
INTRODUCTION
Analysis of myeloma plasma cells using cytogenetics and fluorescence in-situ hybridization (FISH) has formed the basis of genetic subgrouping in myeloma (MM) [1–3]. These investigations generated etiological groups based upon either the presence of a translocation into the immunoglobulin heavy chain (IGH) locus (40%) or hyperdiploidy (HRD). Further analysis identified the overexpression of a D-group cyclin as a key aberration that is uniformly dysregulated as part of a convergent evolutionary pathway integrating all of the genetic events leading to MM [4, 5]. In this respect, translocations into the 11q13 locus directly deregulate CCND1; 6p21 CCND3; and 4p16 (MMSET), 16q23 (MAF), and 20q12 (MAFB) indirectly deregulate CCND2. In contrast, the mechanistic basis for cyclin-D dysregulation in HRD myeloma is less clear [6].
Based on these genetic data, a MM classification incorporating the presence of translocations and D-group cyclin dysregulation, the Translocation Cyclin-D classification (TC) [5] was proposed which recognized eight molecular subgroups. Alternative classification models were also proposed with the most widely used of these, the UAMS classification, recognizing seven molecular subgroups [7]. This classification framework was expanded upon by the HOVON group with additional subgroups defined by secondary features, e.g. NFκB activation and PRL3 expression [8].
Recently, next generation sequencing data has become available, which promises to further refine classification strategies. The spectrum of mutations in MM has been shown to be dominated by mutations in the RAS (43% of patients) and NFκB pathways (17%), and recurrent alterations in genes, including TP53, ATM, and ATR, and secondary translocations into MYC (8q24) that have strong associations with adverse risk [9]. It has also been shown that bi-allelic events in tumor suppressor genes, especially TP53, are associated with proliferation and high risk features which lead to progressive disease [10]. In this work, we investigate the associations between DNA mutation and GEP defined molecular subgroups of MM from a large data set with assessment by gene expression and targeted mutational panels (referred to as FoundationOne Heme, or F1H, below) with particular interest in RAS codon mutations.
RESULTS
Updated TC algorithm
We classify cases throughout according to an updated TC algorithm (TC-6) designed to accurately define molecular subgroups from gene expression data normalized by GCRMA. A detailed description of our methodology is found in the methods section below. This updated TC classification (TC-6) identifies six major subtypes (D1-HRD, D2, CCND1-11q13, CCND3-6p21, MMSET, and MAF) and validated with ≥ 97.9% agreement across iFISH determined translocations on the MRC-IX data for each of t(4;14), t(11;14), and t(14;16) or t(14;20) (Supplementary Table 1). An ordered bar plot illustrates the distinct expression patterns of primary and secondary genes that discriminate subtypes (Supplementary Figure 1). Key copy number abnormalities and GEP70 high risk (HR) are differentially distributed across subgroups, e.g. 1q+, 13q-, and GEP70 HR are enriched in MAF and MMSET subgroups (Table 1 and Supplementary Figure 2A).
Table 1: Clinical and biological features of TC-6 subgroups
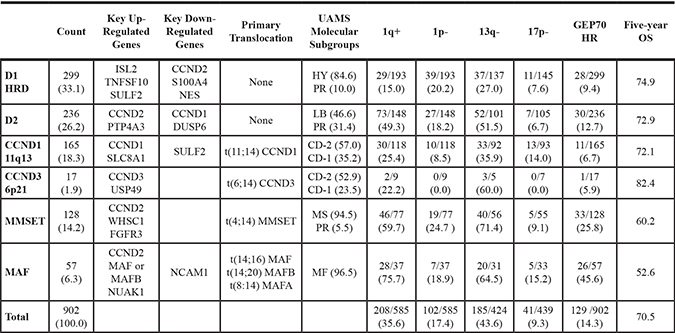
Each TC-6 subgroup has a distinct biology that is reflected in varied distributions of copy number abnormalities (iFISH), GEP70 risk, and overall survival.
Subgroups with over-expression of CCND2 (D2, MMSET, and MAF) had higher rates of 1q+ and 13q-. MAF and MMSET cases have the highest proportions of GEP70 HR and the poorest outcome.
Most frequently altered genes stratified by disease stage
Across our data set of cases with paired gene expression and FoundationOne mutational panels, we observed that NRAS, KRAS, and TP53 were the most commonly mutated genes. In total, 38.6% of all cases had a RAS-RAF mutation (KRAS alone 16.3%, NRAS alone 18.3%, BRAF alone 3.0%, with co-occurrence in 1.1%) and 11.3% had a mutation or deletion in TP53 (Table 2). The rate of TP53 mutation in our data set is elevated when compared to prior studies [9] because of the heterogeneous disease stage, including non-baseline entries, of samples within our data set. Although heterogeneity in disease stage is a potential confounder of subsequent analyses (which we account for by verifying the significance of all main findings in multivariate analyses that include disease stage as a covariate), it also allows us to illustrate directly the association between specific gene alterations and progressive disease. For example, we found that in addition to TP53, CDKN2C and RB1 alterations were also significantly, or nearly significantly in the case of RB1, associated with progressive disease stage implicating these alterations as key markers of late stage disease (p-value < 0.001, 0.056 and 0.025 for TP53, RB1, and CDKN2C, respectively). We also observed that RAS-RAF mutations, especially NRAS mutations of the Q61 codon, were more common at or near relapse than at prior disease stages (20.6% of relapse cases have Q61 NRAS mutation, 12.7% in prior disease stages: p-value = 0.005). None of the remaining key gene alterations were significantly associated with disease stage including mutations previously found to be associated with outcome, e.g. ATM/ATR [9].
