates cell growth-related transcription by indirectly activating transcription factors [7, 9], we used the Transcription Element Search System to analyze a 3,000-bp region around the Shp2 promoter and identify cis-regulatory elements that might explain the effect of HBx on SHP transcription [28]. We identified a putative NF-κB binding site between nucleotides –510 and –500 (GGTAATTTCC; numbering starting at the translation start site) and cloned a fragment from the 5′-flanking region of the Shp2 promoter (spanning nucleotides –1,727 to –151) to the upstream region of the luciferase reporter gene to yield a Shp2–Luc construct. As the HBx protein induces NF-κB activation in liver cells, we first examined the effect of HBx or NF-κB on Shp2 expression. As shown in Figure 2A, HBx or NF-κB overexpression significantly increased Shp2 promoter activity up to 2- or 4- fold, respectively. Expression from the Shp2 promoter was further increased by approximately 5-fold in HBx and NF-κB co-transfected cells. We performed a chromatin immunoprecipitation (ChIP) assay using antibodies against p65 (RelA), an NF-κB protein. The results showed a significant increase in endogenous NF-κB–p65 binding to the Shp2 promoter after HBx overexpression in both HEK293 and Huh7 cells (Figure 2B). To investigate whether NF-κB directly binds to the Shp2 promoter, we performed electrophoretic mobility shift assay (EMSA) using nuclear extracts prepared from Huh7 and Huh7-X cells. We found a shifted band with Huh7-X, but not with Huh7. The shifted band was eliminated by the addition of 10-fold or 100-fold excess of the same unlabeled NF-κB binding DNA of the Shp2 promoter. In contrast, mutated NF-κB probe were unable to out-compete the complexes (Figure 2C).
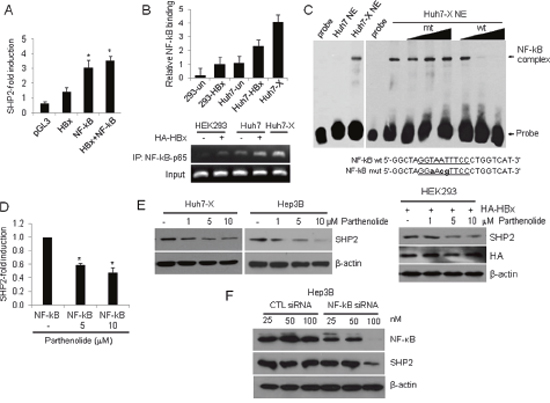
Figure 2: HBx activates the Shp2 promoter via an NF-κB–p65 binding site. (A) SHP2 promoter activities in HEK293 cells after transfection with Shp2–Luc and plasmids expressing HBx, NF-κB, or a combination of both for 48 h. Luciferase activity levels were normalized to Renilla luciferase units. Significance vs. control, *, P < 0.05 (n = 6). (B) ChIP analysis of NF-κB–p65 binding to the Shp2 promoter after HA–HBx overexpression for 24 h. (C) EMSA was performed with Huh7 and Huh7-X nuclear extracts using biotin-labeled probes containing the NF-κB binding site on the Shp2 promoter. Competition assays were performed using 1-fold, 10-fold, and 100-fold molar excesses of unlabeled wild-type (wt) and mutant (mut) oligonucleotides. (D) Shp2 promoter activities in NF-κB-overexpressing HEK293 cells after parthenolide treatment for 18 h. (E) Western blot analyses of SHP2 expression in Huh7-X, Hep3B and HEK293 cells after treatment with parthenolide. (F) Hep3B cells were transfected with a control siRNA or a NF-κB siRNA. After 48 h, the cells were analyzed by Western blot with anti-NF-κB and anti-SHP2 antibodies.
To further confirm the effect of HBx–NF-κB pathway activity on Shp2 expression, Shp2 promoter activity was measured after treatment with increasing amounts of the NF-κB inhibitor parthenolide. The results revealed that both Shp2 promoter activity and SHP2 protein levels were dose-dependently reduced in Huh7-X, Hep3B and HEK293 (Figure 2D, 2E). Next, we assessed the effects of NF-κB knockdown on SHP2 expression in Hep3B cells. Transfection of a NF-κB siRNA resulted in a marked decrease in SHP2 protein levels compared with transfection of a control siRNA (Figure 2F). These data confirm that HBx induces Shp2 expression at the transcription level via the HBx–NF-κB–SHP2 pathway in HBV-infected liver cells.
SHP2 acts as a link between HBx and RAS signaling and a negative regulator of STAT3 signaling in HCC cells
Numerous studies have reported that HBx increases the levels of GTP-bound RAS [13, 14, 29]. However, the mechanism by which HBx activation mediates the RAS pathway is not fully understood. SHP2 competes with RAS–GAP, a terminal protein of the EGFR pathway, for binding to Gab1. We hypothesized that in HBV-positive HCC cells, SHP2 overexpression induced by HBx–NF-κB pathway activation extends RAS activation. To test whether EGF stimulation alters SHP2 binding to the EGFR–Gab1 complex, we treated Huh7-X and Hep3B cells with EGF. EGF-treated Huh7-X and Hep3B cells exhibited the rapid assembly of a ternary complex comprising SHP2, EGFR, and Gab1, as well as ERK activation (Figure 3A). The hepatic inflammatory response activates the IL-6 and STAT3 pathway, accelerating HCC development [30]. Based on this notion, we next treated HBx-overexpressing Hep3B cells with LPS to determine the cell-intrinsic inflammatory responses. As shown in Figure 3B, NF-κB, SHP2, and phospho-ERK levels increased within 5 min of LPS treatment and peaked at 3 h. In contrast, phosphorylated STAT levels were lowest at 3 h, and increased thereafter. We further analyzed the inflammatory response to LPS in Hpe3B cells without HBx overexpression. The induction pattern of the SHP2-ERK pathway and STAT3 signaling was similar to that of HBx overexpressing Hebp3B (Figure 3C); however, SHP2 induction and complete STAT inactivation were noticeably delayed to 6 h in LPS-stimulated Hep3B cells. These results suggest that SHP2 is the missing link between the HBx–NF-κB pathway and RAS signaling. Moreover, an inverse relationship between the HBx–NF-κB–SHP2 pathway and STAT3 signaling exists in HCC cells in vitro.
