
INTRODUCTION
Medulloblastoma is the most common malignant brain tumor of childhood [1]. Despite multimodal therapy approaches including surgery, radiation and chemotherapy, overall survival of patients with medulloblastoma is currently 40–70% and this disease remains a major clinical challenge in pediatric oncology. Deficits in neurocognitive and neuroendocrine function, fertility, hearing and other long-term sequelae are common, and stem from aggressive treatment regimens [2–5]. Development of targeted approaches with fewer side effects is an essential step to improve long-term survival and quality of life in survivors. Since medulloblastomas are molecularly heterogeneous and studies have shown that molecular differences influence therapy response and outcome, therapy reduction should be considered for patients with tumor molecular profiles predicting good outcome as should therapy intensification for patients with profiles predicting poor outcome [6–8]. Four molecular subtypes designated as the WNT subtype, sonic hedgehog (SHH) subtype, Group 3 and Group 4 have been identified with different clinical outcomes [9]. While patients with medulloblastomas belonging to the WNT subtype have a 5-year overall survival of over 90%, 5-year overall survival in patients with a Group 3 medulloblastoma is only 40–60% [8, 10–12], underlining the importance of using this molecular subtyping to select the right therapy. Genetic alterations in the tumor also influence therapy response and outcome, including MYCN or GLI2 amplifications or TP53 mutations [13, 14]. Nevertheless, TP53 tumor suppressor dysfunction is rarely caused by the TP53 mutations identified in medulloblastomas [15, 16], but mostly occurs in WNT and SHH medulloblastoma subtypes and relapsed medulloblastomas [17]. Our group has previously shown that overexpression of MDM2, leading to increased TP53 degradation, is frequently observed in medulloblastomas with wildtype TP53, and that nutlin-3, which inhibits TP53-MDM2 interaction, reduces tumor growth in vitro and in vivo [18]. Nutlin-3 treatment has also been shown to increase the frequency of TP53 mutations in osteosarcomas and colon carcinomas, resulting in resistance [19]. Therefore, a treatment strategy to restore TP53 function in tumors harboring either wildtype or mutated TP53 is required.
The RITA small molecule, whose name stems from reactivation of TP53 and induction of tumor cell apoptosis, binds to the N-terminus of TP53 and induces a conformational change that inhibits interaction with MDM2, resulting in anti-tumor activity in vitro and in vivo [20–23]. Interestingly, this effect was also seen in tumors harboring TP53 mutations [24], questioning the original opinion that RITA works exclusively through blockade of the TP53-MDM2 pathway [20]. Here, we investigated whether RITA could reactivate the TP53 pathway in medulloblastomas independently of TP53 mutational status, therefore, opening new avenues for the successful treatment of medulloblastomas, regardless of their TP53 mutational status.
RESULTS
RITA reduces medulloblastoma cell viability in vitro independent of TP53 status
We set out to explore the effect of RITA on medulloblastoma cell viability in culture and determine whether TP53 mutational status alters RITA efficacy. We treated medulloblastoma cell lines harboring wildtype TP53 (HD-MB03 and ONS-76) or TP53 mutations (DAOY and UW-228-2) with varying concentrations of RITA for 24–72 h, then assessed cell viability. All 4 medulloblastoma cell lines displayed a reduction in cell viability during RITA treatment after 72 h, but the extent of reduction varied between the different cell lines (Figure 1A). Calculating the IC50s for the various medulloblastoma cell lines revealed that the anti-tumor effect of RITA was independent of TP53 mutational status (p = 0.42; Figure 1B). While cells expressing wildtype TP53 displayed IC50s of 19.3 ± 6.4 nM (HD-MB03) and 2.5 ± 0.4 μM (ONS-76), cells harboring TP53 mutations displayed IC50s of 94.0 ± 49.3 nM (DAOY) and 5.4 ± 0.2 μM (UW-228-2). The effect of RITA on the density of the adherently growing cell lines was also observed microscopically, where especially HD-MB03 and DAOY cultures were less dense after 48 h of RITA treatment compared to cells receiving only control medium containing the vehicle, ethanol (Figure 1C). We next examined whether RITA treatment could induce apoptosis in cultured medulloblastoma cell lines. We assessed the fraction of sub-G1 cells in cultures using FACS analysis to detect the apoptotic cell fraction. After 24 h of RITA treatment, the apoptotic fraction in HD-MB03 cultures was expanded by 16-fold (P = 0.004) compared to control cultures (Figure 1D). RITA treatment raised the apoptotic fraction in ONS-76 (P = 0.01) and DAOY (P = 0.03) cultures by 5-fold compared to controls, but had no significant effect on UW-228-2 apoptosis (Figure 1D). To confirm that RITA induced medulloblastoma cell death using an alternative method, we utilized an ELISA to detect mono- and oligonucleosomes in the cytoplasmatic fraction of cell lysates. RITA treatment induced cell death in HD-MB03 cells by 2-fold (P = 0.006) after only 24 h compared to untreated controls (Figure 1E). Data from ONS-76 and DAOY cell lines was too variable for apparent cell death increases to reach significance statistically, and RITA treatment had no significant effect on cell death in UW-228-2 cells in accordance with our data from the FACS analyses. We next investigated whether RITA treatment could halt medulloblastoma cell proliferation by assessing BrdU incorporation. HD-MB03 (P = 0.006) and DAOY (P < 0.0001) cell proliferation was dramatically reduced after 72 h of RITA treatment, whereas proliferation of ONS-76 and UW-228-2 cells remained unaffected (Figure 1F). Taken together, RITA demonstrated variable levels of anti-tumor activity against medulloblastoma cell lines, but variable efficacy did not correlate with the mutational status of TP53.