Table 2: RAS-RAF codon and other key alterations by disease stage
Gene |
Total Mutations |
Total Deletions |
Untreated (n = 182) |
In Treatment |
At or Near Relapse |
Cochran-Armitage Trend Test |
Relapse vs Prior Stage |
|---|---|---|---|---|---|---|---|
RAS-RAF |
311 |
57 (31.3%) |
117 (35.6%) |
137 (46.6%) |
< 0.001 |
0.001 |
|
KRAS |
134 (16.6%) |
25 (13.7%) |
55 (16.7%) |
54 (18.4%) |
0.195 |
0.370 |
|
G12/13 |
66 (8.2%) |
15 (8.2%) |
28 (8.5%) |
23 (7.8%) |
0.841 |
0.872 |
|
Q61 |
68 (8.4%) |
10 (5.5%) |
27 (8.2%) |
31 (10.5%) |
0.053 |
0.136 |
|
NRAS |
151 (18.8%) |
26 (14.3%) |
53 (16.1%) |
72 (24.5%) |
0.003 |
0.002 |
|
G12/13 |
26 (3.2%) |
3 (1.6%) |
11 (3.3%) |
12 (4.1%) |
0.156 |
0.407 |
|
Q61 |
125 (15.5%) |
23 (12.6%) |
42 (12.8%) |
60 (20.4%) |
0.012 |
0.005 |
|
BRAF V600E |
26 (3.2%) |
6 (3.3%) |
9 (2.7%) |
11 (3.7%) |
0.716 |
0.678 |
|
TP53 |
91 (11.3%) |
3 (0.4%) |
12 (6.6%) |
32 (9.7%) |
50 (17.0%) |
< 0.001 |
0.001 |
TRAF3 |
27 (3.4%) |
14 (1.7%) |
6 (3.3%) |
19 (5.8%) |
16 (5.4%) |
0.363 |
0.861 |
FGFR3 |
25 (3.1%) |
3 (1.6%) |
11 (3.3%) |
11 (3.7%) |
0.225 |
0.563 |
|
RB1 |
23 (2.9%) |
15 (1.9%) |
5 (2.7%) |
14 (4.3%) |
19 (6.5%) |
0.056 |
0.111 |
CDKN2C |
6 (0.7%) |
23 (2.9%) |
3 (1.6%) |
10 (3.0%) |
16 (5.4%) |
0.025 |
0.054 |
DNMT3A |
26 (3.2%) |
3 (1.6%) |
13 (4.0%) |
10 (3.4%) |
0.373 |
0.999 |
|
ATM/ATR |
32 (4.0%) |
2 (1.1%) |
20 (6.1%) |
10 (3.4%) |
0.398 |
0.657 |
|
TET2 |
23 (2.9%) |
1 (0.1%) |
4 (2.2%) |
12 (3.6%) |
8 (2.7%) |
0.856 |
0.909 |
BIRC3 |
2 (0.2%) |
21 (2.6%) |
6 (3.3%) |
8 (2.4%) |
9 (3.1%) |
0.955 |
0.965 |
Across the heterogeneous disease stages present in our data set, we observed that RAS-RAF mutations were more common at relapse than at prior disease stage, primarily due to increase of NRAS mutations of Q61 at relapse. In addition, alterations of TP53 are more common at relapse than prior disease stages, and mutation and deletion of RB1 and CDKN2C are significantly or near significantly associated with progressive disease stage.
All rows with either p-value < 0.01 are bolded. Two p-values are reported: a Cochran-Armitage trend test that examines progressive change in distribution across three disease stages and relapse vs prior disease stage Fisher exact test. Mutations and deletions are reported in first two columns, and their combined sum (alterations) is divided across disease stages. For cases with co-occurrence of RAS-RAF mutations, the mutation with highest variant allele frequency is reported here.
Differential distribution of RAS codon and other mutations across TC subgroups
The distribution of RAS codon mutations is not uniform across TC subgroups. Most notably, NRAS mutations, especially at Q61, are common in HRD and t(11;14) MM yet rare in MAF and MMSET (18.5% of D1-HRD (23.5%), D2 (12.3%), and CCND1-11q13 (20.9%) with Q61 NRAS vs 2.2% in MAF (2.1%) and MMSET (2.2%): p-value < 0.001; Table 3 and Figure 1). This differential pattern of Q61 NRAS mutations is independent of disease stage (subgroup-associated p-values remain highly significant in multivariate model including disease stage). The rarity of Q61 NRAS mutations in MMSET and MAF leads to a proportionally increased rate of KRAS and BRAF mutations in these subgroups (68% of MMSET and 80% of MAF cases with RAS-RAF mutations are KRAS or BRAF compared to 48% in D1-HRD, D2, and CCND1-11q13: p-value = 0.004). In addition, subgroups with elevated CCND2 expression (D2, MMSET, and MAF) have fewer RAS-RAF mutations overall compared to cases with elevated CCND1 expression (D1-HRD, CCND1-11q13) (30% of CCND2 high expressers with RAS-RAF mutation, 46% of CCND1 high: p-value < 0.001).
Table 3: RAS-RAF codon and other key alterations by TC-6 subgroup
Gene |
Total Mutations |
Total Deletions |
Most Common Subgroups |
Count (Percentage) |
Least Common Subgroups |
Count (Percentage) |
Fisher Exact Test |
|---|---|---|---|---|---|---|---|
RAS-RAF |
311 (38.6%) |
D1-HRD, CCND1 |
192/414 (46.4%) |
D2, MMSET, MAF |
115/381 (30.2%) |
< 0.001 |
|
KRAS |
134 (16.6%) |
D1-HRD, MAF |
55/261 (21.1%) |
D2, MMSET, CCND1 |
77/534 (14.4%) |
0.020 |
|
G12/13 |
66 (8.2%) |
MAF |
7/48 |
D1-HRD, D2, CCND1, MMSET |
58/747 (7.8%) |
0.162 |
|
Q61 |
68 (8.4%) |
D1-HRD, D2, CCND1 |
59/658 (9.0%) |
MMSET, MAF |
8/137 |
0.309 |
|
NRAS |
151 (18.8%) |
D1-HRD, D2, CCND1 |
140/658 (21.3%) |
MMSET, MAF |
11/137 (8.0%) |
< 0.001 |
|
G12/13 |
26 (3.2%) |
MMSET, MAF |
8/137 |
D1-HRD, D2, CCND1 |
18/658 (2.7%) |
0.107 |
|
Q61 |
125 (15.5%) |
D1-HRD, D2, CCND1 |
122/658 (18.5%) |
MMSET, MAF |
3/137 |
< 0.001 |
|
BRAF V600E |
26 (3.2%) |
MMSET, MAF |
7/137 |
D1-HRD, D2, CCND1 |
17/658 (2.6%) |
0.163 |
|
TP53 |
91 (11.3%) |
3 (0.4%) |
D1-HRD, CCND1, MAF |
62/462 (13.4%) |
D2, MMSET |
31/333 (9.3%) |
0.093 |
TRAF3 |
27 (3.4%) |
14 (1.7%) |
D2, MMSET, MAF |
35/381 (9.2%) |
D1-HRD, CCND1 |
6/414 |
< 0.001 |
FGFR3 |
25 (3.1%) |
MMSET |
25/89 (28.1%) |
D1-HRD, D2, CCND1, MAF |
0/706 |
< 0.001 |
|
RB1 |
23 (2.9%) |
15 (1.9%) |
D2, MMSET |
23/333 (6.9%) |
D1-HRD, CCND1, MAF |
15/462 (3.2%) |
0.019 |
CDKN2C |
6 (0.7%) |
23 (2.9%) |
D2, MMSET |
22/333 (6.6%) |
D1-HRD, CCND1, MAF |
7/462 |
< 0.001 |
DNMT3A |
26 (3.2%) |
D2, CCND1, MMSET |
21/534 (3.9%) |
D1, MAF |
3/261 |
0.044 |
|
ATM/ATR |
32 (4.0%) |
MMSET, MAF |
12/137 (8.8%) |
D1-HRD, D2, CCND1 |
20/658 (3.0%) |
0.006 |
|
TET2 |
23 (2.9%) |
1 (0.1%) |
D2, MAF |
12/292 (4.1%) |
D1-HRD, CCND1, MMSET |
11/503 (2.2%) |
0.129 |
BIRC3 |
2 (0.2%) |
21 (2.6%) |
MMSET |
13/89 (14.6%) |
D1-HRD, D2, CCND1, MAF |
10/706 (1.4%) |
< 0.001 |
RAS-RAF and other key alterations are not distributed evenly across TC-6 subgroups. We have reported the most significant difference in distribution for each specific gene. A full breakdown of mutation and deletion by subgroup can be found in Supplementary Table 2.
