
INTRODUCTION
Colorectal cancer (CRC) is still a major malignancy in the world, which causes significant mortality each year [1–3]. Mitochondrion is vital for regulating cell signaling and survival [4, 5]. Recent studies have developed a small-molecule mitochondrion-interfering compound, named NPC-26 [6]). It disturbs normal mitochondrial functions, causing mitochondrial permeability transition pore (mPTP) opening and reactive oxygen species (ROS) production, and eventually leading to cell death [6, 7]. NPC-26 induces a conversion from elongated to punctate mitochondria, and provokes non-apoptotic cell death which is BAX-/BAK-independent [6]. Further studies have proposed that NPC26-induced cell death is dependent on activation of kinase signaling pathways [6]. To our best knowledge, the potential effect of this compound in CRC cells has not been tested thus far. More importantly, the underlying signaling mechanisms of NPC-26-induced cell death are still vague. Here, we suggest that NPC-26 kills human CRC cells possibly via activating AMP-activated protein kinase (AMPK) signaling.
AMPK is the master energy sensor, which helps to maintain the balance of energy metabolism [8, 9]. Recent research has also proposed an important role of AMPK in mediating cell death (see review [10]). Cancer studies have demonstrated that multiple anti-cancer agents and natural occurring compounds could activate AMPK-dependent cell death pathways [10–21]. AMPK may induce cancer cell death via regulating multiple downstream signal targets, including in-activating cancer-promoting mammalian target of rapamycin (mTOR) signaling [22], phosphorylating pro-death p53 signaling [23] and provoking autophagy [24]. Disruption of mitochondrion was shown to provoke AMPK activation [25]. For instance, Head et al., demonstrated that itraconazole directly binds to and inhibits mitochondrial protein voltage-dependent anion channel 1 (VDAC1), causing ATP depletion and AMP:ATP ratio increase [25]. This would eventually lead to AMPK activation [25]. The current study suggests that AMPK activation also mediates NPC-26-induced killing of CRC cells.
RESULTS
NPC-26 is cytotoxic and anti-proliferative to cultured CRC cells
First, the potential effect of NPC-26 on cultured CRC cells was tested. As demonstrated, treatment with NPC-26 (for 72 hours) in HCT-116 cells dose-dependently inhibited cell survival, which was tested by the CCK-8 OD reduction (Figure 1A). NPC-26, at over 1 μM, significantly decreased CCK-8 OD of HCT-116 cells (Figure 1A). NPC-26’s IC-50, or the concentration that inhibited 50% of cell survival, was 7.31±0.55 μM (Figure 1A). NPC-26 also displayed a time-dependent response in inhibiting HCT-116 cell survival (Figure 1B). It would require 48 hours for 10 μM of NPC-26 to exert a significant effect (Figure 1B). When analyzing cell death, we showed that the number of trypan blue positive cells (“dead” cells) was significantly increased following 1-30 μM of NPC-26 treatment (Figure 1C). Thus, these results indicate that NPC-26 is cytotoxic to cultured HCT-116 cells.
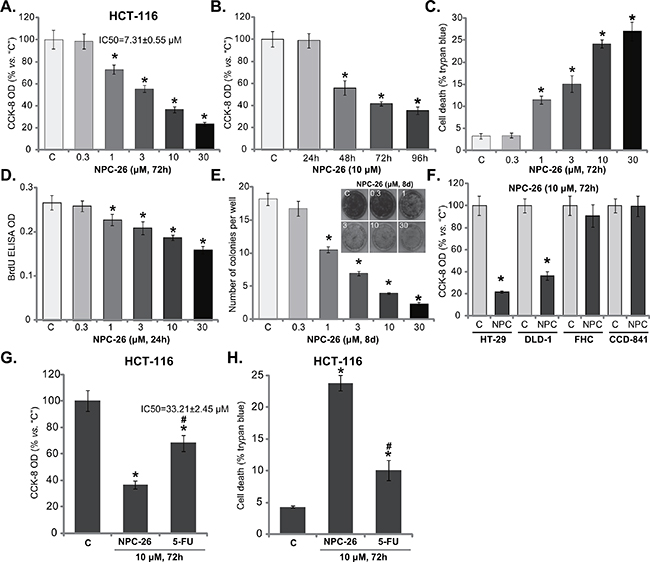
Figure 1: NPC-26 is cytotoxic and anti-proliferative to cultured CRC cells. CRC cell lines (HCT-116, HT-29 and DLD-1) or normal colon epithelial cell lines (FHC and CCD-841) were either left untreated (“C”, same for all figures) or treated with designated concentration of NPC-26 (0.3-30 μM) or 5-Flurouracil (5-FU, 10 μM), cells were further cultivated in conditional medium for indicated time, cell survival A, B, F and G. cell death C and H. and proliferation D and E. were tested by listed assays. Data were expressed as mean ± SD (Same for all figures). For each assay, n=5. Experiments in this figure were repeated five times, and similar results were obtained each time. * p <0.05 vs. “C”. # p <0.05 vs. “NPC-26” (G and H).