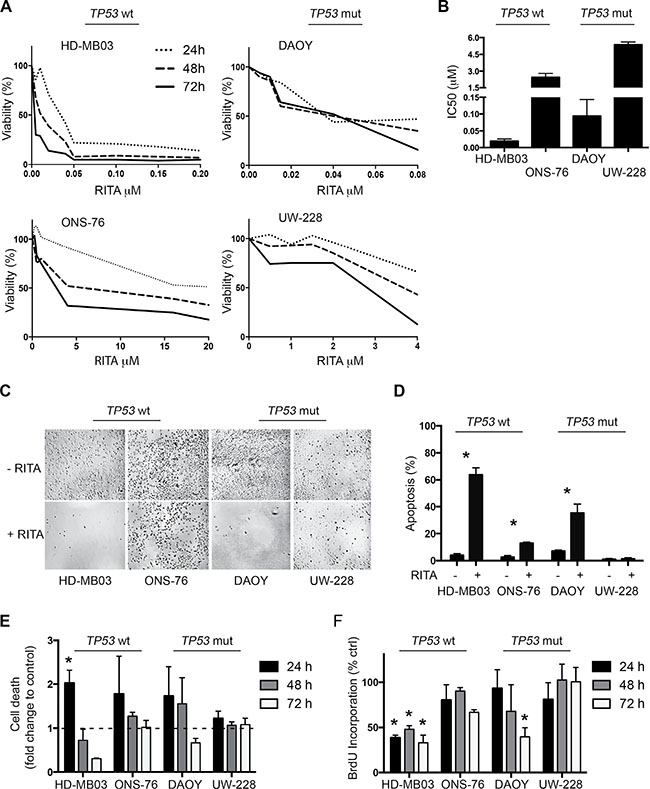
Figure 1: RITA treatment reduces cell viability and increases apoptosis in medulloblastoma cell lines with and without TP53 mutations. (A) Medulloblastoma cell viability was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolinum bromide (MTT) assay in a time- and dose-dependent manner. (B) IC50 calculation of the data shown in A. (C) Microscopic pictures of the four adherent cell lines treated with 1 μM RITA for 48 h. Negative control: ethanol. (D) Assessment of apoptosis in medulloblastoma cell lines after 24 h of treatment with 1 μM RITA or ethanol by sub-G1 fraction FACS analysis. * = P < 0.05 in Student’s t-test. (E, F) Cell death and proliferation analysis of medulloblastoma cell lines treated with 1 μM RITA for 24, 48 and 72 h using ELISA and BrdU Assay. Ethanol served as a negative control. * = P < 0.05 in Student’s t-test.