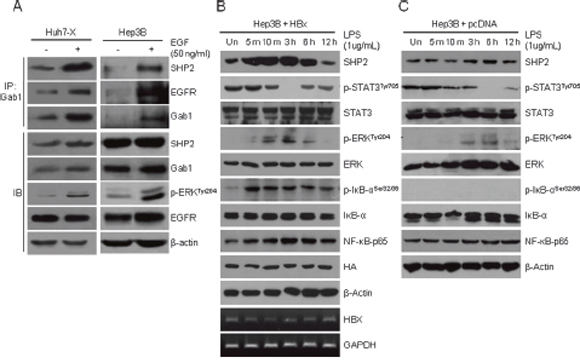
Figure 3: EGFR–SHP2–Gab1 complex-mediated ERK signaling and STAT3 signaling are inversely active in hepatocellular carcinoma cells. (A) A ternary complex comprising SHP2, EGFR, and Gab1 was immunoprecipitated (IP) using anti-Gab1 antibodies after EGF treatment for 10 min. Endogenous levels of SHP2, Gab1, EGFR, and phosphorylated ERK proteins were determined by immunoblotting (IB). (B) HBx-overexpressing Hep3B cells were stimulated with LPS (1 μg/mL). The relative amounts of total SHP2, p-STAT3, STAT3, p-ERK, ERK, p-IKB, NF-κB-p65 and HA were determined by Western blotting at the indicated times. β-Actin was used as the loading control. The expression of the HBx gene was detected by RT-PCR. GAPDH served as a control. (C) Hep3B cells were stimulated with LPS (1 μg/mL) and analyzed as described in panel (B).
High expression of SHP2 and NF-κB–p65 proteins in HBV-infected liver tissues
Formation of the Gab1/SHP2 complex is crucial for liver regeneration. Liver-specific SHP2 or Gab1 knockout mice display defective liver regeneration after partial hepatectomy [31]. Jiang et al. reported that SHP2 acts as a tumor suppressor protein and that decreased SHP2 expression is a poor prognostic marker in HCC [32]. To further confirm the existence of the HBx–NF-κB–SHP2 pathway, we obtained 23 pairs of tumor tissues and surrounding non-neoplastic tissues from patients with HBV-associated HCC and analyzed SHP2 expression via Western blot analysis. SHP2 expression was significantly lower in tumor tissues compared with surrounding non-neoplastic tissues in the 17 pairs examined (Figure 4A). We further analyzed 6 pairs of tissues in terms of the activity of the NF-κB–SHP2 and STAT3 signaling pathways. Notably, HCC-surrounding non-neoplastic tissues expressed significantly higher levels of NF-κB–p65, SHP2, and phospho-ERK than those of matched HCC tissues (Figure 4B). Bard-Chapeau et al. reported that increased inflammatory signaling through STAT3 could exacerbate chemical-induced HCC development [26]. In addition, HBV infection greatly exacerbates the NF-κB-mediated host immune response, and active NF-κB subsequently triggers massive inflammatory responses via IL-6–JAK–STAT3 signaling [33, 34]. Based on these observations, we next examined STAT3 activation in patient tissue samples. In contrast to in vitro data suggesting an inverse relationship between HBx–NF-κB–SHP2 activity and STAT3 signaling, HCC-surrounding non-neoplastic tissues exhibited increased IL-6–JAK–STAT3 signaling compared with matched tumor tissues. Decreased levels of both phospho-STAT3 and NF-κB in tumor tissues were also confirmed by enzyme-linked immunosorbent assay (ELISA; Figure 4C). These data suggest that the anti-parallel NF-κB–SHP2 and STAT signaling pathways are competitively active in precancerous inflammatory liver tissues, but their activities are lost simultaneously upon transformation of liver cells during HCC development.
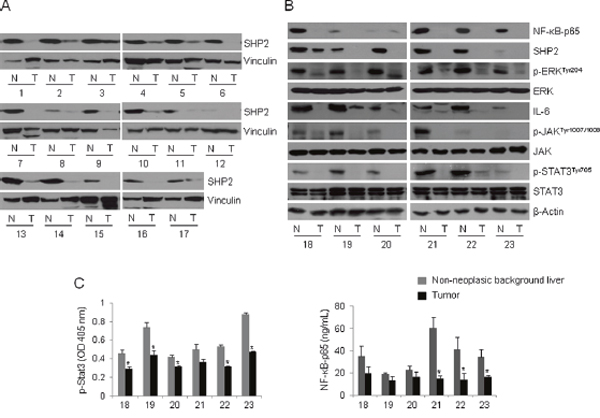
Figure 4: The NF-κB–SHP2–ERK and IL-6–JAK–SHP2 pathways are concomitantly increased in HBV-associated liver tissues. (A) Whole tissue lysates were prepared from 17 pairs of HBV-infected tumors (T) and matched non-neoplastic tissues (N), and analyzed by Western blot with anti-SHP2 antibodies. Vinculin served as a loading control. (A) NF-κB-p65, SHP2, p-ERK, ERK, IL-6, p-JAK, JAK, p-STAT3 and STAT3 levels were assessed by Western blotting in T/N tissues prepared as described in A. β-Actin was used as a loading control. (B) The whole tissue lysates described in (A) were assayed to determine the levels of p-STAT3 and NF-κB-p65. Columns represent the means of three independent experiments; bars indicate standard deviations (* P < 0.05). The numbers denote tissue numbers.