We also tested the potential role of NPC-26 on CRC cell proliferation. BrdU ELISA assay was performed, and results showed that NPC-26 dose-dependently decreased the BrdU ELISA OD in HCT-116 cells (Figure 1D), indicating its anti-proliferative activity. Note that for the BrdU assay, cells were treated with NPC-26 for only 24 hours, when no significant cytotoxicity was yet noticed (Figure 1B). Further studies showed that NPC-26 at 1-30 μM dramatically decreased the number of proliferative HCT-116 colonies, again confirming its anti-proliferative activity (Figure 1E). Therefore, NPC-26 is also anti-proliferative to HCT-116 cells.
CCK-8 assay was again utilized to test the potential activity of NPC-26 to other CRC cells. Results showed that 10 μM of NPC-26 significantly inhibited survival of two other established CRC cell lines: HT-29 and DLD-1 (Figure 1F). On the other hand, same NPC-26 treatment (10 μM, 72 hours) failed to affect the survival of two normal colon epithelial cell lines: FHC and CCD-841 (Figure 1F). Thus, it appears that NPC-26 is only cytotoxic to the cancerous cells, which is proposed by other studies [7]. 5-Flurouracil (5-FU) in a very common chemotherapeutic drug for CRC treatment [26]. We showed that 10 μM of NPC-26 was more potent than same concentration of 5-FU in inhibiting HCT-116 cell survival (Figure 1G) and inducing cell death (Figure 1H). The IC-50 for 5-FU (72 hours treatment) was 33.21±2.45 μM (Figure 1G), which was also significantly higher than that of NPC-26 (7.31±0.55 μM).
NPC-26-induced killing of CRC cells requires AMPK activation
As discussed, recent studies have shown that activation of AMPK by a number of different agents could kill CRC cells [13, 14, 27–29]. Thus, the possible involvement of AMPK signaling in NPC-26’s actions was tested. Western blot assay results in Figure 2A demonstrated that treatment with NPC-26 (at 1-30 μM) in HCT-116 cells induced significant AMPK activation, the latter was tested by phosphorylation (“p-”) of AMPKα1 (T172) and its major downstream target acetyl-CoA carboxylase (ACC, Ser-79) [9]. NPC-26 again displayed a dose-dependent response in activating AMPK (Figure 2A, quantification).
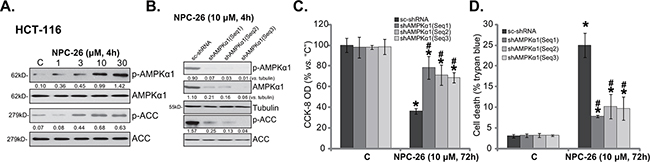
Figure 2: NPC-26-induced killing of CRC cells requires AMPK activation. HCT-116 cells were treated with designated concentration of NPC-26 for 4 hours, expression of listed proteins was shown A. Puromycin-selected HCT-116 cells, expressing listed shRNA, were treated with/out NPC-26 (10 μM) for applied time, listed proteins were shown B.; Cell survival C. and cell death D. were tested. For each assay, n=5. Experiments in this figure were repeated three times, and similar results were obtained each time. AMPKα1/ACC phosphorylation (vs. total protein, or vs. tubulin when mentioned) was quantified (A and B). * p <0.05 vs. “C”. # p <0.05 vs. “sc-shRNA”.
Genetic strategy was then applied to interfere AMPK activation. First, three different AMPKα1 shRNAs with non-overlapping sequences [“shAMPKα1(Seq1/2/3)”] [30] were utilized to stably knockdown AMPKα1. As demonstrated, the three shRNAs indeed dramatically downregulated AMPKα1 in HCT-116 cells (Figure 2B). NPC-26-induced AMPK activation, or p-AMPKα1/p-ACC, was almost completely blocked in AMPKα1-silenced cells (Figure 2B). Consequently, NPC-26-induced cytotoxicity, tested by CCK-8 OD reduction (Figure 2C) and trypan blue staining increase (Figure 2D), was attenuated in AMPKα1-silenced HCT-116 cells. Notably, these AMPKα1 shRNAs alone didn’t affect HCT-116 cell survival/death (Figure 2C and 2D).
