
INTRODUCTION
Acute myeloid leukemias (AMLs) are heterogeneous tumor blood diseases characterized by deregulated cell proliferation, survival and differentiation of hematopoietic/stem progenitor cells [1]. One of the most aggressive subtypes of AML is characterized by the presence of translocation involving the mixed lineage leukemia gene (MLL, or KMT2A). MLL encodes an H3K4 methyltransferase that forms multiprotein chromatin-modifying complexes required in controlling transcriptional program necessary for the development and maintenance of hematopoiesis [2, 3]. Translocations that include MLL count more than 60 different fusion partners, which have been identified in AML, acute lymphoid leukemia, and biphenotypic or chemotherapy-related leukemias [4]. In pediatric and adult AML, the most common translocation juxtaposes the N-terminal portion of the MLL protein to the C-terminal fragment of the AF9 fusion partner in the t(9;11)(p22;q23) generating the oncogenic MLL-AF9 fusion protein [5–7]. MLL translocations contribute to leukemogenesis subverting self-renewal program and block of hematopoietic differentiation [5, 8]. Transformation by MLL-AF9 induced specifically aberrant expression of several transcriptional target genes involved in stem cell self-renewal, maintenance and repression of differentiation-associated genes [5, 9–10]. Among these MLL targets genes, such as HOXA9 and MEIS1, it is well established the crucial role played in MLL-induced leukemia [11, 12], however many other of the genes strongly deregulated by MLL fusion proteins remain poorly characterized.
Zinc finger protein 521 (ZNF521) is a C2H2-type zinc finger transcription factor containing an amino-terminal motif that binds to the nucleosome remodeling and histone deacetylase (NuRD) complex, which is associated with transcriptional repression and conserved among other zinc finger proteins, including Friend of GATA (FOG1 and FOG2), BCL11A, and SALL family members [13]. Initially identified for its expression restricted to human CD34+ progenitor cells [14], ZNF521 has been shown to repress erythroid differentiation by inhibiting GATA-1 activity [15], and to block B-lymphoid differentiation in primary hematopoietic progenitors by antagonizing early B-cell factor 1 (EBF1) [16].
Deregulated expression of ZNF521 mRNA has been observed in medulloblastoma, lymphoblastic lymphoma and acute leukemia [17–19]. Recently, knock-in mice models for E2A-HLF and E2A-PBX1 involving fusion genes in B-lineage acute lymphoblastic leukemia (B-ALL) have demonstrated that enhanced expression of Zfp521, the murine counterpart of human ZNF521, cooperates to leukemia transformation, and an upregulation of ZNF521 was found in human B-ALL samples bearing E2A-HLF or E2A-PBX1 fusion oncogenes. Therefore, an altered expression of ZNF521 may be an important cofactor contributing to hematopoietic cell transformation. Recently, high expression of ZNF521 has been observed in pediatric AML, particularly in those cases carrying MLL gene rearrangements [20, 21]; however the role of ZNF521 in MLL-rearranged AML is currently unknown. In this study, we examined the role of ZNF521 in MLL-rearranged AML cell lines and primary ex vivo MLL-AF9-expressing cells and showed that depletion of ZNF521 impaired AML progression by inducing myeloid differentiation.
RESULTS
ZNF521 is aberrantly overexpressed in pediatric MLL-rearranged AML
Previously, by use of microarray analysis we found a frequent ZNF521 overexpression in pediatric AML with MLL rearrangements [21]. To validate these data and analyze the relationship between ZNF521 expression and distinct MLL-fusion genes, we performed quantitative real-time PCR (qRT-PCR) in an independent cohort of 50 pediatric AML patients (16 MLL-rearranged and 34 non-MLL-rearranged; Supplementary Table 1) and 7 normal bone marrow (BM) controls. We found that ZNF521 was expressed at significantly higher level in AML patients with MLL rearrangements compared to non-rearranged AML and normal controls (P < 0.001, Figure 1A), The analysis of ZNF521 expression between the most frequent MLL rearrangements detected in pediatric AML did not reveal significant difference based on MLL fusion partners (data not shown). In addition, we analyzed the expression of ZNF521 in 6 MLL-rearranged and 6 non-MLL-rearranged human leukemic cell lines. Similarly, leukemic cell lines with MLL rearrangements, with the exception of those carrying MLL-AF4 fusion transcripts, showed significantly higher ZNF521 mRNA levels compared to cell lines with other abnormalities (P < 0.05, Figure 1B). Thus, our data indicate that ZNF521 is likely involved in MLL-mediated transformation in AML.