All rows with p-values < 0.01 are bolded. Mutations and deletions are reported in first two columns, and their combined sum (alterations) is divided across the TC-6 subgroups. CCND3-6p21 subgroup is not included in this subgroup analysis due to sample size restraints. For cases with co-occurrence of RAS-RAF mutations, the mutation with highest variant allele frequency is reported here.
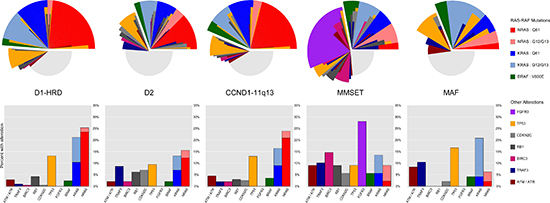
Figure 1: Distribution of RAS-RAF codon and other key alterations across TC-6 subgroups. NRAS mutations at Q61 are commonly seen in D1-HRD, D2, and CCND1-11q13 subgroups but rare among the MMSET and MAF subtypes. FGFR3 mutations are exclusive to the MMSET subtype and other genes are altered at a higher frequency in particular subgroups, e.g. MMSET cases are enriched for BIRC3 alterations and CCND2 expressing subgroups (D2, MMSET, and MAF) are enriched for TRAF3 alterations. Stacked bar plots represent frequencies of key alterations reported as percentages within each TC-6 subgroup. For the small number of cases (1.1%) with co-occurrence of RAS-RAF mutations, only the mutation with highest variant allele frequency (VAF) was used. For the radial alteration plots, each individual slice corresponds to the mutational profile of one specific subject.
We also investigated whether specific RAS codon mutations were associated with expression of CD20 (MS4A1) in t(11;14) myeloma as this is a common divisor of this subtype [11,12]. We found that across all t(11;14) cases, those with RAS-RAF mutations were to some extent less likely to overexpress CD20 (44.3% with RAS-RAF mutations expressed CD20, 60.2% expressed without RAS-RAF mutation; p-value = 0.032). This negative association between RAS-RAF mutation and CD20 expression was found primarily in cases with Q61 NRAS mutations as only 33% (14/42) of such cases overexpressed CD20. When excluding these cases, we observed no significant difference in the rate of CD20 overexpression among remaining cases (RAS-RAF mutated excluding Q61 NRAS vs non-RAS-RAF mutated cases: p-value = 0.708). Thus t(11;14) cases with RAS-RAF mutations appear less likely to overexpress CD20, especially those with NRAS mutations at Q61.
Overall, we observed an increased frequency in the rate of NRAS mutations, especially at Q61, when compared to other cancers. Our data set confirms prior work in MM that identified NRAS mutations as more common than KRAS and an increased frequency of NRAS mutations at Q61 [13]. An enrichment of mutations at Q61 was not observed in cases with KRAS mutations (NRAS: 83% Q61, 17% G12/G13 compared to KRAS: 51% Q61, 49% G12/G13, p-value < 0.001; Table 2). While mutations at Q61 compose 60% of NRAS mutations in other cancers [14], we find this rate to be over 80% in MM likely due to the increased rate of NRAS mutations at Q61 observed in HRD and t(11;14) myeloma—subgroups that comprise the majority of MM.
Other key mutations were enriched in specific subgroups. For example, BIRC3 was frequently altered in MMSET but rarely in other subgroups (14.6% of MMSET with alteration, 1.4% of others: p-value < 0.001; Table 3). In addition, CDKN2C and RB1 were more frequently altered in the D2 and MMSET subgroups (p-values 0.019 and < 0.001, respectively). Mutations and deletions of TRAF3, a gene in the alternative NFκB pathway [15,16], were enriched in high expressers of CCND2 (9.2% of D2, MMSET, and MAF, 1.4% of D1-HRD and CCND1-11q13: p-value < 0.001). A full compendium of gene mutation and deletion counts both overall and split across TC-6 and UAMS molecular subgroups is available in Supplementary Table 2.