AMPKα1 mutation inhibits NPC-26-induced killing of HCT-116 cells
The above shRNA results imply that AMPK activation mediates NPC-26-induced cytotoxicity against HCT-116 cells. To further support this hypothesis, a dominant negative AMPKα1 (“dn-AMPKα1”, T172D) construct [27, 31, 32] was introduced to the HCT-116 cells. Via puromycin selection, two stable HCT-116 cell lines with this construct were established [“dn-AMPKα1 (L1/2)”]. Western blot assay results in Figure 3A confirmed expression of dn-AMPKα1 (Flag-tagged) in the stable cells. Significantly, dn-AMPKα1 expression almost completely blocked NPC-26-induced AMPK activation (Figure 3A). As a result, NPC-26-induced HCT-116 cell death was also attenuated (Figure 3B). Interestingly, treatment with NPC-26 (10 μM, 4h) induced significant AMPK activation in HT-29 cells (Figure 3C), but not in the FHC colon epithelial cells (Figure 3D). As a matter of fact, expression of total AMPKα1 and ACC was also extremely low in the epithelial cells (Figure 3C and 3D). These results could at least in part explain why these epithelial cells were not killed by NPC-26 (Figure 1F). The above AMPKα1 shRNA and mutation experiments were also repeated in HT-29 cells, and similar results were obtained (Data not shown). Together, these results suggest that NPC-26-induced killing of CRC cells requires AMPK activation.
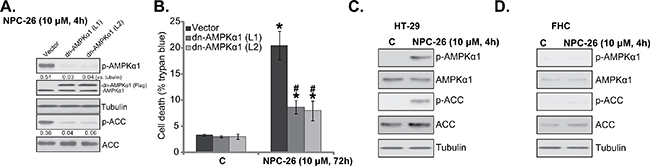
Figure 3: AMPKα1 mutation inhibits NPC-26-induced killing of HCT-116 cells. Puromycin-selected HCT-116 cells, expressing dominant negative AMPKα1 (“dn-AMPKα1”, T172D) or empty vector, were treated with/out NPC-26 (10 μM) for applied time, listed proteins were shown A.; Cell death B. were tested. HT-29 cells or FHC colon epithelial cells were treated with designated concentration of NPC-26 for 4 hours, expression of listed proteins was shown C. and D. For each assay, n=5. Experiments in this figure were repeated three times, and similar results were obtained each time. AMPKα1/ACC phosphorylation (vs. total protein, or vs. tubulin when mentioned) was quantified (A). * p <0.05 vs. “C”. # p <0.05 vs. “Vector”.
NPC-26 disrupts mitochondrial function, causing AMPK activation
A very recent study by Dong et al., [7] demonstrated that NPC-26 disrupts normal mitochondrial function, leading to mPTP opening and ROS production in pancreatic cancer cells. Here, NPC-26 treatment (10 μM) in HCT-116 cells similarly induced mitochondrial depolarization (JC-10 intensity increase, indicating mPTP opening [7]) (Figure 4A) and significant ROS production (Figure 4B). Thus, normal mitochondrial functions could also be disrupted by NPC-26 in HCT-116 cells. To study the link between mitochondrial dysfunction and AMPK activation in NPC-26-treated cells, pharmacological strategy was applied. As demonstrated in Figure 4C, NPC-26-induced AMPK activation was largely inhibited with co-treatment of ROS scavengers (NAC and MnTBAP [33]) or mPTP blocker cyclosporin A (CsA) [34] and sanglifehrin A (SfA) [35]. Remarkably, above inhibitors also alleviated NPC-26-induced killing of HCT-116 cells (Figure 4D and 4E). Treatment with these inhibitors alone didn’t affect AMPK activation and HCT-116 cell survival/death (Data not shown). These results suggest that NPC-26 induces mitochondrial dysfunction, which possibly leads to AMPK activation and subsequent cell death.
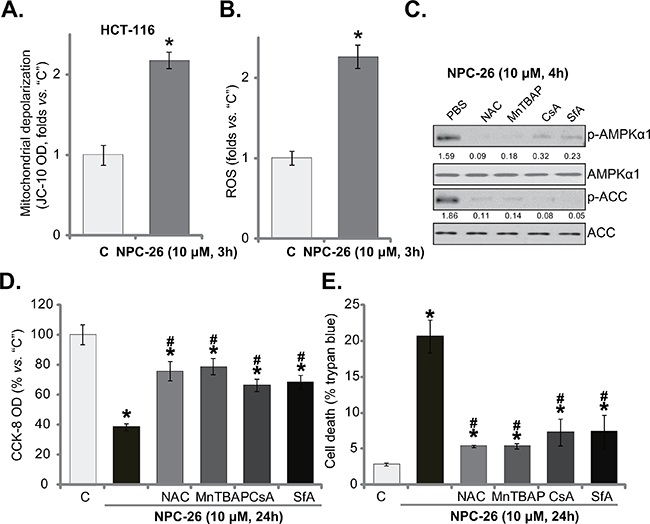
Figure 4: NPC-26 disrupts mitochondrial function, causing AMPK activation. HCT-116 cells were treated NPC-26 (10 μM) for 3 hours, mitochondrial depolarization A. and cellar ROS content B. were tested. HCT-116 cells were pretreated for 30 min with NAC (500 μM), MnTBAP (10 μM), cyclosporin A (CsA, 0.5 μM), or sanglifehrin A (SfA, 2.5 μM), followed by NPC-26 (10 μM) treatment for indicated time period; Expression of listed proteins were shown C.; Cell survival D. and cell death E. were also tested. For each assay, n=5. Experiments in this figure were repeated three times, and similar results were obtained each time. AMPKα1/ACC phosphorylation (vs. total protein) was quantified (C). * p <0.05 vs. “C”. # p <0.05 vs. NPC-26 only.