RITA restores TP53 activity in medulloblastoma cell lines with and without TP53 mutations
To investigate whether RITA treatment reactivates TP53 signaling in medulloblastoma cells, we assessed expression of TP53 pathway components in the 4 medulloblastoma cell lines with and without RITA treatment. An increase in TP53 and CDKN1A expression was detected in HD-MB03, ONS-76 and DAOY cells after 48 h of RITA treatment (Figure 2A). Likewise, CDKN1A expression was also elevated in these 3 cell lines after RITA treatment (HD-MB03 3-fold higher, P = 0.003; ONS-76 7.2-fold higher, P = 0.002; DAOY 6.7-fold higher, P < 0.001; Figure 2B). Interestingly, RITA treatment induced MDM2 expression only in cell lines with wildtype TP53 (HD-MB03 1.7-fold higher, P = 0.003; ONS-76 2.4-fold higher, P = 0.01; Figure 2C). We also enforced expression of a TP53 dominant negative mutant (dn-p53) in the medulloblastoma cell line most responsive to RITA treatment (HD-MB03) and the ONS-76 cell line, which was less sensitive to RITA treatment (Supplementary Figure 1). Enforced dn-p53 expression did not affect proliferation in either cell line (HD-MB03 wt vs HD-MB03 dn-p53 p = 0.47, ONS-76 wt vs ONS-76 dn-p53 p = 0.18; data not shown). We determined the IC50s for RITA treatment after dn-p53 expression compared to parental cell lines (Figure 3A). Enforced dn-p53 expression did not significantly influence the effect of RITA treatment, as measured by the IC50s (HD-MB03 vs HD-MB03 dn-p53, P = 0.07; ONS-76 vs ONS-76 dn-p53, P = 0.33), demonstrating that TP53 mutational status does not strongly impact the anti-tumor activity of RITA. We next examined whether enforced dn-p53 expression influenced the effect of RITA on apoptosis and cell proliferation. No differences in proliferation and cell death were detectable between transfected and non-transfected cell lines (Figure 3B, 3C), but increased CDKN1A was observed (Supplementary Figure 2). Taken together, our results support the hypothesis that RITA treatment activates TP53 signaling in medulloblastoma cell lines regardless of TP53 mutational status.
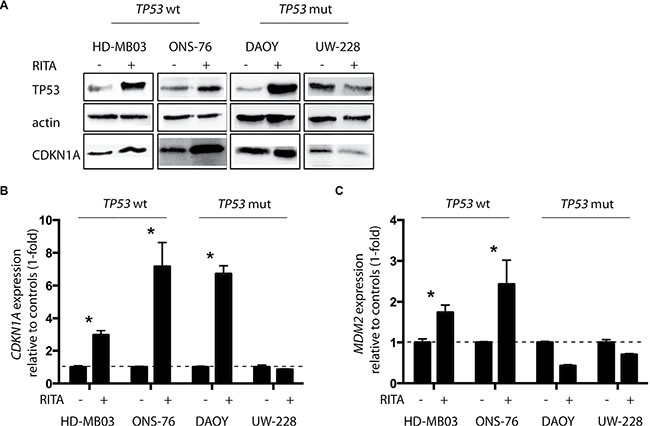
Figure 2: RITA restores TP53 activity in medulloblastoma cell lines with and without TP53 mutations. (A) TP53 and CDKN1A protein expression after 48 h of RITA treatment assessed by western blot. Ethanol served as a negative control. wt = wildtype, mut = mutation. (B, C) Relative expression of CDKN1A and MDM2 mRNA after 48 h of RITA treatment measured by real-time polymerase chain reaction (PCR). Ethanol served as a negative control. * = P < 0.05 in Student’s t-test.
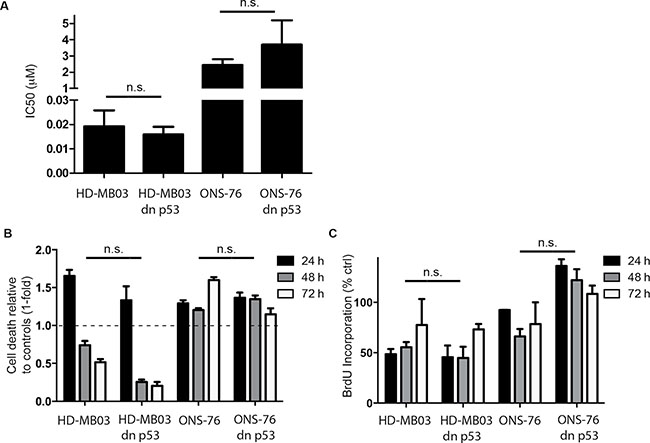
Figure 3: RITA reduces viability and proliferation in medulloblastoma cell lines expressing dominant-negative TP53 (dn-p53). (A) Determination of IC50s from HD-MB03 or ONS-76 with and without dn-p53 using cell viability data obtained by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolinum bromide (MTT) assay. (B, C) Cell death and proliferation analysis of medulloblastoma cell lines treated with 1 μM RITA for 24, 48 and 72 h using ELISA and BrdU Assay. Ethanol served as a negative control.