Patterns of differential expression associated with RAS-RAF mutations
Differential expression analysis revealed many genes to have patterns of expression highly associated with the presence of RAS-RAF and FGFR3 mutations. Probes for DUSP6, DKK1, SPRED2, COBLL1, and ETV5 were the most significantly associated with presence of RAS-RAF or FGFR3 mutations across all cases (Supplementary Table 3 and Figure 2A–2E). Additional analyses revealed that for specific subgroups, certain genes were highly associated with the presence of RAS-RAF mutations. For example, sprouty-related protein gene SPRED2, established as a negative regulator of MAPK/ERK signaling [17,18], was positively associated with the presence of RAS-RAF mutations, especially in the CCND1-11q13 subgroup. This differential expression pattern of SPRED2 in CCND1-11q13 cases provides a nearly dichotomous split of RAS-RAF mutations at a certain threshold of SPRED2 expression, i.e. 81.4% of cases with SPRED2 expression above and 8.7% of cases with SPRED2 expression below had RAS-RAF mutations (79/97 cases with SPRED2 expression above 4.65 versus 9/104 below; p-value < 0.001; Figure 2C). In addition, the expression of the oncogene RRAS2 has a dichotomous distribution across D1-HRD cases, i.e. differential clusters of low and high expressers, where high expression was negatively associated with presence of RAS-RAF mutations (17% of high RRAS2 expressers with RAS-RAF mutations, 64% in low expressers; p-value < 0.001; Figure 2F). We note that these patterns of gene expression associated with RAS-RAF mutations are capable of further subtyping molecular subgroups of myeloma, especially in the D1-HRD, D2, and CCND1-11q13 subgroups where significant gene expression patterns were observed (Supplementary Figure 3).
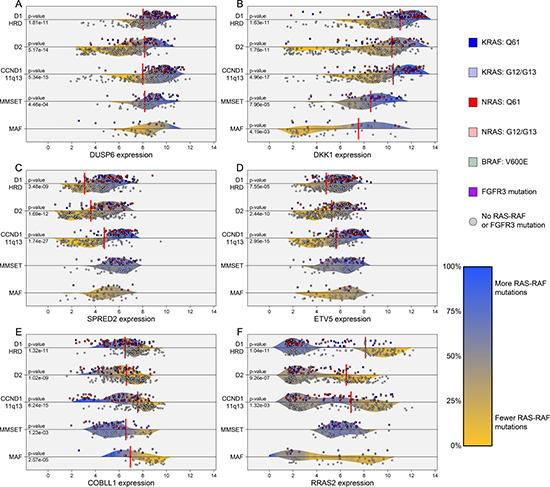
Figure 2: Differential gene expression patterns associated with presence of RAS-RAF mutations across TC-6 subgroups. The gene expression levels of DUSP6, DKK1, SPRED2, ETV5, and COBLL1 were highly associated with the presence of RAS-RAF or FGFR3 mutations across all cases (A–E). COBLL1 expression is negatively associated with presence of RAS-RAF mutation while remaining genes are positively associated. The RAS viral oncogene, RRAS2, has a dichotomous expression pattern in the D1-HRD subgroup where high expressers of RRAS2 have few RAS-RAF mutations (F). Density curves are colored according to localized rate of RAS-RAF/FGFR3 mutation and represent probability distributions for mutated and non-mutated cases, separated by dividing horizontal lines. Vertical red lines indicate optimal thresholds of significance according to Fisher exact tests, limited to interior 60% of percentiles, where only highly significant p-values were reported (< 0.005). CCND3-6p21 subgroup not included due to sample size restraints (n =10). Gene expression represents log2 of GCRMA normalized data using the following probe or average of probes: DUSP6 – 208891_at, 208892_s_at, 208893_s_at; DKK1 – 204602_at; SPRED2 – 212458_at; ETV5 – 203349_s_at; COBLL1 – 203641_s_at, 203642_s_at; and RRAS2 – 212589_at, 212590_at.
We noted that two key genes observed to be significantly differentially expressed according to presence of RAS-RAF mutations were included in previous studies that defined the molecular subgroups of MM. Most notably, DUSP6, a negative regulator of MAPK/ERK signaling [19], is one of the top under-expressed genes in the LB (low bone) subgroup of the UAMS molecular subgroups and also under-expressed in the PRL3 subgroup of the HOVON model. We observed DUSP6 expression to be positively associated with the presence of RAS-RAF mutation, thus these prior models of MM subtyping unknowingly identified cohorts that were negatively enriched for the presence of RAS-RAF mutations. We validated this by classifying our F1H cohort according to UAMS molecular subgroups and observed LB cases to have the lowest rate of RAS-RAF mutation (17.3%), while CD-1 (40.6%), HY (48.1%), and PR (43.8%) subgroups had significantly higher rates (LB RAS-RAF mutation rate vs other molecular subtypes: p-value = 0.001). In addition, the expression of DKK1, a known Wnt-signaling antagonist [20], was previously described as up-regulated in HY and down-regulated in MF subgroups in the UAMS molecular subgroups. We observed similar patterns of expression in D1-HRD and MAF subgroups, and its positive association with the presence of RAS-RAF mutations (Figure 2B). Overall, due to the abundance of RAS-RAF mutations and distinct associated patterns of gene expression, gene-expression based subtyping in MM is likely to incorporate these clear transcriptional relationships either knowingly or unknowingly.
NFκB signaling inversely associated with RAS-RAF mutations
NFκB signaling, according to the 11-gene NFκB signature [21], varied across the TC subgroups with significantly elevated levels in the MAF subgroup (p-values < 0.001 in TT and F1H; 0.17 in MRC-IX; Supplementary Figure 2B). Furthermore, NFκB signaling was negatively associated with presence of RAS-RAF and FGFR3 mutations across all subgroups except for MAF (p-value < 0.001 for D1-HRD, D2, CCND1-11q13, and MMSET subgroups combined; p-value = 0.843 in MAF; Figure 3A).
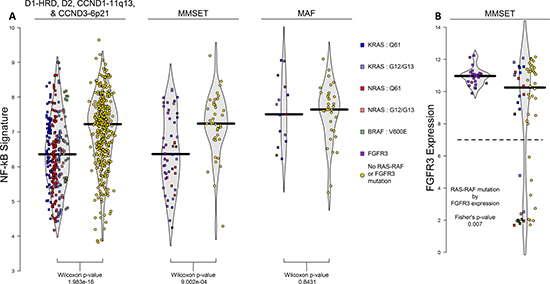
Figure 3: NFκB signature across TC-6 subgroups and FGFR3 expression in MMSET subgroup. The 11-gene NFκB signature is significantly associated with presence of RAS-RAF or FGFR3 mutations in all TC-6 subgroups excluding MAF where cases without RAS-RAF mutations have higher levels of NFκB activation (A). When examining interaction of FGFR3 expression and RAS-RAF mutation in MMSET, we observed that 56% (10/18) cases with low FGFR3 expression have RAS-RAF mutations compared to 21% of remaining cases (15/71) (B).