NPC-26 inhibits HCT-116 tumor growth in SCID mice
In order to test the anti-tumor activity of NPC-26 in vivo, the severe combined immuno-deficient (SCID) mice bearing HCT-116 xenograft tumor model was applied. Results in Figure 5A demonstrated that i.p. injection of NPC-26 (25 mg/kg body weight, daily) [7] dramatically suppressed growth of HCT-116 tumors with scramble control shRNA (“sc-shRNA”, or control tumors). Significantly, NPC-26-induced anti-tumor activity in vivo was remarkably weakened against HCT-116 tumors with AMPKα1 shRNA (“Seq-1”) (Figure 5A), indicating that AMPK activation could also be required for NPC-26’s actions in vivo. Indeed, when analyzing tumor tissue samples, we found that NPC-26 administration (12 hours after initial administration) induced significant AMPK activation, or p-AMPK/p-ACC, in control tumors (Figure 5B, left panel), which was absent in tumors expressing AMPKα1 shRNA (Figure 5B, left panel). Tumors with AMPKα1 shRNA also showed extremely low expression of total AMPKα1 (Figure 5B, left panel). Proliferating cell nuclear antigen (PCNA) expression is a well-established marker of proliferation. Cyclin D1 is important for cell proliferation [36, 37]. Here, we showed that PCNA and Cyclin D1 were both downregulated in NPC-26-treated control tumors, but not in AMPKα1-silenced tumors (Figure 5B, right panel). IHC staining assay further confirmed AMPK activation [p-AMPKα1 (Thr-172) staining] by NPC-26 in control tumors, but not in the AMPKα1 shRNA-expressing tumors (Figure 5C). Notably, expression of AMPKα1 shRNA alone didn’t affect HCT-116 tumor growth in SCID mice. Results in Figure 5D demonstrated that the above NPC-26 administration didn’t affect the body weight of experimental mice, indicating that the regimens were relatively safe [7].
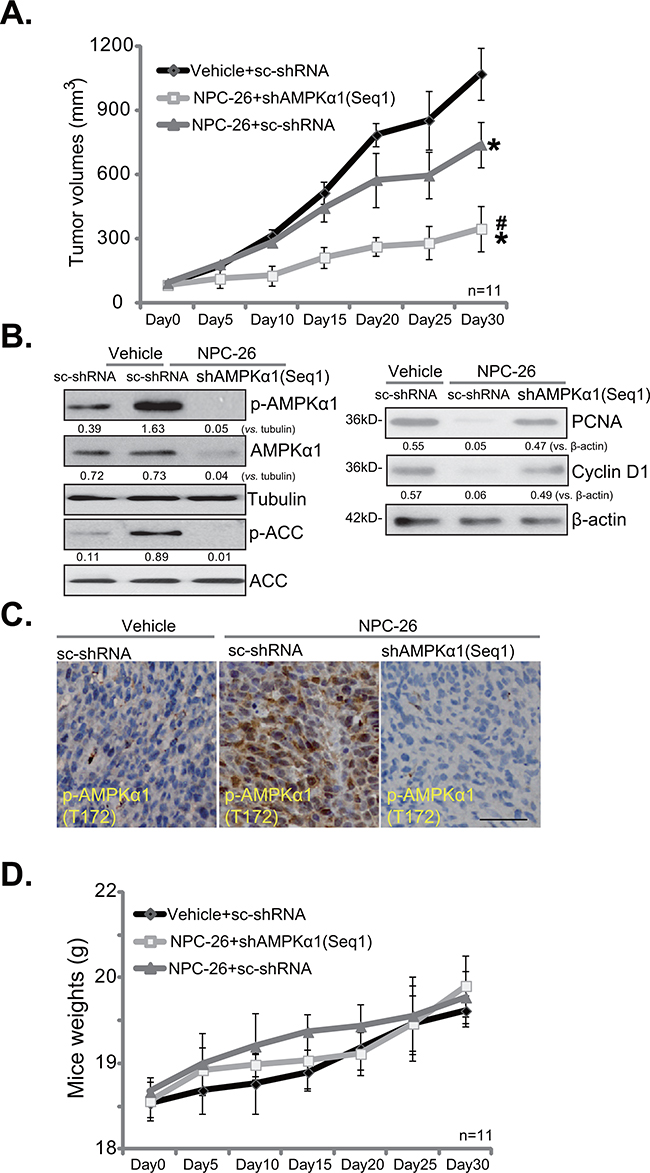
Figure 5: NPC-26 inhibits HCT-116 tumor growth in SCID mice. Same amount (5 million per mouse) of HCT-116 cells expressing scramble control shRNA (“sc-shRNA”) or AMPKα1 shRNA (“Seq1”) were inoculated to the SCID mice to establish xenografted tumors. Mice were administrated with NPC-26 (25 mg/kg, daily, i.p.) or saline (“Vehicle”) for a total of 30 days, tumor volume A. and mice body weight D. were recorded every 5 days. Twelve hours after initial NPC-26 administration, one tumor of each group was isolated, and expression of listed proteins was tested by Western blot assay B. or IHC staining assay C. * p <0.05 vs. “Vehicle”. # p <0.05 vs. NPC-26 treatment of “sc-shRNA” tumors. Bar=100 μm (C).