High SHP2 expression in non-neoplastic livers with advanced fibrosis
Because our earlier results indicating a loss of SHP2 in HCC tissues relative to HBV-positive paired background tissues were not obtained from a random cohort, we performed an additional immunohistochemical analysis of SHP2 expression in 162 tissues from patients with HBV-associated HCC (Supplementary Table 1). Patient demographics are described in Table 1. SHP2 was strongly and diffusely expressed in the cytoplasm of both tumor cells and surrounding non-neoplastic liver cells in patients with HCC. Representative levels of SHP2 immunoreactivity in tumor cells and background liver cells are shown in Figure 5A. Notably, the SHP2 expression levels were lower in tumors than in matched non-neoplastic surrounding tissues (P < 0.001) (Figure 5B). In addition, SHP2-positive tumors tended to associate with a lack of microvascular invasion (P = 0.016) (Table 2). No associations were observed between SHP2 expression and other clinicopathologic factors including patient age, sex, etiology, tumor size, serum alpha-fetoprotein level, and histological grade. In the survival analysis, SHP2 positivity was not associated with overall (P = 0.092) or recurrence-free (P = 0.744) survival.
Table 1: Clinicopathologic characteristics of 162 patients with resectable hepatocellular carcinoma
Characteristics |
Patients |
|
|---|---|---|
Number |
% |
|
All patients |
162 |
100 |
Age (years) |
||
Median, years (SD) |
53 (8.8) |
|
Range |
26–71 |
|
Sex |
||
Male |
120 |
74.1 |
Female |
42 |
25.9 |
Etiology |
||
HBV |
162 |
100 |
BCLC stage |
||
1 |
152 |
93.8 |
2 |
10 |
6.2 |
Fibrosis stage |
||
Early (I-II) |
26 |
16.0 |
Advanced (III-IV) |
136 |
84.0 |
Tumor size |
||
<5 cm |
121 |
74.7 |
≥5 cm |
41 |
25.3 |
AFP level (ng/ml) |
||
Normal (<10) |
57 |
35.2 |
Elevated (≥10) |
105 |
64.8 |
Edmondson grade |
||
I-II |
108 |
66.7 |
III-IV |
54 |
33.3 |
Microvascular invasion |
||
No |
112 |
69.1 |
Yes |
50 |
30.9 |
SD, standard deviation; BCLC, Barcelona clinic liver cancer; AFP, alpha-fetoprotein
Table 2: Relationship between SHP2 expression and clinicopathologic parameters in 162 patients with surgically resectable HBV-associated hepatocellular carcinoma
Parameter |
Total no. |
SHP2 (−) |
SHP2 (+) |
P-value* |
|---|---|---|---|---|
Age, years (median) |
162 |
53 |
53 |
0.758 |
Sex (%) |
||||
Male |
120 |
85 (70.8) |
35 (29.2) |
0.201 |
Female |
42 |
34 (81.0) |
8 (19.0) |
|
Fibrosis stage (%) |
||||
Early (I-II) |
26 |
15 (57.7) |
11 (42.3) |
0.047 |
Advanced (III-IV) |
136 |
104 (76.5) |
32 (23.5) |
|
Tumor size (%) |
||||
<5 cm |
121 |
89 (73.5) |
32 (26.5) |
0.962 |
≥5 cm |
41 |
30 (73.2) |
11 (26.8) |
|
AFP level (ng/ml) (%) |
||||
Normal (<10) |
57 |
40 (70.2) |
17 (29.8) |
0.486 |
Elevated (≥10) |
105 |
79 (75.2) |
26 (24.8) |
|
Edmondson grade (%) |
||||
I-II |
108 |
75 (69.4) |
33 (30.6) |
0.102 |
III-IV |
54 |
44 (81.5) |
10 (18.5) |
|
Microvascular invasion (%) |
||||
No |
112 |
76 (67.9) |
36 (32.1) |
0.0168 |
Yes |
50 |
43 (86.0) |
7 (14.0) |
*P-value was calculated using the two-tailed Student’s t-test.
HBV, hepatitis B virus; AFP, alpha-fetoprotein
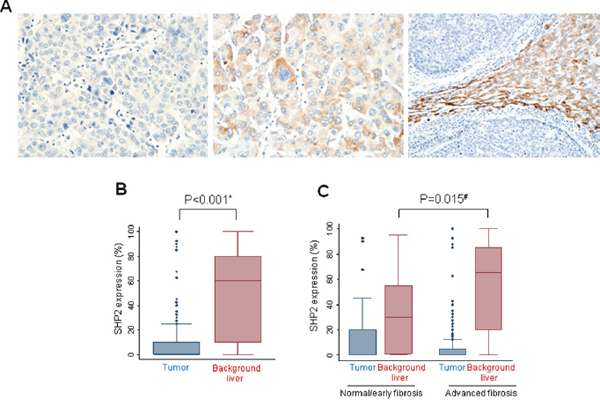
Figure 5: Immunohistochemical detection of SHP2 expression in human HCCs. (A) SHP2-negative tumor cells (left), SHP2-positive tumor cells (middle), and SHP2-negative tumor cells on a SHP2-positive liver background (right). (B) SHP2 expression is lower in tumor cells than in matched non-neoplastic liver cells. (C) SHP2 expression tends to be higher in surrounding tissues with advanced fibrosis. *, Wilcoxon matched-pairs signed ranks test and #, Wilcoxon rank sum test.