Unique mutational features of MMSET myeloma
MMSET cases are unique as they can overexpress and have activating mutations in FGFR3. Overall, 25 MMSET cases had FGFR3 mutations (28.1%), 25 had a RAS-RAF mutation (28.1%), and 39 had neither (43.8%); co-occurrence of FGFR3 and RAS-RAF mutations was not observed (Figure 1), consistent with both RAS-RAF and FGFR3 mutations activating similar pathways and, therefore, being functionally redundant. The FGFR3 locus is lost in 26% of MMSET cases [22], and consistent with this, we see lower expression of FGFR3 in 20% (18 of 89) of MMSET cases in the F1H dataset. Over half of these cases (56%) had a RAS-RAF mutation, while only 21% of cases with high FGFR3 expression had RAS-RAF mutations, indicating that FGFR3 expression is more likely to be lost in the presence of a RAS-RAF mutation (p-value = 0.007; Figure 3B). In addition, both FGFR3 and RAS-RAF mutated cases had lower NFκB signaling than those without either mutation (Figure 3A).
Outcome of RAS-RAF codon mutations across subgroups
Despite a relatively short follow-up and heterogeneity in disease stage across our data set, we observed highly significant patterns in outcome associated with specific RAS-RAF codon mutations. Among subgroups with significant enrichment of Q61 NRAS mutations, we observed Q61 NRAS mutations to be associated with a favorable outcome in the t(11;14) subgroup, and a less favorable outcome in the D1-HRD and D2 subgroups (p-value = 0.001, independent of disease stage; Supplementary Figure 4).
For the MMSET subgroup, cases with RAS-RAF mutations had an inferior outcome compared to cases with FGFR3 mutations or those without RAS-RAF mutations (p-value = 0.004, independent of disease stage). In addition, only 1/25 cases (4%) with an FGFR3 mutation also had a CDKN2C or RB1 alteration, while 28% of cases with RAS-RAF mutations had one or more of these alterations (p-value = 0.049). This is in keeping with alterations in CDKN2C and RB1 being indicators of late stage disease [23,24], thus the absence of these alterations in FGFR3 mutated cases suggests FGFR3 mutations as early events and RAS-RAF mutations as markers of late stage disease in t(4;14) MM.
Alterations of TP53, CDKN2C, and RB1 associated with adverse outcome
A conditional inference tree analysis of all possible DNA variants against OS showed that alterations in TP53, CDKN2C, and RB1 were significant prognostic factors across the F1H data set when excluding cases at or near relapse (Figure 4A). The subset of 66 non-relapse cases (12.9% of all non-relapse cases) with one or more of these adverse alterations had an extremely poor prognosis with an 18-month OS rate of 34.1%—statistically similar to the rate observed in GEP70 HR cases (p-value = 0.157; Figure 4B-4C). The combination of GEP70 risk and presence of adverse DNA alterations stratified all non-relapse cases into three arms with distinct clinical course: 32 cases (GEP70 HR and any adverse alteration) with an extremely poor outcome (17.3% OS at 18-months), 81 cases (GEP70 HR or any adverse alteration) with an intermediate outcome (60.3% OS rate at 18-months), and 398 cases (no adverse alterations and GEP70 LR) with a standard outcome (86.5% OS at 18-months) (p-value < 0.001; Figure 4D). Overall, this subset of alterations (TP53, CDKN2C, and RB1) is significantly associated with progressive disease stage where 25.9% of cases at or near relapse had one or more of these alterations compared to 12.9% in prior disease stages (p-value < 0.001, Table 2).
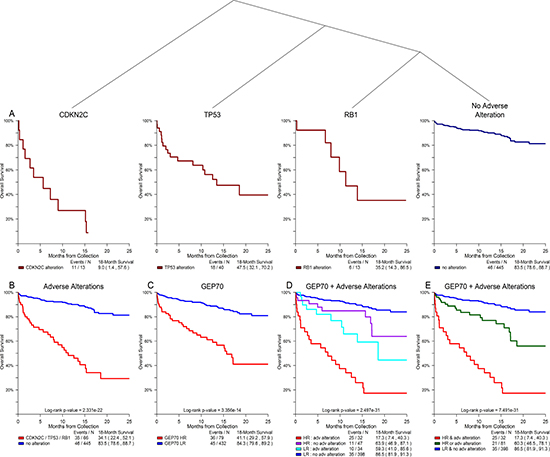
Figure 4: Alterations of TP53, CDKN2C, and RB1 showed significant association with overall survival in non-relapse clinical setting. Conditional inference tree analysis of all possible DNA alterations against outcome revealed that alterations in TP53, CDKN2C, and RB1 were significantly associated with an adverse outcome for all cases with disease stages prior to relapse (A). The subset of cases with one or more of these adverse alterations had an 18-month OS rate that was statistically similar to that of GEP70 HR cases (B, C). Combining GEP70 risk and the presence of adverse alterations identified three cohorts with distinct clinical course: poor, intermediate, and standard outcomes (D, E).
In order to define a mechanistic basis for HR, we examined patterns of gene expression associated with these adverse alterations. We observed strong associations between CDKN2C deletions and low expression of FAF1, and that high expression of CDKN2C was associated with alterations of RB1. We also found cases exhibiting these expression patterns across two NDMM data sets, and observed significant associations with proliferation and outcome for cases exhibiting either expression pattern. Cases with either a low FAF1 expression signature (associated with CDKN2C deletion) or high CDKN2C expression signature (associated with RB1 alteration) had an 18-month OS rate of 26.9% compared to 73.5% in cases without either signature (p-value < 0.001; Figure 5Aii). In addition, cases with either adverse expression signature had elevated proliferation indexes, according to the 50-gene proliferation signature [25] (p-value < 0.001; Figure 5Aiii). These patterns of gene expression, outcome, and proliferation were validated in both the TT and MRC-IX datasets of NDMM cases (Figure 5B–5C).