DISCUSSION
AMPK plays a pivotal role in regulating a number of key cellular functions, from energy metabolism, cell mitosis, apoptosis to autophagy [9, 38]. However, whether AMPK is pro-survival or pro-death is still debatable. It is now known that sustained or intensified AMPK activation will inhibit cell growth and promote cancer cell death [13, 39, 40]. As a matter of fact, a number of anti-cancer agents were shown to kill cancer cells via activating AMPK-dependent signalings [10–21].
Under certain circumstances, it has yet been proposed that AMPK activation could also be pro-survival [41, 42]. The difference might be due to the intensity of AMPK activation. Low level of AMPK activation might promote cell survival, but intensified AMPK activation could promote cell death via regulating its downstream signalings (p53, mTOR inhibition and autophagy etc.). In fact, the activity of AMPK could increase over 100-fold on phosphorylation of a conserved threonine residue (Thr-172) within the activation loop at α1 subunit [38, 43]. In the current study, we showed that NPC-26 induced significant AMPKα1 phosphorylation at Thr-172, indicating a profound AMPK activation. Remarkably, AMPKα1 shRNA knockdown or Thr-172 dominant negative mutation not only abolished NPC-26-induced AMPK activation, but also attenuated CRC cell death. Thus, AMPK activation by NPC-26 is indeed pro-death in CRC cells. Notably, NPC-26 was non-cytotoxic to normal colon epithelial cells, where AMPK was also not significantly provoked. Further studies showed that NPC-26 disrupted mitochondrial function, causing mPTP opening and ROS production, which served as the upstream signal for AMPK activation. Remarkably, ROS scavengers (NAC or MnTBAP) and mPTP blockers (CsA or SfA) almost completely blocked NPC-26-induced AMPK activation.
It should be noted that mitochondria in cancer cells are structurally and functionally different from those in normal (“epithelial”) cells, which are often highly-active in malignant cells to participate in metabolic reprogramming and cell activities [4, 5]. Intriguingly, existing literatures have also reported that certain key mPTP components are up-regulated in cancer cells. For example, the ATP synthase c subunit was upregulated in human breast cancer cells [44]. VDAC-1 over-expression was also observed in several cancer cells [7, 45]. Unique upregulation of mPTP components and high mitochondrial activity in cancer cells could explain why only cancer cells, but not the epithelial cells, were killed by NPC-26 treatment. As a matter of fact, we found that NPC-26 failed to induce ROS production, AMPK activation and significant cytotoxicity in two normal colon epithelial cell lines (FHC and CCD-841). The selective cytotoxicity of NPC-26 to cancerous cells has been reported early as well [7].
Intriguingly, AMPK blockage, via AMPKα1 shRNA knockdown or Thr-172 dominant negative mutation, didn’t completely abolished NPC-26-mediated killing of CRC cells (Figure 2 and 3). Meanwhile, ROS scavengers (NAC or MnTBAP) as well as mPTP blockers (CsA and SfA) only alleviated, but didn’t abolish NPC-26’s cytotoxicity (Figure 4). It is possible that these interfering strategies didn’t result in complete inhibition of the targeted pathways (AMPK, ROS and mPTP). It is more likely that other signalings besides AMPK may also contribute to NPC-26’s actions in CRC cells. Therefore, further studies will be needed to explore the relationship between AMPK and these other pathways in mediating NPC-26’s actions in CRC cells. It will also be important to further characterize the underlying mechanism of NPC-26-induced AMPK activation. In summary, we propose that NPC-26 kills CRC cells possibly via activating AMPK signaling. NPC-26 might have translational value for the treatment of CRC.