When patients were divided into early fibrosis and advanced fibrosis groups, a tendency toward lower SHP2 expression was observed in tumor cells in the advanced fibrosis group than in tumor cells in the early fibrosis group (P = 0.045). Inversely, SHP2 expression in background liver cells was much higher in the advanced fibrosis group than in the early fibrosis group (P = 0.015, Figure 5C). These results strongly suggest that SHP2 expression is significantly associated with inflammatory responses leading to fibrosis during hepatocarcinogenesis.
A gradual increase in SHP2 expression in accordance with increases in inflammation and fibrosis
To evaluate SHP2 expression according to the degrees of inflammation and fibrosis, tissue microarrays of 48, 21, and 51 patients with normal liver, chronic hepatitis, and dysplastic nodules, respectively, were subjected to IHC staining for SHP2 (Supplementary Table 2). The characteristics of the 120 patients are listed in Table 3. Normal liver tissues biopsied from healthy donors were negative for hepatitis A, B, and C viruses and chronic liver disease. All patients with HBV-associated chronic hepatitis exhibited variable degrees of inflammation and fibrosis, but not cirrhosis; however, HBV-positive patients with dysplastic nodules had cirrhotic background liver tissues. SHP2 expression was higher in chronic hepatitis tissues than in normal liver tissues (P = 0.057), and the highest levels observed in cirrhotic tissues (P < 0.001) (Figure 6A). In a further evaluation of cirrhotic tissues, SHP2 expression levels were lower in dysplastic nodules than in matched non-neoplastic cirrhotic background liver tissues, with mean values of 40 and 70, respectively (P = 0.301) (Figure 6B). Accordingly, the expression of phosphorylated STAT3 (active STAT3) was decreased in dysplastic nodules compared with chronic hepatitis tissues and completely lost in hepatocellular carcinoma tissue (Table 4), which is similar to the pattern of SHP2 expression. These data indicate a gradual increase in SHP2 expression during chronic liver inflammation and fibrosis, followed by a decrease during the early developmental stage of HCC.
Table 3: Clinicopathologic characteristics of patients without hepatocellular carcinoma
Characteristics |
Normal |
Chronic hepatitis |
Dysplastic nodule |
|---|---|---|---|
Mean age (range), years |
26 (15–54) |
47 (19–67) |
54 (41–71) |
Sex (%) |
|||
Male |
39 (81.3) |
15 (71.4) |
40 (78.4) |
Female |
9 (18.7) |
6 (28.6) |
11 (21.6) |
Hepatitis B virus (%) |
|||
Positive |
0 (0) |
21 (100) |
51 (100) |
Negative |
48 (100) |
0 (0) |
0 (0) |
AFP level (%) |
|||
Normal (<10) |
- |
- |
23 (45.1) |
Elevated (≥10) |
- |
- |
28 (54.9) |
Cirrhosis (%) |
|||
Present |
0 (0) |
0 (0) |
51 (100) |
Absent |
48 (100) |
21 (100) |
0 (0) |
AFP, alpha-fetoprotein
Table 4: Expression of p-STAT in HBV-infected liver
p-STAT |
Chronic |
Dysplastic |
Hepatocelluar |
P-value* |
|---|---|---|---|---|
Positive |
9 (42.8%) |
2 (3.9%) |
0 (0.0%) |
<0.001 |
Negative |
12 (57.2%) |
49 (96.1%) |
36 (100.0%) |
*P-value was calculated using the Fisher exact test.
p-STAT, phoshorylated STAT
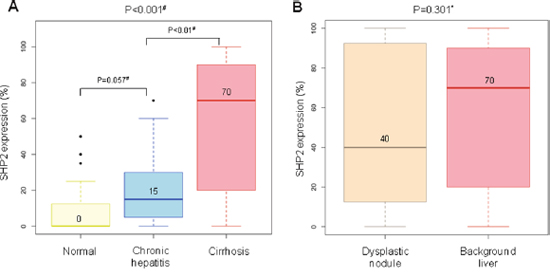
Figure 6: Representative levels of SHP2 immunoreactivity in livers without hepatocellular carcinoma. (A) SHP2 expression tends to increase gradually from normal livers to cirrhotic livers. (B) SHP2 expression is lower in dysplastic nodules than in matched non-neoplastic cirrhotic liver tissues. *, Wilcoxon matched-pairs signed ranks test and #, Wilcoxon rank sum test. The number above the histogram represents the mean value.
DISCUSSION
NF-κB acts major roles in liver damage induced by chronic inflammation and subsequent hepatocyte proliferation to replace damaged tissue [35–38]. Here we report for the first time the existence of an HBx–NF-κB–SHP2 pathway in the liver. The levels of SHP2 and NF-κB expression, as well as STAT3 phosphorylation, were significantly higher in non-neoplastic surrounding tissues than in matched HBV-associated HCC tissues. SHP2 expression gradually increased from normal liver tissues to those with chronic hepatitis and cirrhosis, decreased in dysplastic nodules, and was lost in HCCs. Of note, the combination of increased HBx–NF-κB–SHP2 pathway activity and increased STAT3 signaling in non-neoplastic background liver tissues suggests that these two signals are necessary for preventing HCC progression.