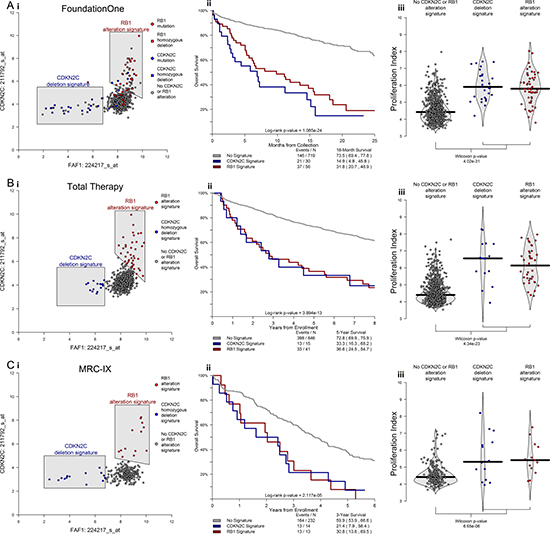
Figure 5: CDKN2C and RB1 alterations elicit distinct patterns of gene expression that have significant impact on proliferation and clinical outcome in newly diagnosed MM. Differential expression analysis revealed highly significant patterns of expression associated with alteration in CDKN2C and RB1. Namely that cases with deletion of CDKN2C have lower expression of FAF1 and that cases with alterations of RB1 have higher expression of CDKN2C (Ai). Cases with either of these distinct expression patterns had inferior outcomes (Aii) and higher 50-gene proliferation scores (Aiii). Identical patterns of inferior outcome and elevated proliferation were observed in both the Total Therapy and MRC-IX data sets of NDMM (B, C). In total, alterations in CDKN2C and RB1 offer mutually exclusive paths that culminate in similar high-risk behavior.
We note that within the F1H data set, CDKN2C alterations (29 cases) and RB1 alterations (38 cases) did not co-occur in any single case. This observation, along with the disparate patterns of gene expression, is consistent with alterations in CDKN2C and RB1 offering mutually exclusive paths to elevated proliferation and high-risk behavior.
DISCUSSION
We show for the first time a clear difference in mutational spectrum across the molecular subgroups of MM defined by an updated TC algorithm. Specifically, we show that NRAS mutations at Q61 are common in HRD and t(11;14) myeloma but rare in MMSET and MAF. This heterogeneity in RAS codon mutations parallels the diversity in primary translocation and cyclin D initiating events in myeloma. These primary events yield diverse genetic backgrounds that likely influence the rate, type, and impact of the acquisition of secondary mutations and deletions. Overall, this analysis aims to describe the unique distribution and clinical impact of key mutations across the molecular subtypes of myeloma while highlighting the importance of defining RAS mutations at the codon level.
Both primary TC event and RAS-RAF mutation have distinct resultant patterns in gene expression. We used the gene expression patterns associated with cyclin D and translocation events to generate an updated model that defines all cases according to six primary TC events. This model is unique from prior TC models in that it uses additional secondary genes, e.g. SLC8A1 up-regulation in t(11;14) or DSG2 up-regulation in t(4;14) myeloma, incorporated into a support vector machine (SVM) classification model to determine optimal subgroups in a simultaneous rather than dichotomous step-wise fashion. This model is distinct from the molecular subtyping models because it groups cytogenetically similar cases together rather than allowing secondary events to determine subgroup, e.g. cases with t(4;14) and t(11;14) may classify as PR (proliferative) in UAMS molecular subgroups. We also observed strong patterns of gene expression associated with the presence of RAS-RAF mutations. Most notably, DKK1, DUSP6, and SPRED2 were positively associated with the presence of RAS-RAF mutations. This pattern was strongest in subgroups with higher rates of NRAS mutations at Q61, i.e. D1-HRD, D2, and CCND1-11q13. We suspect that the increased expression of MAPK/ERK antagonists (DUSP6 and SPRED2) associated with Q61 NRAS mutations is a result of a regulating transcriptional response attempting to counteract activating RAS mutations, i.e. a repeating negative feedback mechanism.
We show that all non-MAF cases lacking a RAS-RAF or FGFR3 mutation have increased NFκB signaling. This reciprocal relationship between NFκB signaling and activating RAS-RAF mutations is consistent with a functional similarity of signaling via these pathways and, therefore, functional redundancy. We propose, based on this observation, that MM is characterized by growth signaling delivered primarily via MAPK or NFκB pathways in a mutually exclusive fashion. The MAF subgroup is unique in this respect as all cases have elevated NFκB signaling irrespective of their RAS mutational status, likely indicative of their unique genetic background [26].
DNA alterations of TP53, CDKN2C, and RB1 were identified as key markers of progressive disease and associated with adverse outcome in a non-relapse MM setting. We also presented the unique interaction between deletion of CDKN2C and alteration of RB1 where both were mutually exclusive with distinct downstream signals in gene expression, yet each impart similar paths to elevated proliferation and adverse outcome. As both CDKN2C and RB1 interact with G1/S cell cycle checkpoint through CDK4/6 [27–29], they are potentially targetable through CDK4/6 inhibition [30], for which it has been shown in other cancers that co-deletion of CDKN2C and CDKN2A with functional presence of RB1 increases sensitivity in cell lines [31]. A better understanding of the interaction of these two key prognostic markers in MM could open new possibilities for targeted therapy. Overall, this work aims to serve as a fundamental step in the transition from panel-based DNA assessment and GEP-based expression analysis to RNA-Seq and whole genome assessments that fully examine the complete mutational landscape of MM.
MATERIALS AND METHODS
A set of 805 UAMS GEP samples underwent targeted sequencing using the FoundationOne Heme (F1H) mutational assay (Foundation Medicine, Cambridge, MA) annotated for known and likely mutations and deletions with variant allele frequencies at or above five percent. Additionally, KRAS, NRAS, and BRAF mutations were limited to missense mutations annotated for known activating mutations: codons G12, G13, and Q61 for K and N RAS and V600E for BRAF. Additional mutations in non RAS-RAF genes included all varieties of short variants: frameshift, missense, nonsense, etc. This series has a median follow-up of 13.5 months with samples collected at various disease stages including 23% prior to treatment, 41% in treatment, and 36% at or near relapse (within 90 days +/– of progression event). This data set was analyzed for associations in mutation and deletion associated with TC subgroups.
An additional data set of 902 UAMS Myeloma Institute gene expression profiling (GEP) samples from newly diagnosed (NDMM) patients accrued to Total Therapy (TT) trials between 2000 and 2010 had a median follow-up of over 10 years and was used to develop an updated TC classification model. iFISH data for 1q+, 1p–, 13q–, and 17p– gathered at baseline were available but not in full. Patients gave written informed consent for bone marrow sampling and the research was approved by the institutional review board of UAMS. This data set of 902 NDMM cases was used to train the updated TC algorithm.