RITA treatment has anti-tumoral activity against human medulloblastoma cells grown as xenografts in mice
To test the anti-tumoral activity of RITA in an in vivo model of medulloblastoma, we treated mice with established HD-MB03 xenografts once daily with RITA then assessed its efficacy. The treatment regimen was well tolerated, and did not cause weight loss or otherwise alter the physical status or behavior of the mice, or produce obvious signs of toxicity. The growth of HD-MB03 xenografts was significantly slowed in mice treated with RITA (n = 13) compared to mice treated with vehicle control (n = 10; Figure 4A). After 7 days of treatment, mean tumor volume progressed only to 324 mm3 in RITA-treated mice compared with the mean tumor volume of 588 mm3 for vehicle-treated mice (P = 0.04). With a primary end point defined as a tumor volume of 1000 mm3, the survival of the mice treated with RITA was significantly prolonged compared to mice treated with vehicle control (P = 0.04; Figure 4B). In a different, more intensive treatment model, RITA treatment was administered twice daily for 3 days and tumors were subsequently analyzed. Intensified RITA treatment did not increase CDKN1A expression, but increased MDM2 expression (P = 0.01) and activated the TP53 pathway in the xenograft tumor tissue (Figure 4C–4F), which corresponded well with our results for RITA treatment of medulloblastoma cell lines grown in vitro. Immunohistochemical analysis of the xenograft tumors indicated RITA treatment increased apoptotic activity from increased levels of cleaved caspase 3 (P = 0.003, Figure 4E) and restored TP53 levels (P < 0.0001; Figure 4F). Thus, RITA activated the TP53 pathway in vivo, decreased xenograft tumor growth and increased mouse host survival.
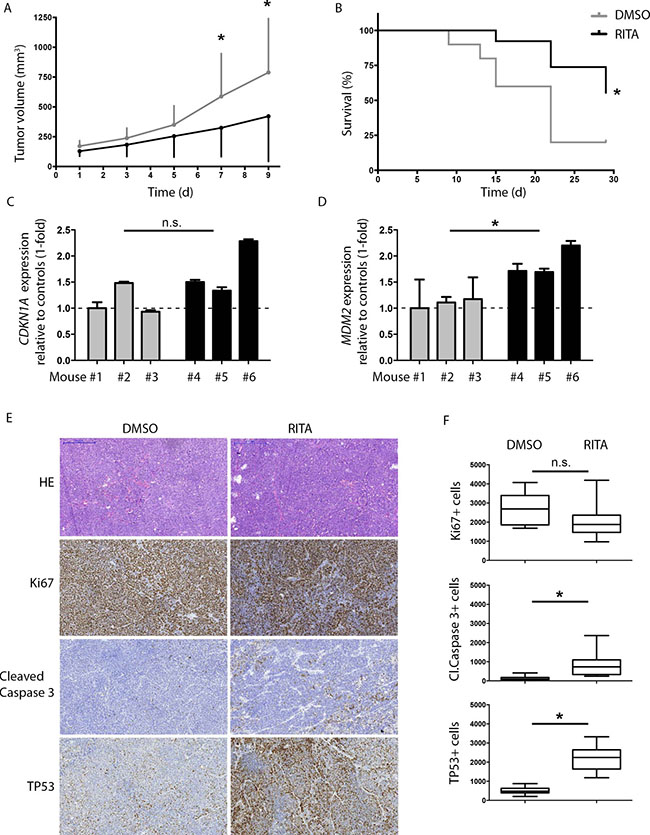
Figure 4: RITA treatment has anti-tumoral activity against medulloblastoma tumor xenografts in mice. (A) Tumor growth response to intraperitoneal administration of RITA (n = 13), compared to DMSO (n = 10) in mice. * = P < 0.05 in Student’s t-test. d = days. (B) Kaplan–Meier survival plot of treated cohorts from A. (C, D) Relative expression of CDKN1A and MDM2 mRNA in tumors harvested after 3 days of DMSO (mouse#1, 2 and 3) or RITA (mouse#4, 5, 6) treatment. mRNA levels were measured using real-time polymerase chain reaction (PCR). Ethanol served as a negative control. * = P < 0.05 in Student’s t-test. (E, F) Immunohistochemical analysis of proliferation (Ki67), apoptosis (cleaved caspase 3) and TP53 in tumors from mice treated for 3 days with RITA (n = 3) or DMSO (n = 3). * = P < 0.05 in Student’s t-test.
DISCUSSION
We report here that RITA shows anti-tumor activity in vitro against medulloblastoma cell lines independent of TP53 status as well as in vivo against a xenograft tumor formed using a medulloblastoma cell line lacking TP53 mutation. RITA exerted its anti-tumor effect and re-activated the TP53 pathway.
Reactivation of TP53 functionality has recently been shown to be achievable by different methods [15]. RITA, for example, binds to the MDM2 binding site of TP53, thereby blocking TP53 ubiquitination by MDM2. RITA has been demonstrated to have anti-tumor activity against cell lines derived from colon carcinoma, osteosarcoma, fibrosarcoma and myeloma [20, 25]. Importantly, even tumors that displayed resistance against standard chemotherapies remained sensitive to RITA treatment [26]. Here we demonstrated that RITA reduces cell viability and increases apoptosis of medulloblastoma cell lines. RITA treatment also induced transcript and protein expression of the TP53 downstream target, CKDN1A, demonstrating reactivation of TP53 function. Interestingly, MDM2 expression was only induced in cell lines that did not harbor TP53 mutations, indicating activation of the MDM2-TP53 feedback loop.