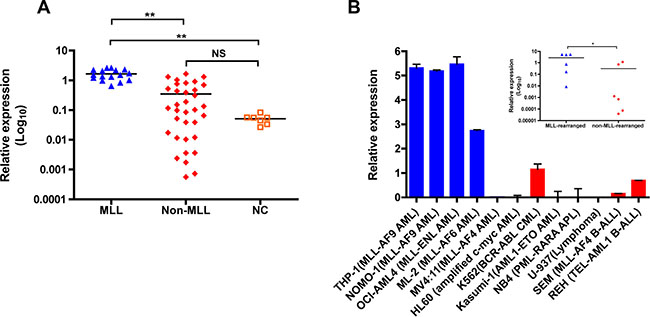
Figure 1: ZNF521 is aberrantly overexpressed in MLL-rearranged AML. (A) qRT-PCR for the expression of ZNF521 in 16 MLL-rearranged AML (MLL), 34 non-MLL-rearranged AML (Non-MLL) and 7 normal control (NC). The results are normalized to GAPDH and analyzed by 2−ΔCt method. NS, not significant, **P < 0.001, kruskal-Wallis test. (B) qRT-PCR analysis of ZNF521 expression in a representative panel of 12 human leukemic cell lines normalized to GAPDH and analyzed by 2−ΔCt method. Data are represented as mean ± SD of three independent experiments. y axis is linear. Inset, dot plots of mean ZNF521 mRNA levels in MLL-rearranged and non-MLL-rearranged cell lines from data presented in (B). *P < 0.05, Mann–Whitney U-test.
ZNF521 depletion reduces cell viability and causes cell cycle arrest without inducing apoptosis of MLL-rearranged AML cell lines
To determine whether ZNF521 is functionally important in MLL-rearranged AML, we first examined the effects of ZNF521 knockdown on the cell proliferation using a panel of human MLL-rearranged AML cell lines, including, THP-1, NOMO-1 (both expressing MLL-AF9), ML-2 (expressing MLL-AF6) and OCI-AML4 (expressing MLL-ENL). To suppress ZNF521, we used GFP-tagged lentiviral vectors expressing anti-ZNF521 shRNAs (ZNF004 and ZNF710) or a non-targeting shRNA sequence (shScram). After assessing transduction efficiency by flow cytometry (range 30–80%) (Supplementary Figure 1 and data not shown), GFP-positive cells were sorted and maintained under standard cell culture conditions for subsequent analysis. As expected, in all four cell lines downregulation of ZNF521 varied between 60% and 75% compared to ZNF521 mRNA expression in shScram-transduced cells, and this correlated with a decrease in ZNF521 protein amount (Supplementary Figure 2). In addition, ZNF521 knockdown progressively reduced viability of all the transduced cell lines (Figure 2A), and it inhibited colony formation ability of MLL-rearranged cells, measured 2 weeks after transduction (Figure 2B). In order to get a deeper insight, cell cycle analysis and apoptosis induction were assessed in GFP-positive MLL-rearranged cells. At day 7, we observed an accumulation of cells in G1 phase (17%–77%) in three out of four cell lines (THP-1, NOMO-1 and ML-2) expressing anti-ZNF521 shRNAs. This was most likely due to S phase reduction (from 29% to 65%) rather than G2/M alterations (Figure 2C). However, annexin V/DAPI assay measured at day 4 and day 7 demonstrated that ZNF521 knockdown did not caused increased apoptosis (Figure 2D), suggesting that ZNF521 may be involved in proliferation and differentiation of MLL-rearranged cells rather than in cell survival. To substantiate this hypothesis, GFP-sorted transduced THP-1 and ML-2 cells were collected on glass slides by cytospin and stained with antibodies against p21 (CDKN1A) and p27 (CDKN1B) cell cycle inhibitors [22]. At day 7, we observed an increase of both p21 and p27 protein expression in ZNF521 knockdown cells, suggesting a prolonged G1/S transition as the main reason for the aforementioned cell cycle arrest (Supplementary Figure 3). Taken together, these findings indicate that ZNF521 expression is essential in the growth potential of MLL-rearranged AML cell lines.
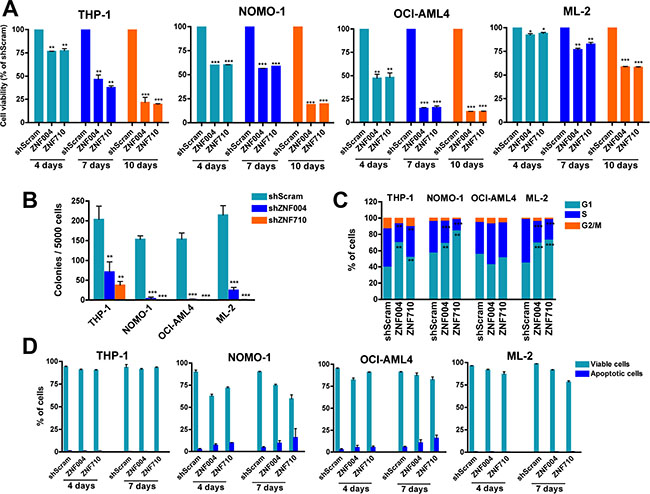
Figure 2: ZNF521 depletion impairs cell proliferation, induces cell cycle arrest but not apoptosis in MLL-rearranged cell lines. (A) MTT cell viability assay in the MLL leukemic cells THP-1, NOMO-1, OCI-AML4 and ML2 transduced with ZNF521 shRNAs (ZNF004 or ZNF710) or non-targeting scramble control (shScram). GFP+ cells were sorted 4 days after transduction and placed in appropriate medium. Graphs show percentage of GFP+ cells measured at day 4, day 7 and day 10, normalized to the percentage of shScram cells. Data are represented as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.001, ***P < 0.0001, t-test. (B) Colony formation of GFP+ cells transduced with ZNF521 shRNAs or shScram. Error bars represent mean ± S.D. of three independent experiments. **P < 0.001, ***P < 0.0001, t-test. (C) Cell cycle distribution at day 7 of ZNF521 knockdown cells and control shScram of gated GFP+ cells. Data are represented as mean ± SD of three independent experiments. **P < 0.001, ***P < 0.0001, t-test. (D) Percentage of apoptotic cells (Annexin V+/DAPI- and Annexin V+/DAPI+) measured after 4 and 7 days post-transduction of gated GFP+ cell population. Data are represented as mean ± SD of three independent experiments.