Gab1/SHP2 complex formation promotes ERK signaling by sequestering RasGAP, a RAS signal-terminating protein and thereby blocking its binding to Gab1 [39]. The Gab1/SHP2 complex can be detected immediately after hepatectomy, and liver-specific Gab1 or Shp2 knockout mice display similar defects in liver regeneration [31]. In our study, we found that the Shp2 (Ptpn11) promoter contains a putative NF-κB binding site (5′-GGTAATTTCC-3′) located 500-bp upstream of the translation initiation site. Transiently expressed HBx or NF-κB induced Shp2 promoter activity, and NF-κB bound to the Shp2 promoter in the ChIP assay. These results are the first to establish a link between the HBx–NF-κB and EGFR–Gab1/SHP2–RAS pathways. Depending on the magnitude of NF-κB activity, SHP2 could promote the compensatory proliferation of surviving hepatocytes via ERK pathway activation in damaged hepatic tissues.
NF-κB activation after HBV infection leads to the secretion of pro-inflammatory cytokines, including IL-1β, TNFα, and IL-6, in hepatocytes. Among these, IL-6, an IL-6–JAK–STAT3 pathway component, has a specific and critical function in HCC development [34, 40]. In our experiments, we observed concomitant NF-κB and SHP2 overexpression in non-neoplastic tissues adjacent to HBV-associated tumors. Increased IL-6–JAK–STAT3 signaling was also detected in those tissues (Figure 4A, 4B). NF-κB activation is an early event in hepatocarcinogenesis [41]. The NF-κB-activating kinase IKKβ acts as an early suppressor of chemically induced liver tumorigenesis by inhibiting hepatocyte death and compensatory cell proliferation [42]. Here we propose a model for SHP2 expression in which NF-κB and SHP2 progressively accumulate during the early stages of tumor development in liver tissues with a heterogeneous background, inflammation, and/or compensatory hepatocyte proliferation. After HCC initiation, SHP2 depletion could skew the balance between proliferation and inflammation and lead to an inflammatory phenotype involving STAT3, which might accelerate HCC development, as reported by Bard-Chapeau et al [26]. The HBx–NF-κB–SHP2 pathway might not be essential for the proliferation of hepatocytes that undergo malignant conversion; therefore, SHP2 expression might decline after HCC development. In this respect, we believe that SHP2 is a tumor suppressor, although its role is largely determined by NF-κB activity (Figure 7).
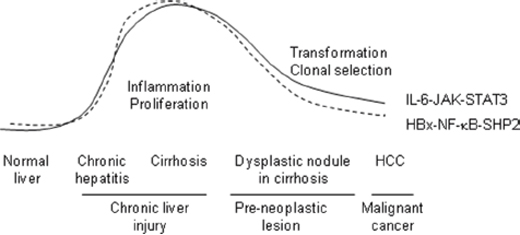
Figure 7: HCC-promoting HBx–NF-κB–SHP2 and IL-6–JAK–STAT3 pathways in the human liver. The activity levels of both pathways concomitantly increase during chronic inflammation and then gradually decline during the malignant transformation of hepatocytes in dysplastic nodules. Constant crosstalk is observed between IL-6–JAK–STAT3-mediated inflammatory responses and compensatory hepatocyte proliferation via the NF-kB–SHP2–EGFR pathway after viral infection. SHP2 depletion skews this balance and leads to accelerated HCC development.
Recently, Jiang et al. reported differences in SHP2 expression between tumor (T) and adjacent non-tumor (NT) tissues and divided HCCs into two groups depending on the level of SHP2 expression: T < NT and T ≥ NT. In tumors, a strong decrease in SHP2 expression (T < NT) was associated with short overall survival [32]. We also found that SHP2 expression was lower in the 162 HCC tissues than in the matched non-neoplastic surrounding tissues (P < 0.001) (Figure 5B), but did not detect a relationship between SHP2 expression and the overall patient survival duration (P = 0.092). Instead, we found strong associations of SHP2 overexpression with a lack of microvascular invasion (P = 0.016) (Table 2) and the presence of advanced background tissue fibrosis (P = 0.015) (Figure 5C). SHP2 expression levels were very low in normal liver tissues from HBV-free individuals, but they gradually increased in HBV-infected livers in accordance with increased inflammation and fibrosis. In addition, SHP2 expression tended to decrease or disappear in tumors and increase in fibrotic background tissues. Taken together, our results indicate that SHP2, as a target of NFκB, is likely to be a marker of inflammation, the expression of which can be either maintained or increased during fibrosis and the early stages of HCC development. It is important to note that most studies of HCC development have involved specific conditions (such as LPS treatment, chemically induced HCC animal models, or knockout mice), which focus on the process of HCC development. Our in vitro data show the competition between HBx–NF-κB–SHP2 activity and STAT3 signaling; in contrast, our in vivo data are indicative of the consequences of competition between the two pathways.
In summary, we showed that SHP2 expression was activated by the HBx–NF-κB pathway. In patients with HCC, a loss of SHP2 expression was associated with suppressed NF-κB–SHP2–ERK pathway activity and accelerated HCC development, whereas SHP2 overexpression in parallel with increased STAT3 activity was associated with fibrosis promotion during the early stages of HCC development. To our knowledge, this is the first report to demonstrate the existence of an HBx–NF-κB–SHP2 pathway in HBV-infected livers and the simultaneous activation of SHP2 and STAT3 pathways in inflammatory livers, leading to the spontaneous development of HCC.