Existing in vitro data present some controversy about whether RITA requires intact TP53 activity to induce its anti-tumor effects. While earlier studies demonstrated that the anti-tumor effect was stronger in cell lines possessing wildtype TP53 than in cell lines harboring TP53 mutations [26, 27], more recent publications showed RITA produced an anti-tumor effect independent of TP53 status [24, 25, 28]. Here, the lowest IC50 concentrations of RITA were obtained against the DAOY medulloblastoma cell line, which harbors a TP53 mutation, and the HD-MB03 medulloblastoma cell line, harboring a non-functional SNP but otherwise wildtype TP53. RITA produced an anti-tumor effect in 3 of the 4 tested medulloblastoma cell lines, independent of their TP53 status. The IC50s obtained in our study were in the same range as those described by other groups. To further investigate whether sensitivity to RITA was dependent or independent of TP53 status, we enforced expression of a dominant-negative TP53 mutant. This did not abrogate the RITA effect, indicating that an intact TP53 pathway is not needed for medulloblastoma cell sensitivity to RITA. This is in line with results reported by Krajewski and colleagues, who demonstrated that RITA does not block the MDM2 binding site, suggesting RITA works independently of the TP53-MDM2 interaction [29]. One possible TP53-MDM2 pathway-independent anti-tumor effector mechanism could be the JNK or p38 pathway that was shown to be activated by RITA before [28, 30]. In summary, our data added further evidence for the hypothesis that RITA functions independently of the TP53 status.
Translation to the clinic for therapeutic use also requires in vivo evidence. Here we initially evaluated the efficacy of RITA treatment in a medulloblastoma xenograft mouse model using a dosage regimen previously shown to be safe for control mice and effective against neuroblastoma tumor xenografts [31]. RITA inhibited growth of medulloblastoma tumor xenografts and significantly prolonged mouse survival without causing visible signs of toxicity in the animals. These findings were consistent with previous studies of RITA in xenograft models of colon carcinoma, neuroblastoma, and mesothelioma [20, 22, 31, 32]. Interestingly, while RITA triggered CDKN1A and MDM2 transcription in the HD-MB03 cell line, which harbors wildtype TP53, in vitro, only MDM2 transcription was increased in xenograft tumors derived from HD-MB03 cells in mice receiving 3 days of the more intensive treatment in our study. Nevertheless, short-term RITA treatment was sufficient to increase TP53 expression and levels of cleaved caspase 3 in the xenografts, indicating activation of the TP53 pathway leading to apoptosis.
Our data support that the TP53 pathway can be re-activated by RITA treatment in medulloblastoma xenograft tumors, and that targeting the MDM2-TP53 axis using RITA may represent a potentially attractive therapeutic strategy for medulloblastomas independent of their TP53 status. Medulloblastoma has a great biological heterogeneity as displayed in the currently used 4-subgroup classification defining risk and treatment strategy. Our data underline the importance of performing comprehensive genetic profiling early on during treatment and at relapse to detect different genetic variants, although it has been shown that the initial subgroup is maintained in relapse MB [33]. It is known that TP53 mutations occur in all 4 subgroups or in relapsed medulloblastoma that did not have a TP53 mutation at first diagnosis. Therefore patients can benefit from targeting the MDM2-TP53 axis independent of the subgroup the tumor belongs to. Our data underline that further molecular and cell surface markers should be used to define even more detailed risk group definitions helping to find the optimal therapy for the patient [34, 35]. RITA displayed promising anti-tumoral activity against medulloblastoma cells both in vitro and in vivo. RITA treatment prevented TP53 degradation by MDM2 in medulloblastoma cells effecting a re-activation of the TP53 pathway. In addition to this, RITA must also have reactivated TP53 activity independently of MDM2 and TP53 status, since RITA triggered CDKN1A transcription in medulloblastoma cells harboring either wildtype or mutated TP53. Ultimately, clinical testing of RITA in patients with medulloblastoma is necessary to demonstrate its usefulness therapeutically for the treatment of this highly aggressive pediatric malignancy. In light of the fact that about 30% of patients with medulloblastoma experience relapses and that relapse tumors are often resistant to chemotherapeutic agents, RITA might improve efficacy of standard relapse protocols as an additional component. Since the current treatment does not target the MDMD2-TP53 axis, RITA could provide another point of tumor attack to possibly improve overall survival in children with relapsed medulloblastoma.