MATERIALS AND METHODS
Chemicals, reagents, and antibodies
NPC-26 was a gift from Dr. Dong’s group [33]. N-acetyl cysteine (NAC) and Mn (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), a superoxide dismutase mimetic, were provided by Sigma Chemicals (Sigma, St. Louis, MO). mPTP blockers cyclosporin A (CsA) or sanglifehrin A (SfA) and 5-Flurouracil (5-FU) were also obtained from Sigma Chemicals. Antibodies for tubulin and AMPK signaling proteins were purchased from Cell Signaling Tech (Shanghai, China).
Cell culture
HCT-116, DLD-1 and HT-29 CRC cell lines were provided by Dr. Lu’s group [27, 46]. Cells were maintained in DMEM medium (with 10% FBS). Two normal colon epithelial cell lines, FHC and CCD-841, were purchased from the Cell Bank of Shanghai Institute of Biological Science (Shanghai, China). The above epithelial cells were also cultivated in DMEM medium.
Cell survival assay
To test cell survival, cell counting kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) assay kit was performed based on the attached manual. CCK-8 absorbance optic density (OD) was recorded at 450 nm.
Trypan blue staining assay
After treatment, trypan blue (0.2%)-stained cells was counted by the Countess automatic cell counter (Invitrogen, Shanghai, China). The cell death percentage (%) was calculated by the number of the trypan blue cells divided by the total cell number.
BrdU incorporation assay
To test cell proliferation, 5-bromo-2’-deoxyuridine (BrdU) incorporation assay was applied. Briefly, BrdU (10 μM, Roche, Shanghai, China) was pre-added. Following treatment of cells, BrdU incorporation was determined in enzyme-linked immunosorbent assay (ELISA) format. ELISA OD at 450 nm was tested.
Colony formation assay
HCT-116 cells with applied NPC-26 treatment were re-suspended in DMEM medium containing 0.5% agar (Sigma), which were then plated onto a pre-solidified six-well plate. Afterwards, cells were cultured in NPC-26-containing medium for 8 days. The remaining colonies were counted manually.
Western blot assay
Cell or HCT-116 tumor tissue samples were incubated in lysis buffer described [27, 46]. Quantified protein lysates (30 μg/lane) were separated by 8-10% of SDS-PAGE gels, and were transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were then blocked with 10% milk, and incubated with designated primary and secondary antibodies. The blots were then subjected to ECL detection. Indicated protein band was quantified of total gray via ImageJ software (NIH).
Mitochondrial depolarization assay
As described previously [7], mitochondrial depolarization, indicating mPTP opening, was tested by the commercial JC-10 dye (Invitrogen) [7]. Following the applied treatment, CRC cells were incubated with JC-10 dye (2 μg/mL). Afterwards, the green fluorescence intensity was tested immediately via a fluorescence microplate reader (Titertek Fluoroscan, Germany) [7].
ROS detection
Following treatment, cells were stained with CellRox Orange Reagent (5 μM, Invitrogen) at 37°C for 30 min. ROS content was detected by the above fluorescence microplate reader (Titertek Fluoroscan, Germany).
AMPKα1 short hairpin RNA (shRNA) knockdown
Three AMPKα1 shRNAs [31] with non-overlapping sequences (“Seq-1/2/3”) were designated by Genepharm (Shanghai, China). The short hairpin sequences for AMPKα1 were : 5’-GCAGAAGTTT GTAGGGCAATT-3’(shAMPKα1-Seq1) [47], 5’-GCATAA TAAGTCACAGCCAAA-3’ (shAMPKα1-Seq2) [48] and 5’CTCCAAGACCAGGAAGTCATACAATAGAA-3’(shAMPKα1-Seq3) [49]. The AMPKα1 shRNAs were packed into GV248 lentiviral vector containing puromycin resistance gene. The lentiviral AMPKα1 shRNA was added to HCT-116 cells for 12 hours. Afterwards, puromycin (5 μg/mL) was added to select stable colonies for 5-6 passages. AMPKα1 expression in the stable cells was determined by Western blot assay. Control cells were infected with same amount of lentiviral scramble shRNA.
AMPK dominant negative mutation
Dominant-negative (dn) mutant of AMPKα1 (AMPKα1-T172A) construct was provided by Dr. Lu’s group [27, 31, 32]. The dn-AMPKα1 or the empty vector (each 0.2 μg/mL per well) was transfected to cancer cells via Lipofectamine 2000, and stable cells were again selected by puromycin (5 μg/mL).
Mice xenograft assay
The protocols using 6-8 week-old severe combined immunodeficient (SCID) mice (weighting 18-19g) were approved by the IACUC of all authors’ institutions. Exponentially growing HCT116 cells (5*106 cells per mouse), expressing scramble control shRNA or AMPKα1 shRNA (“Seq1”), were subcutaneously (s.c.) injected into SCID mice. Within three weeks, the xenografted tumors were established around 100 mm3 in volume. The SCID mice were then treated as described. Tumor volumes, recorded every 5 days, were calculated using the formula: (mm3) = (A2 × B)/2: A and B were the shortest and the longest diameter, respectively. Mice body weights were also recorded.