For all GEP data, plasma cells were CD138-purified from bone marrow aspirates and processed on U133 Plus 2.0 microarrays (Affymetrix, Santa Clara, CA) [32]. CEL files were normalized using GCRMA [33] for application of updated TC algorithm. MAS5 normalization was also performed when necessary, e.g. for calculation of GEP70 and UAMS molecular subtypes. All expression data was normalized using R Bioconductor and transformed to the UAMS TT2 and TT3 NDMM standard according to a variant of M-ComBat [34].
Updated TC classifier
An updated GEP-based TC classifier was developed (TC-6) which reflects the primary molecular events in MM. The model was trained on the NDMM TT set of 902 cases using a support vector machine (SVM) [35] where translocation groups were identified according to clear gene expression spikes with remaining non-translocated cases classified according to cyclin-D dysregulation. The model uses 24 probes chosen for their power to discriminate translocation and cyclin-D dysregulated subgroups and is available online at http://github.com/SteinCK/TC-6 for public use.
We have simplified the original eight to six primary subgroups, classifying non-translocated MM into two groups according to deregulation of D-group cyclin (D1 or D2) without a hybrid class with expression in both, i.e. D1 + D2. This reflects our inability to elicit a cluster with expression in both CCND1 and CCND2 that is a distinct entity apart from the D1 and D2 subgroups. Additional analysis of NGS copy number data also supports the existence of a singular D1-HRD subgroup, where a homogeneous cohort with copy number gains of chromosomes 5, 9, 15, 19, and, uniquely, 11, emerges that also identifies as D1-HRD according to TC-6 classification [36].
The methods used to perform subgroup determinations were also updated considerably from original TC methodology in that determinations are now performed simultaneously with an advanced classification system (SVM) rather than in a dichotomous step-wise fashion using simple binary thresholds. This allows for more sophisticated discrimination for each individual case that weighs the entire composition of gene expression to form an optimal subgroup determination rather than individual probes with binary outcomes. We also address sample purity and contamination concerns in our methodology by including healthy, normal plasma cell (NPC) sample controls as a distinct subgroup in model training. Thus across our heterogeneous data set we limited our analyses to samples that failed to cluster with the NPC contamination cluster. In addition, we built this classifier on GCRMA rather than MAS5 normalized expression data as GCRMA normalization has been shown to improve overall accuracy in comparative studies [37,38]. This updated TC-6 model validated well on an external set of 259 GEP samples from the MRC-IX trial [39,40], where it accurately predicted iFISH translocation designations (Supplementary Table 1).
All statistical analyses were performed in R with primary use of the survival [41], party [42], glmnet [43], and e1071 [44] packages. Differential expression analysis of associations between mutation and GEP data was performed with limma [45]. All p-values reported for contingency tables, continuous variable, and survival comparisons were determined by Fisher’s exact tests, Wilcoxon signed rank tests, and log-rank tests, respectively.
Authors’ contributions
GJM and CKS initiated concept and design of investigation, CKS led and performed all analyses and was primary writer of manuscript, CP SC LR NW AC and AB consulted on additional analyses, GJM BW and FED also helped compose the manuscript, GJM and BB provided study materials, TM and JH for their role at FoundationOne Medicine, and GJM supervised project. All authors read and provided approval of final manuscript.
ACKNOWLEDGMENTS
We thank the patients and staff of the Myeloma Institute.
CONFLICTS OF INTEREST
GJM has received honoraria from Bristol-Myers Squibb, Celgene, Takeda-Millenium, Janssen, and Amgen, served in a consulting or advisory role for Bristol-Myers Squibb, Celgene, Takeda-Millenium, and Janssen, and has received research funding from Celgene and Janssen. FED has received honoraria from Bristol-Myers Squibb, Celgene, and Takeda-Millenium, served in a consulting or advisory role for Bristol-Myers Squibb, Celgene, and Takeda-Millenium. CP has participated in advisory boards and spoken at meetings for Celgene and Takeda-Millenium, and received travel support to attend meeting from Celgene. Bart Barlogie has received research funding from Celgene and Millennium, is a consultant to Celgene and Millennium, and a co-inventor on patents and patent applications related to use of GEP in cancer medicine.The remaining authors declare no relevant conflicts of interest.
FUNDING
This work was supported in part by P01 CA 55819 grant from the National Cancer Institute.
REFERENCES
1. Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel LP, Chesi M, Davies EF, Drach J, Greipp RP, Kirsch RI, Kuehl MW, Hernandez MJ, Minvielle S. et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004; 64:1546–58.
2. Cremer FW, Bila J, Buck I, Kartal M, Hose D, Ittrich C, Benner A, Raab SM, Theil CA, Moos M, Goldschmidt H, Bartram RC, Jauch A. Delineation of distinct subgroups of multiple myeloma and a model for clonal evolution based on interphase cytogenetics. Genes Chromosomes Cancer. 2005; 44:194–203.
3. Fonseca R, Debes-Marun CS, Picken BE, Dewald WG, Bryant CS, Winkler MJ, Blood E, Oken MM, Santana-Dávila R, González-Paz N. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. 2003; 102:2562–2567.
4. Bergsagel PL, Kuehl MW, Zhan F, Sawyer J, Barlogie B, Shaughnessy J, Cyclin D. dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005; 106:296–303.
5. Bergsagel PL, Kuehl MW. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005; 23:6333–8.
6. Specht K, Haralambieva E, Bink K, Kremer M, Mandl-Weber S, Koch I, Tomer R, Hofler H, Schuuring E, Kluin MP. Different mechanisms of cyclin D1 overexpression in multiple myeloma revealed by fluorescence in situ hybridization and quantitative analysis of mRNA levels. Blood. 2004; 104:1120–1126.
7. Zhou Y, Barlogie B, Shaughnessy DJ. The molecular characterization and clinical management of multiple myeloma in the post-genome era. Leukemia. 2009; 23:1941–56.
8. Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A, Bertsch U, Buijs A, Stevens-Kroef M, Beverloo BH, Vellenga E, Zweegman S, Kersten JM, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. 2010; 116:2543–53.
9. Walker BA, Boyle ME, Wardell PC, Murison A, Begum BD, Dahir MN, Proszek ZP, Johnson CD, Kaiser FM, Melchor L, Aronson IL, Scales M, Pawlyn C, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol, 2015.
10. Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens WO, Tytarenko R, Bauer AM, Meissner T, Deshpande S. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood. 2016; 128:1735–1744.
11. Robillard N, H. Avet-Loiseau, Garand R, Moreau P, Pineau D, Rapp MJ, Harousseau JL, Bataille R. CD20 is associated with a small mature plasma cell morphology and t (11; 14) in multiple myeloma. Blood. 2003; 102:1070–1071.