MATERIALS AND METHODS
Cell lines and RITA treatment
The human medulloblastoma cell lines DAOY, HD-MB03, ONS-76, UW-228-2 (all adherently growing), were grown in RPMI 1640 (Lonza) supplemented with 10% fetal calf serum (GE Healthcare), penicillin (100 U/ml) and streptomycin (100 μg/ml; Life Technologies). Medium for HD-MB03 cells was additionally supplemented with 1% non-essential amino acids (Life Technologies) and 1% amphotericin B (GE Healthcare). Medium for ONS-76 cells was additionally supplemented with 1% L-glutamine (Life Technologies). All cell lines were authenticated by short tandem repeat DNA typing by the German Collection of Microorganisms and Cell cultures (Braunschweig, Germany) prior to the experiments. The genetic profile of the UW-228-2 cell line has not been previously described. Supplementary Table 1 lists all gene mutations detected in UW-228-2 and provided by the German Collection of Microorganisms and Cell cultures. RITA (Cayman Chemicals) was dissolved in ethanol and stored as a 10 mmol/L stock solution at –20°C. Cells were exposed to 0–8 μM RITA for the period indicated, with the final ethanol concentration kept constant in each experiment. For rescue experiments, HD-MB03 and ONS-76 cells were transfected with an expression construct for dominant negative TP53 (donated by Prof. M. Eilers) [36]. Selection of transfected cells was achieved by addition of puromycin (1 g/ml; Life Technologies) to the medium.
Western blot analysis
Protein lysates were extracted from cells treated with 1 μM RITA for 24, 48, and 72 h, lysed 30 min on ice in radioimmunoprecipitation assay (RIPA) Buffer supplemented with Complete Protease Inhibitor Cocktail Tablets and PhosSTOP Phosphatase Inhibitor Cocktail Tablets (Roche). 50 μg proteins were separated on 10% SDS-PAGE then transferred to Amersham-Hybond™-C Extra (GE Healthcare) membranes. Transferred membranes were blocked in 5% milk powder in Tris-buffered saline and Tween 20 and then incubated for 12 to 24 h, using the following antibodies and dilutions: CDKN1A (1:500, cat ab-7960, Abcam), TP53 (1:500, cat#sc-71817, Santa Cruz Biotechnology), MDM2 (1:1000, IF2, cat#33-7100, Life Technologies), beta-actin (1:2000, cat#A5541, Sigma-Aldrich), GAPDH (1:2000, cat#MAB374, Merck Millipore), and HRP-conjugated anti-rabbit IgG (1:2000; GE Healthcare) or HRP-conjugated anti-mouse IgG (1:2000; GE Healtcare) was added for 1 h at room temperature. Proteins were visualized using Amersham ECL Plus™ western blotting detection reagents (GE Healthcare) and the UVchem Detection Device (Biometra).
Gene expression analysis
TP53, MDM2 and CDKN1A expression was monitored using real-time polymerase chain reaction (PCR) using Assays on Demand (Applied Biosystems-Life Technologies). Expression values were normalized to the geometric mean of GAPDH. For all these experiments, total RNA was isolated from cells with use of the RNeasyMini kit (Qiagen) and cDNA synthesis was performed using the SuperScript® reverse transcription kit (Life Technologies).
dnP53 was measured using the following primer: 5′GGAGCACTAAGCGAGCACTG3′ (sen) and 5′-TATGG CGGGAGGTAGACTGA-3′ (rev) and normalized to endogenous P53 expression.
Cell viability, proliferation and cycle analysis
Cells were seeded onto 96-well plates (5– 7.5 × 10³ per well) in triplicate, incubated for 6 h to permit surface adherence, and treated with 1 μM RITA for 24, 48 and 72 h. Medium was replaced daily, RITA and ethanol concentrations were constant throughout the experiment. Cell viability was analyzed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolinum bromide (MTT) assay (Roche) and IC50 was calculated using GraphPad Prism 5.0 (GraphPad Incorporation). Apoptosis was assessed using the Cell Death ELISA (Roche). Cell proliferation was determined using the Bromdesoxyuridin (BrdU) ELISA (Roche). All three assays were performed according to the manufacturer´s protocol. For cell cycle analysis, cells were incubated for 15 min with propidium iodide (10 μg/ml) and cellular content was analyzed using a FC500 flow cytometer (Beckman Coulter).
Immunohistochemistry
Xenograft tumors were formalin fixed, paraffin embedded, and 5 μm sections were cut from each block. Immunohistochemical staining was conducted as described before [18]. Primary antibodies used were: TP53 (1:250, cat#ab4060, Abcam), Ki67 (1:500, cat#ab16667, Abcam) and cleaved caspase 3 (1:200, cat#9661, Cell Signaling Technology). The slides were developed using the EnVision (Dako).