MATERIALS AND METHODS
Materials
EGF was obtained from Cell Signaling Technology (Beverly, MA, USA). An NF-κB inhibitor, parthenolide, was obtained from Sigma-Aldrich (P0667; St. Louis, MO, USA) and prepared in dimethyl sulfoxide.
Cell culture
The HCC cell lines Huh7 and Hep3B were obtained from the National Cancer Institute (Bethesda, MD, USA). All cells were maintained in DMEM (Invitrogen-Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml; Invitrogen-Gibco) at 37°C in a humidified 5% CO2 incubator. Huh7-X cells overexpressing HBx were maintained in DMEM containing 200 mg/ml of G418 (Duchefa Biochemie, Haarlem, Netherlands) [43].
Western blot analysis
Whole-cell lysates were prepared in a cell lysis buffer (Cell Signaling Technology) containing a protease (Tech & InnovationTM, Bucheon, Korea) and phosphatase (sc-45065; Santa Cruz Biotechnology, Dallas, TX, USA) inhibitor cocktail. Proteins were separated on 10% or 12% SDS-PAGE gels, transferred to polyvinylidene fluoride membranes using an iBlotTM dry blotting system (Invitrogen), and analyzed using primary antibodies against the following proteins: SHP2 (sc-280; Santa Cruz), ERK (sc-154; Santa Cruz), phospho-ERK (Tyr204) (sc-7383; Santa Cruz), HA (H6908; Sigma-Aldrich), Gab1 (06-579; Millipore, Billerica, MA, USA), GAP (06-157; Upstate Biotechnology, Charlottesville, VA, USA), EGFR (2232; Cell Signaling Technology), STAT3 (9132; Cell Signaling), phospho-STAT3 (Tyr705) (9131; Cell Signaling), and β-actin (A5441; Sigma-Aldrich) or vinculin (v9131; Sigma-Aldrich). Blots were developed using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA).
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA (1 μg) was extracted using the NucleoSpin® RNA II kit (740955; Macherey-Nagel, Duren, Germany) and reverse-transcribed using SuperScript® II Reverse Transcriptase (Invitrogen). qRT-PCR was performed on a Bio-Rad iQ5 machine (Hercules, CA, USA) with SYBR Green (S7563; Invitrogen) and the following primer sets: HBx, 5′-GGACTCTACCGTCCCCTTCT-3′ and 5′-CGCTGAGAGTCCAAGAGTCC-3′, yielding a 203-bp fragment; SHP2, 5′-ATACGACGTTGGTGGAGGAG-3′ and 5′-TAAGGGGCTGCTTGAGTTGT-3′, yielding a 116-bp fragment; and GAPDH, 5′-GAAGGTGAAGGTCGG AGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′, yielding a 226-bp product. The thermal cycling conditions comprised an initial denaturation step at 95°C for 5 min and 40 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, followed by a final extension step at 72°C for 7 min. The expression of HBx or SHP2 mRNA was normalized to that of GAPDH.
Luciferase assays
HEK293 cells were plated in 12-well culture plates and transfected with 0.5 μg of Shp2 promoter-luciferase reporter plasmids (Shp2-Luc: 5′-flanking region of the human Shp2 promoter spanning nucleotides –1,727 to –151, with numbering starting at the position of the translation start site) or the control pGL3-Basic Vector (Promega, Madison, WI, USA) alone or in combination with 0.5 μg of a plasmid encoding HA–HBx or NF-κB, together with 0.01 μg of Renilla luciferase plasmid (pRL-SV40). Lipofectamine 2000 (Invitrogen) was used for transfection. The cells were harvested 48 h after transfection, and firefly luciferase activity was measured in three independent experiments. Data were normalized to Renilla luciferase activity.
ChIP assay
Approximately 106 Huh7 or HEK293 cells were plated onto 10-cm plates and transfected with 6 μg of HBx expression vectors. After a 24-h transfection step, the cells were cross-linked with 1% formaldehyde for 10 min at room temperature. ChIP assays were performed using an antibody against NF-κB–p65 (sc-372; Santa Cruz). Immunoprecipitated DNA was analyzed by PCR using primers spanning the NF-κB binding site: 5′-TGGCTTAAGCGAACCTCTTG-3′ and 5′-CGCTCCACTCATTAGCTGTG-3′. The GAPDH gene was used as an internal control and amplified using the following primers: 5′-TACTAGCGGTTTTACGGGCG-3′ and 5′-TCGAACAGGAGGAGCAGAGAGCGA-3′.
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared from Huh7 and Huh7-X cells using NE-PER Nuclear and Cytoplasmic Extraction Reagents (78833, Thermo Scientific, Rockford, IL) and EMSA was performed using a LightShift® Chemiluminescent EMSA Kit (20148, Thermo Scientific) according to the manufacturer’s instructions. Eight micrograms of nuclear extracts were pre-incubated for 20 min at room temperature in a 20-μL reaction solution containing 1× binding buffer, 50 ng/μL poly (dI-dC), and 20 pmol of biotin-labeled probes complementary to the NF-κB–binding site (GGTAATTTCC) in the Shp2 promoter. Complexes were resolved in 6% non-denaturing polyacrylamide gels. For competition experiments, the nuclear extracts were pre-incubated for 10 min in the presence of a 1-, 10-, or 100-fold excess of the following unlabeled double-stranded oligonucleotides: wild-type NF-κB, 5′-GGCTAGGTAATTTCCCTGGTCAT-3′; mutated NF-κB, 5′-GGCTAGGaAcgTTCCCTGGTCAT-3′ (NF-κB–binding sites are underlined, and mutated sites are in lowercase).
siRNA transfection
Hep3B cells were transfected with 25, 50 or 100 nmol/L siRNA targeting NF-κB (#6261, Cell Signaling) or with control siRNA (D-001210-01; GE Healthcare Dharmacon, Lafayette, CO) using Oligofectamine (Invitrogen). After 48 h, the cells were harvested.