12. Zhan F, Huang Y, Colla S, Stewart PJ, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, et al. The molecular classification of multiple myeloma. Blood. 2006; 108:2020–8.
13. Chng WJ, Gonzalez-Paz N, Price-Troska T, Jacobus S, Rajkumar SV, Oken MM, Kyle RA, Henderson KJ, Van Wier S, Greipp P, Van Ness B, Fonseca R. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008; 22:2280–4.
14. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer research. 2012; 72:2457–2467.
15. Demchenko YN, Kuehl MW. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010; 1:59–68. doi: 10.18632/oncotarget.109.
16. Demchenko YN, Glebov KO, Zingone A, Keats JJ, Bergsagel LP, Kuehl MW. Classical and/or alternative NF-κB pathway activation in multiple myeloma. Blood. 2010; 115:3541–3552.
17. Shukla A, Rai K, Shukla V, Chaturvedi KN, Bociek GR, Pirruccello JS, Band H, Lu R, Joshi SS. Sprouty 2: a novel attenuator of B-cell receptor and MAPK-Erk signaling in CLL. Blood. 2016; 127:2310–21.
18. Haydn JM, Hufnagel A, Grimm J, Maurus K, Schartl M, Meierjohann S. The MAPK pathway as an apoptosis enhancer in melanoma. Oncotarget. 2014; 5:5040–5053. doi: 10.18632/oncotarget.2079.
19. Li C, Scott AD, Hatch E, Tian X, Mansour LS. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007; 134:167–176.
20. Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD Jr. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. New England Journal of Medicine. 2003; 349:2483–2494.
21. Annunziata CM, Davis ER, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt ME, Tan B, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007; 12:115–30.
22. Keats JJ, Reiman T, Maxwell AC, Taylor JB, Larratt ML, Mant JM, Belch RA, Pilarski ML. In multiple myeloma, t (4; 14)(p16; q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003; 101:1520–1529.
23. Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, Middha S, Asmann Y, Schmidt J, Braggio E. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012; 120:1060–1066.
24. Morgan GJ, Walker BA, Davies EF. The genetic architecture of multiple myeloma. Nature Reviews Cancer. 2012; 12:335–348.
25. Hose D, Rème T, Hielscher T, Moreaux J, Messner T, Seckinger A, Benner A, Shaughnessy DJ, Barlogie B, Zhou Y, Hillengass J, Bertsch U, Neben K, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011; 96:87–95.
26. Walker BA, Wardell PC, Murison A, Boyle ME, Dahir MN, Proszek ZP, Melchor L, Pawlyn C, Kaiser FM, Johnson CD. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nature communications. 2015. 6.
27. Sherr CJ, Roberts MJ. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 1999; 13:1501–1512.
28. Harbour JW, Luo XR, Dei Santi A, Postigo AA, Dean CD. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999; 98:859–869.
29. Lim S, Kaldis P, Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013; 140:3079–3093.
30. O’Leary B, Finn RS, Turner CN. Treating cancer with selective CDK4/6 inhibitors. Nature Reviews Clinical Oncology. 2016.
31. Wiedemeyer WR, Dunn FI, Quayle NS, Zhang J, Chheda GM, Dunn PG, Zhuang L, Rosenbluh J, Chen S, Xiao Y. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proceedings of the National Academy of Sciences. 2010; 107:11501–11506.
32. Zhan F, J. Hardin, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, Anaissie E, Morris C, Muwalla F, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002; 99:1745–57.
33. Wu J, Irizarry R, MacDonald J, Gentry J. Gcrma: background adjustment using sequence information. R package version. 2012. 2200.
34. Stein CK, Qu P, Epstein J, Buros A, Rosenthal A, Crowley J, Morgan G, Barlogie B. Removing batch effects from purified plasma cell gene expression microarrays with modified ComBat. BMC Bioinformatics. 2015; 16:63.
35. Burges CJ, A tutorial on support vector machines for pattern recognition. Data mining and knowledge discovery. 1998; 2:121–167.
36. Walker BA, Samur KM, Mavrommatis K, Ashby C, Wardell PC, Ortiz M, Towfic F, Stein KC, Bauer AM, Amatangelo M. The Multiple Myeloma Genome Project: Development of a Molecular Segmentation Strategy for the Clinical Classification of Multiple Myeloma. Blood. 2016; 128:196–196.
37. Vardhanabhuti S, Blakemore JS, Clark MS, Ghosh S, Stephens JR, Rajagopalan D. A comparison of statistical tests for detecting differential expression using Affymetrix oligonucleotide microarrays. Omics: a journal of integrative biology. 2006; 10:555–566.
38. Irizarry RA, Wu Z, Jaffee AH. Comparison of Affymetrix GeneChip expression measures. Bioinformatics. 2006; 22:789–794.
39. Morgan GJ, Davies EF, Gregory MW, Bell ES, Szubert JA, Navarro Coy N, Cook G, Feyler S, Johnson RP, Rudin C, Drayson TM, Owen GR, Ross MF, et al. Cyclophosphamide, thalidomide, and dexamethasone as induction therapy for newly diagnosed multiple myeloma patients destined for autologous stem-cell transplantation: MRC Myeloma IX randomized trial results. Haematologica. 2012; 97:442–50.
40. Morgan GJ, Davies EF, Gregory MW, Bell ES, Szubert JA, Cook G, Drayson TM, Owen GR, Ross MF, Jackson HG, Child AJ. Long-term follow-up of MRC Myeloma IX trial: Survival outcomes with bisphosphonate and thalidomide treatment. Clin Cancer Res. 2013; 19:6030–8.
41. Therneau TM, Grambsch MP. Modeling Survival Data: Extending the Cox Model. 2000, New York: Springer.
42. Hothorn T, Hornik K, Zeileis A. Unbiased recursive partitioning: A conditional inference framework. Journal of Computational and Graphical statistics. 2006; 15:651–674.
43. Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of Statistical Software. 2010; 33:1–22.
44. Meyer D, Dimitriadou E, Hornik KA. Weingessel and Leisch F. e1071: Misc Functions of the Department of Statistics (e1071), TU Wien. 2014.
45. Ritchie ME, Phipson B, Wu D, Hu Y, Law WC, Shi W, Smyth KG. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015; 43. doi: 10.1093/nar/gkv007.