Growth of xenograft tumors in nude mice
HD-MB03 medulloblastoma cells were cultured to 80% confluence, harvested, and suspended in 200 μL Matrigel (BD Biosciences) for subcutaneous inoculation (1.5 × 107 cells per mouse) into the left flank of 8-week-old female athymic NCR (nu/nu) mice. After reaching a tumor size between 100 and 300 mm³, mice were (randomly) assigned to two groups. The first group was the standard treatment group in which the mice were treated intraperitoneally either with RITA (0.3 mg/mouse/day) or with a vehicle control over multiple days in order to analyze the effect of RITA on tumor growth and survival of the mice. The second group of mice received for 3 days an extensive i.p. treatment of RITA twice daily (2 × 0.6 mg/mouse/day) or the vehicle control. In this group the mice were euthanized after the 3-day treatment period and tumors were harvested for immunohistochemical and gene expression analysis to investigate the immediate effect of RITA on the tumor. Each group was further randomly assigned to either RITA or vehicle control groups. RITA or vehicle control was administered with 10% Dimethylsulfoxid (DMSO) in PBS (Gibco®, Life Technologies). Tumor growth was monitored daily using a caliper, and tumor volume was calculated using the formula (breadth × length × height)/2. Mice were euthanized by cervical dislocation when tumor size exceeded 1000 mm³. The tumor was removed and a third was formalin fixed and paraffin embedded, the other two thirds were snapped frozen in liquid nitrogen and then stored at –80°C. The mice receiving intensive treatment were euthanized 12 h after the last injection. All experiments were performed in accordance with the Council of Europe guidelines for accommodation and care of laboratory animals and protocols were approved (reference number 87–51.04.2010.A158) by the North Rhine-Westphalia State Environment Agency (Landesamt für Natur, Umwelt und Verbraucherschutz NRW).
Statistical analysis
Graph Pad Prism was used to conduct statistical analysis, calculate IC50 concentrations, design graphs, and perform Kaplan-Meier survival analysis with log-rank statistics for the mouse cohorts. Data are presented as means ± SD. The Student t test was conducted as a two-sided unpaired test with a confidence interval of 95%. Results with a P value of less than 0.05 were considered statistically significant. MDM2 and CDKN1A mRNA expression levels were calculated using biogazelle (www.biogazelle.com). Ki67, cleaved caspase 3 and TP53 protein expression was assessed analyzing 5 immunohistochemically stained slides of each of the tumors using the ImageJ program [37].
ACKNOWLEDGMENTS
We thank N. Solomentsew for excellent technical assistance and K. Astrahantseff for proofreading the manuscript. A. Künkele is participant in the BIH - Charité Clinical Scientist Program funded by the Charité - Universitätsmedizin Berlin and the Berlin Institute of Health.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. McNeil DE, Cote TR, Clegg L, Rorke LB. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol. 2002; 39:190–4.
2. Silber JH, Radcliffe J, Peckham V, Perilongo G, Kishnani P, Fridman M, Goldwein JW, Meadows AT. Whole-brain irradiation and decline in intelligence: the influence of dose and age on IQ score. J Clin Oncol. 1992; 10:1390–6.
3. Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004; 5:399–408.
4. Laughton SJ, Merchant TE, Sklar CA, Kun LE, Fouladi M, Broniscer A, Morris EB, Sanders RP, Krasin MJ, Shelso J, Xiong Z, Wallace D, Gajjar A. Endocrine outcomes for children with embryonal brain tumors after risk-adapted craniospinal and conformal primary-site irradiation and high-dose chemotherapy with stem-cell rescue on the SJMB-96 trial. J Clin Oncol. 2008; 26:1112–8.
5. Armstrong GT, Liu Q, Yasui Y, Huang S, Ness KK, Leisenring W, Hudson MM, Donaldson SS, King AA, Stovall M, Krull KR, Robison LL, Packer RJ. Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009; 101:946–58.
6. Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002; 415:436–42.
7. Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011; 29:1424–30.
8. Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, Clifford SC, Pietsch T, A.O. von Bueren, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of SWT H, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012; 123:473–84.
9. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012; 123:465–72.
10. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011; 29:1408–14.
11. Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D, Ellison DW. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006; 5:2666–70.
12. Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, Taylor RE, Bailey S, Clifford SC, et al. Medulloblastoma: clinicopathological correlates of WSH N, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011; 121:381–96.
13. Pfaff E, Remke M, Sturm D, Benner A, Witt H, Milde T, von Bueren AO, Wittmann A, Schottler A, Jorch N, Graf N, Kulozik AE, Witt O, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 2010; 28:5188–96.
14. Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, Milde T, Bourdeaut F, Ryzhova M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014; 25:393–405.