Immunoprecipitation experiment
Huh7-X and Hep3B cells were treated with EGF (50 ng/ml) for 10 min after serum starvation for 24 h. The cells were lysed in CHAPS lysis buffer containing 150 mM NaCl, 10 mM HEPES pH 7.5, 1% CHAPS, and protease inhibitors. Gab1 was precipitated from cell extracts during an overnight incubation with anti-Gab1 antibodies, after which 20 μl of protein A/G PLUS-agarose beads (sc-2003; Santa Cruz) were added. The immunoprecipitates were centrifuged and subjected to Western blot analysis with SHP2, EGFR, or Gab1 antibodies.
ELISA
The levels of NF-κB-p65 and phospho-STAT3 (p-Tyr705) in tumors and background non-neoplastic liver tissues from patients with HCC were analyzed using the NBP2-29661 (Novus Biologicals, Littleton, CO, USA) and RAB0447 ELISA kits (Sigma, St. Louis, MO, USA), respectively. The tissues were lysed with a RIPA cell lysis buffer supplemented with phosphatase inhibitors. To detect NF-κB-p65 and phospho-STAT3 (p-Tyr705) proteins, ELISA plates were coated with a capture antibody and incubated overnight at 4°C. The wells were subsequently washed, blocked for 1 h, and incubated for 2 h with standards or samples. After extensive washing, the wells were incubated with a detection antibody followed by an HRP-conjugated secondary antibody for 1 h each. Color reactions were obtained by adding a substrate, followed by a stop solution. The absorbance of the color reactions was read at 450 nm using an absorbance spectrometer (Victor X3; PerkinElmer, Waltham, MA, USA).
Isolation of proteins from HBV-infected HCC tissues
This work was approved by the Institutional Review Board of Asan Medical Center, and all patients provided written informed consent. A total of 23 pairs of HCC and peripheral non-neoplastic tissues from patients with HBV-associated HCC and their associated data were provided by Asan Bio-Resource Center [2011-2(31)], Korea Biobank Network at Asan Medical Center. All tissues were lysed with RIPA buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 0.1% NP-40, 0.1% SDS], followed by three freeze-thaw cycles and vortexing for 10 min at 4°C. After centrifugation at 16,100 ×g for 30 min, a volume of the upper layer, corresponding to 30 μg of the protein, was analyzed by Western blotting.
Tissue microarrays
A total of 18 tissue microarrays were constructed using formalin-fixed, paraffin-embedded tissues from surgically resected or ultrasound-guided needle biopsy specimens. Hematoxylin and eosin-stained slides were examined by two pathologists (EY and HJK) to identify tumor and normal tissues and evaluate inflammation and fibrosis. Two tumor cores measuring 2 mm in diameter were arrayed from the corresponding paraffin blocks into a recipient block containing holes created using an arraying machine (TMArrayer; Pathology Devices, Westminster, MD, USA).
IHC
IHC staining was performed on formalin-fixed, paraffin-embedded tissue sections using a Benchmark automatic immunostaining device (Ventana Medical Systems, Tucson, AZ, USA). Briefly, 5-μm-thick tissue sections were transferred onto adhesive slides and dried at 62°C for 30 min. After incubation in 3% H2O2 to block endogenous peroxidase activity, tissue sections were subjected to standard heat-mediated epitope retrieval in ethylene diamine tetraacetic acid (pH 8.0) for 32 min. The samples were incubated with primary antibodies against SHP2 (1:75; Abcam, Cambridge, UK) and phospho-STAT3 (Tyr705) (1:200, Cell signaling) for 16 min, respectively. SHP2– and phospho-STAT3–antibody complexes were detected with the OptiView DAB IHC Detection Kit (Ventana Medical Systems). Immunostained sections were counterstained with hematoxylin, dehydrated in ethanol, and cleared in xylene. The percentage of stained cells (0–100%) was used as an estimate of the SHP2 expression level. Tissue samples exhibiting diffuse cytoplasmic staining with/without nuclear staining in >10% of tumor cells or background non-neoplastic liver cells were considered SHP2 positive. Cases exhibiting strong nuclear staining in >10% of tumor cells or background non-neoplastic liver cells were considered positive for phospho-STAT3.
Statistics
Values are presented as means ± standard deviations (n = 3 or 4). Comparisons between groups were analyzed using two-tailed Student’s t-tests or the Wilcoxon rank sum test as appropriate. The relationship between SHP2 positivity and clinicopathologic factors was determined using the chi square test. Survival analysis was determined according to the Kaplan–Meier method, and survival curves were compared using the log-rank test. The Wilcoxon-matched pairs signed rank test was used to evaluate patients with matched tumor and normal tissues. Statistical significance (P < 0.05) is indicated in figures by asterisks (*). Statistical analyses were performed using the R program (v2.14.2, www.r-project.org, Vienna Austria) and the Stata/IC statistical software package (v12; StataCorp Ltd., College Station, TX, USA).