Immunohistochemistry (IHC) staining
The staining was performed on the cryostat sections (4 μm) of HCT-116 tumors using the standard protocol [32]. p-AMPKα1 (Thr-172, 1: 50) antibody was utilized. DAB was applied to stain the positive staining.
Statistical analysis
All values were expressed as the mean ± standard deviation (SD). A p-value, calculated by ANOVA, of less than 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work is supported by Shanghai medical key subject construction project (ZK2015B15), Shanghai weak discipline construction plan (2016ZB0202) and Shanghai Minhang District science and technology research plan (2015MHZ039). The funders have no role in design, in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
CONFLICTS OF INTEREST
The listed authors have no conflicts of interest.
Author contributions
All authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to draft the manuscript.
REFERENCES
1. Schmoll HJ, Stein A. Colorectal cancer in 2013: Towards improved drugs, combinations and patient selection. Nat Rev Clin Oncol. 2014; 11:79-80.
2. Kuipers EJ, Rosch T, Bretthauer M. Colorectal cancer screening--optimizing current strategies and new directions. Nat Rev Clin Oncol. 2013; 10:130-142.
3. Brouquet A, Nordlinger B. Metastatic colorectal cancer outcome and fatty liver disease. Nat Rev Gastroenterol Hepatol. 2013; 10:266-267.
4. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, Bazhin AV. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J Cell Physiol. 2016.
5. Costantini P, Jacotot E, Decaudin D, Kroemer G. Mitochondrion as a novel target of anticancer chemotherapy. J Natl Cancer Inst. 2000; 92:1042-1053.
6. Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe'er D, Shaik F, Bulinski JC, Stockwell BR. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc Natl Acad Sci U S A. 2011; 108:E771-780.
7. Dong YY, Zhuang YH, Cai WJ, Liu Y, Zou WB. The mitochondrion interfering compound NPC-26 exerts potent anti-pancreatic cancer cell activity in vitro and in vivo. Tumour Biol. 2016.
8. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009; 9:563-575.
9. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011; 13:1016-1023.
10. Vakana E, Altman JK, Platanias LC. Targeting AMPK in the treatment of malignancies. J Cell Biochem. 2012; 113:404-409.
11. Cao C, Lu S, Kivlin R, Wallin B, Card E, Bagdasarian A, Tamakloe T, Chu WM, Guan KL, Wan Y. AMP-activated protein kinase contributes to UV- and H2O2-induced apoptosis in human skin keratinocytes. J Biol Chem. 2008; 283:28897-28908.
12. Chen MB, Shen WX, Yang Y, Wu XY, Gu JH, Lu PH. Activation of AMP-activated protein kinase is involved in vincristine-induced cell apoptosis in B16 melanoma cell. J Cell Physiol. 2011; 226:1915-1925.
13. Kang MR, Park SK, Lee CW, Cho IJ, Jo YN, Yang JW, Kim JA, Yun J, Lee KH, Kwon HJ, Kim BW, Lee K, Kang JS, Kim HM. Widdrol induces apoptosis via activation of AMP-activated protein kinase in colon cancer cells. Oncol Rep. 2012; 27:1407-1412.
14. Kim YM, Hwang JT, Kwak DW, Lee YK, Park OJ. Involvement of AMPK signaling cascade in capsaicin-induced apoptosis of HT-29 colon cancer cells. Ann N Y Acad Sci. 2007; 1095:496-503.
15. Sauer H, Engel S, Milosevic N, Sharifpanah F, Wartenberg M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-Jun N-terminal kinase. Int J Oncol. 2012; 40:501-508.
16. Shao JJ, Zhang AP, Qin W, Zheng L, Zhu YF, Chen X. AMP-activated protein kinase (AMPK) activation is involved in chrysin-induced growth inhibition and apoptosis in cultured A549 lung cancer cells. Biochem Biophys Res Commun. 2012; 423:448-453.
17. Sun H, Yu T, Li J. Co-administration of perifosine with paclitaxel synergistically induces apoptosis in ovarian cancer cells: more than just AKT inhibition. Cancer Lett. 2011; 310:118-128.
18. Wang B, Wang XB, Chen LY, Huang L, Dong RZ. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem Biophys Res Commun. 2013; 437:1-6.
19. Yang L, Zheng LY, Tian Y, Zhang ZQ, Dong WL, Wang XF, Zhang XY, Cao C. C6 ceramide dramatically enhances docetaxel-induced growth inhibition and apoptosis in cultured breast cancer cells: a mechanism study. Exp Cell Res. 2015; 332:47-59.
20. Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, Wang QJ, Yang Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 2010; 285:40461-40471.
21. Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH, Ai X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Biophys Res Commun. 2012; 419:741-747.
22. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003; 115:577-590.
23. Nieminen AI, Eskelinen VM, Haikala HM, Tervonen TA, Yan Y, Partanen JI, Klefstrom J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc Natl Acad Sci U S A. 2013; 110:E1839-1848.
24. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13:132-141.
25. Head SA, Shi W, Zhao L, Gorshkov K, Pasunooti K, Chen Y, Deng Z, Li RJ, Shim JS, Tan W, Hartung T, Zhang J, Zhao Y, Colombini M, Liu JO. Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells. Proc Natl Acad Sci U S A. 2015; 112:E7276-7285.
26. Chung KY, Saltz LB. Adjuvant therapy of colon cancer: current status and future directions. Cancer J. 2007; 13:192-197.
27. Lu PH, Chen MB, Ji C, Li WT, Wei MX, Wu MH. Aqueous Oldenlandia diffusa extracts inhibits colorectal cancer cells via activating AMP-activated protein kinase signalings. Oncotarget. 2016; 7:45889-45900. doi: 10.18632/oncotarget.9969.
28. Chen MB, Zhang Y, Wei MX, Shen W, Wu XY, Yao C, Lu PH. Activation of AMP-activated protein kinase (AMPK) mediates plumbagin-induced apoptosis and growth inhibition in cultured human colon cancer cells. Cell Signal. 2013; 25:1993-2002.
29. Chen MB, Wei MX, Han JY, Wu XY, Li C, Wang J, Shen W, Lu PH. MicroRNA-451 regulates AMPK/mTORC1 signaling and fascin1 expression in HT-29 colorectal cancer. Cell Signal. 2014; 26:102-109.
30. Chen L, Chen Q, Deng G, Kuang S, Lian J, Wang M, Zhu H. AMPK activation by GSK621 inhibits human melanoma cells in vitro and in vivo. Biochem Biophys Res Commun. 2016; 480:515-521.
31. Lv G, Zhu H, Zhou F, Lin Z, Lin G, Li C. AMP-activated protein kinase activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Biochem Biophys Res Commun. 2014; 453:13-18.
32. Chen MB, Jiang Q, Liu YY, Zhang Y, He BS, Wei MX, Lu JW, Ji Y, Lu PH. C6 ceramide dramatically increases vincristine sensitivity both in vivo and in vitro, involving AMP-activated protein kinase-p53 signaling. Carcinogenesis. 2015; 36:1061-1070.
33. Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, Marrack PC. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999; 10:735-744.
34. Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999; 160:226-234.
35. Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002; 277:34793-34799.
36. Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol. 2008; 182:509-517.
37. Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008; 27:1106-1113.
38. Hardie DG, Ross FA, Hawley SA. AMP-Activated Protein Kinase: A Target for Drugs both Ancient and Modern. Chem Biol. 2012; 19:1222-1236.
39. Chen MB, Shen WX, Yang Y, Wu XY, Gu JH, Lu PH. Activation of AMP-activated protein kinase is involved in vincristine-induced cell apoptosis in B16 melanoma cell. J Cell Physiol. 2010; 226:1915-1925.
40. Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH, Ai X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Biophys Res Commun. 2012.
41. Narbonne P, Roy R. Caenorhabditis elegans dauers need LKB1/AMPK to ration lipid reserves and ensure long-term survival. Nature. 2009; 457:210-214.
42. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012; 485:661-665.
43. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012; 13:251-262.
44. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, Sneddon S, Hulit J, Howell A, Lisanti MP. Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. 2012; 11:4390-4401.
45. Shoshan-Barmatz V, Ben-Hail D, Admoni L, Krelin Y, Tripathi SS. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim Biophys Acta. 2015; 1848:2547-2575.
46. Chen MB, Yang L, Lu PH, Fu XL, Zhang Y, Zhu YQ, Tian Y. MicroRNA-101 down-regulates sphingosine kinase 1 in colorectal cancer cells. Biochem Biophys Res Commun. 2015; 463:954-960.
47. Zhang L, Jouret F, Rinehart J, Sfakianos J, Mellman I, Lifton RP, Young LH, Caplan MJ. AMP-activated protein kinase (AMPK) activation and glycogen synthase kinase-3beta (GSK-3beta) inhibition induce Ca2+-independent deposition of tight junction components at the plasma membrane. J Biol Chem. 2011; 286:16879-16890.
48. Huang CH, Tsai SJ, Wang YJ, Pan MH, Kao JY, Way TD. EGCG inhibits protein synthesis, lipogenesis, and cell cycle progression through activation of AMPK in p53 positive and negative human hepatoma cells. Mol Nutr Food Res. 2009; 53:1156-1165.
49. Chi PI, Huang WR, Lai IH, Cheng CY, Liu HJ. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/eIF2alpha signaling pathways. J Biol Chem. 2013; 288:3571-3584.