15. Benard J, Douc-Rasy S, Ahomadegbe JC. TP53 family members and human cancers. Hum Mutat. 2003; 21:182–91.
16. Saylors RL, 3rd, Sidransky D, Friedman HS, Bigner SH, Bigner DD, Vogelstein B, Brodeur GM. Infrequent p53 gene mutations in medulloblastomas. Cancer Res. 1991; 51:4721–3.
17. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A, Ryan SL, Poon E, Robinson SP, et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. 2015; 27:72–84.
18. Kunkele A, De Preter K, Heukamp L, Thor T, Pajtler KW, Hartmann W, Mittelbronn M, Grotzer MA, Deubzer HE, Speleman F, Schramm A, Eggert A, Schulte JH. Pharmacological activation of the p53 pathway by nutlin-3 exerts anti-tumoral effects in medulloblastomas. Neuro Oncol. 2012; 14:859–69.
19. Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011; 30:4678–86.
20. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004; 10:1321–8.
21. Enge M, Bao W, Hedstrom E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell. 2009; 15:171–83.
22. Grinkevich VV, Nikulenkov F, Shi Y, Enge M, Bao W, Maljukova A, Gluch A, Kel A, Sangfelt O, Selivanova G. Ablation of key oncogenic pathways by RITA-reactivated p53 is required for efficient apoptosis. Cancer Cell. 2009; 15:441–53.
23. Zhao CY, Szekely L, Bao W, Selivanova G. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res. 2010; 70:3372–81.
24. Zhao CY, Grinkevich VV, Nikulenkov F, Bao W, Selivanova G. Rescue of the apoptotic-inducing function of mutant p53 by small molecule RITA. Cell Cycle. 2010; 9:1847–55.
25. Surget S, Descamps G, Brosseau C, Normant V, Maiga S, Gomez-Bougie P, Gouy-Colin N, Godon C, Bene MC, Moreau P, Le Gouill S, Amiot M, Pellat-Deceunynck C, RITA (Reactivating p53 and Inducing Tumor Apoptosis) is efficient against TP53abnormal myeloma cells independently of the p53 pathway. BMC Cancer. 2014; 14:437.
26. Jones RJ, Bjorklund CC, Baladandayuthapani V, Kuhn DJ, Orlowski RZ. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol Cancer Ther. 2012; 11:2243–53.
27. Kazemi A, Safa M, Shahbazi A. RITA enhances chemosensivity of pre-B ALL cells to doxorubicin by inducing p53-dependent apoptosis. Hematology. 2011; 16:225–31.
28. Weilbacher A, Gutekunst M, Oren M, Aulitzky WE, van der Kuip H, RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38. Cell Death Dis. 2014; 5:e1318.
29. Krajewski M, Ozdowy P, D’Silva L, Rothweiler U, Holak TA. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat Med. 2005; 11:1135–6; author reply 1136–7.
30. Saha MN, Jiang H, Yang Y, Zhu X, Wang X, Schimmer AD, Qiu L, Chang H. Targeting p53 via JNK pathway: a novel role of RITA for apoptotic signaling in multiple myeloma. PLoS One BMC Cancer. 2012;7:p. e30215.
31. Burmakin M, Shi Y, Hedstrom E, Kogner P, Selivanova G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin Cancer Res. 2013; 19:5092–103.
32. Di Marzo D, Forte IM, Indovina P, Di Gennaro E, Rizzo V, Giorgi F, Mattioli E, Iannuzzi CA, Budillon A, Giordano A, Pentimalli F. Pharmacological targeting of p53 through RITA is an effective antitumoral strategy for malignant pleural mesothelioma. Cell Cycle. 2014; 13:652–65.
33. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA, Schuller U, Gururangan S, McLendon R, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013; 14:1200–7.
34. Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, Kool M, Dufour C, Vassal G, Milde T, Witt O, von Hoff K, Pietsch T, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016; 131:821–31.
35. Liang L, Aiken C, McClelland R, Morrison LC, Tatari N, Remke M, Ramaswamy V, Issaivanan M, Ryken T, Del Bigio MR, Taylor MD, Werbowetski-Ogilvie TE. Characterization of novel biomarkers in selecting for subtype specific medulloblastoma phenotypes. Oncotarget. 2015; 6:38881–900. doi: 10.18632/oncotarget.6195.
36. Ferrandiz N, Martin-Perez J, Blanco R, Donertas D, Weber A, Eilers M, Dotto P, Delgado MD, Leon J. HCT116 cells deficient in p21(Waf1) are hypersensitive to tyrosine kinase inhibitors and adriamycin through a mechanism unrelated to p21 and dependent on p53. DNA Repair (Amst). 2009; 8:390–9.
37. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9:671–5.