ACKNOWLEDGMENTS
This work was supported by grant number NRF-2013R1A1A2062598 from the National Research Foundation of Korea and grant number 2014-309 from the Asan Institute for Life Sciences.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007; 132: 2557-2576.
2. Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, Brechot C, Harris CC. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002; 181-182: 43-47.
3. Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol. 2014; 20: 11630-11640.
4. Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. 2011; 26: 144-152.
5. Zheng Y, Chen WL, Louie SG, Ou JH. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology. 2007; 45: 16-21.
6. Luedde T, Schwabe RF. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011; 8: 108-118.
7. Seto E, Mitchell PJ, Yen TS. Transactivation by the hepatitis B virus X protein depends on AP-2 and other transcription factors. Nature. 1990; 344: 72-74.
8. Natoli G, Avantaggiati ML, Chirillo P, Costanzo A, Artini M, Balsano C, Levrero M. Induction of the DNA-binding activity of c-jun/c-fos heterodimers by the hepatitis B virus transactivator pX. Mol Cell Biol. 1994; 14: 989-998.
9. Nakatake H, Chisaka O, Yamamoto S, Matsubara K, Koshy R. Effect of X protein on transactivation of hepatitis B virus promoters and on viral replication. Virology. 1993; 195: 305-314.
10. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004; 78: 12725-12734.
11. Liu LP, Hu BG, Ye C, Ho RL, Chen GG, Lai PB. HBx mutants differentially affect the activation of hypoxia-inducible factor-1alpha in hepatocellular carcinoma. Br J Cancer. 2014; 110: 1066-1073.
12. Maguire HF, Hoeffler JP, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991; 252: 842-844.
13. Doria M, Klein N, Lucito R, Schneider RJ. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995; 14: 4747-4757.
14. Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A. 1994; 91: 10350-10354.
15. Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007; 128: 369-381.
16. Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006; 25: 6706-6716.
17. Yun C, Um HR, Jin YH, Wang JH, Lee MO, Park Lee JH, Cho H. NF-kappaB activation by hepatitis B virus X (HBx) protein shifts the cellular fate toward survival. Cancer Lett. 2002; 184: 97-104.
18. Lim KH, Choi HS, Park YK, Park ES, Shin GC, Kim DH, Ahn SH, Kim KH. HBx-induced NF-kappaB signaling in liver cells is potentially mediated by the ternary complex of HBx with p22-FLIP and NEMO. PLoS One. 2013; 8: e57331.
19. Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004; 103: 2325-2331.
20. Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003; 34: 148-150.
21. Zheng H, Alter S, Qu CK. SHP-2 tyrosine phosphatase in human diseases. Int J Clin Exp Med. 2009; 2: 17-25.
22. Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008; 27: 179-192.
23. Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology. 2008; 53: 389-402.
24. Cunnick JM, Mei L, Doupnik CA, Wu J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem. 2001; 276: 24380-24387.
25. Liu Y, Rohrschneider LR. The gift of Gab. FEBS Lett. 2002; 515: 1-7.
26. Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, Fang DD, Han T, Bailly-Maitre B, Poli V, Varki NM, Wang H, Feng GS. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011; 19: 629-639.
27. Li S, Hsu DD, Wang H, Feng GS. Dual faces of SH2-containing protein-tyrosine phosphatase Shp2/PTPN11 in tumorigenesis. Front Med. 2012; 6: 275-279.
28. Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998; 26: 362-367.
29. Cross JC, Wen P, Rutter WJ. Transactivation by hepatitis B virus X protein is promiscuous and dependent on mitogen-activated cellular serine/threonine kinases. Proc Natl Acad Sci U S A. 1993; 90: 8078-8082.
30. Yu H, Pardoll D, Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009; 9: 798-809.
31. Bard-Chapeau EA, Yuan J, Droin N, Long S, Zhang EE, Nguyen TV, Feng GS. Concerted functions of Gab1 and Shp2 in liver regeneration and hepatoprotection. Mol Cell Biol. 2006; 26: 4664-4674.
32. Jiang C, Hu F, Tai Y, Du J, Mao B, Yuan Z, Wang Y, Wei L. The tumor suppressor role of Src homology phosphotyrosine phosphatase 2 in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2012; 138: 637-646.
33. Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013; 4: 176-185.
34. He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011; 21: 159-168.
35. Arsura M, Cavin LG. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005; 229: 157-169.
36. Chung C, Park SG, Park YM, Joh JW, Jung G. Interferon-gamma sensitizes hepatitis B virus-expressing hepatocarcinoma cells to 5-fluorouracil through inhibition of hepatitis B virus-mediated nuclear factor-kappaB activation. Cancer Sci. 2007; 98: 1758-1766.
37. Pal S, Bhattacharjee A, Ali A, Mandal NC, Mandal SC, Pal M. Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. J Inflamm (Lond). 2014; 11: 23.
38. Sun B, Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene. 2008; 27: 6228-6244.
39. Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol. 2003; 13: 122-130.
40. Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. Interferon: cellular executioner or white knight? Curr Med Chem. 2007; 14: 1279-1289.
41. Liu P, Kimmoun E, Legrand A, Sauvanet A, Degott C, Lardeux B, Bernuau D. Activation of NF-kappa B, AP-1 and STAT transcription factors is a frequent and early event in human hepatocellular carcinomas. J Hepatol. 2002; 37: 63-71.
42. He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010; 17: 286-297.
43. Gu JM, Lim SO, Oh SJ, Yoon SM, Seong JK, Jung G. HBx modulates iron regulatory protein 1-mediated iron metabolism via reactive oxygen species. Virus Res. 2008; 133: 167-177.