Depletion of ZNF521 induces myeloid differentiation of MLL-rearranged AML cell lines
Given that ZNF521 can regulate lineage progression of different cell types, including hematopoietic cells [14–16], we analyzed whether ZNF521 depletion might influence differentiation in MLL-rearranged leukemic cells. Flow cytometry analysis of CD11b and CD14 myeloid markers was then performed on GFP-positive cells and revealed a change of these markers in 3 out of 4 cell lines transduced with ZNF521 shRNAs (Figure 3A). The phenotypic changes were also sustained by a more mature macrophage-like morphology observed in all these cell lines upon ZNF521 depletion as compared with transduced control cells (Figure 3B). Additionally, maturation induced by ZNF521 depletion was also supported by upregulation of C/EBPA and PU.1 mRNA levels, two myeloid differentiation markers (Figure 3C). Furthermore, a downregulation of ZNF521 expression occurred in response to treatment with all-trans retinoid acid (ATRA) and with Securinine, two differentiation agents administered to THP-1 and NOMO-1 AML cells, respectively (Supplementary Figure 4). In particular, ATRA and Securinine, previously tested on these cell lines by others [23, 24], were able to reduce ZNF521 mRNA and protein expression, and stimulate MLL-rearranged cell differentiation, supporting the hypothesis that ZNF521 is required to maintain those cells in an undifferentiated state.
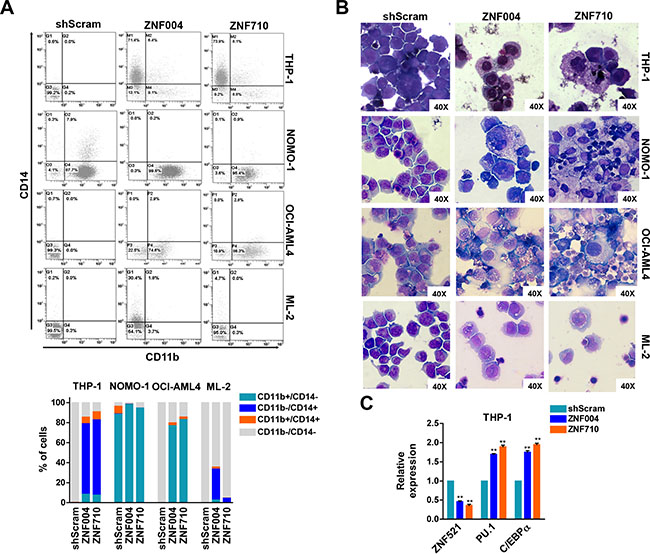
Figure 3: ZNF521 depletion induces myelomonocytic differentiation in MLL-rearranged cell lines. (A) Representative flow cytometry dot plots of gated GFP+ cells analyzed for CD11b and CD14 expression after 7 days of transduction. The mean percentage of CD11b+/CD14−, CD11b−/CD14+, CD11b+/CD14+ and CD11b−/CD14− cells of three biological replicates are shown below. (B) Representative Wright-Giemsa staining of cytospin preparations at day 7 of THP-1, NOMO-1, OCI-AML4 and ML2 GFP+ cells transduced with ZNF521 shRNAs or shScram. Original magnification, × 40. (C) qRT-PCR on THP-1 GFP+ cells for the expression of ZNF521, PU.1 and C/EBPα at day 7 post transduction with ZNF521 shRNAs or shScram. The results are relative to shScram-transduced cells, normalized to GAPDH and analyzed by 2−ΔΔCt method. Data are represented as mean ± SD of three independent experiments. **P < 0.01, t-test.
Effects of ZNF521 depletion in patient-derived AML xenograft cells
To extend our findings to primary cells containing MLL-AF9 oncogene, we transduced ZNF521 shRNAs in ex vivo cells obtained from patient-derived xenografts (Figure 4A). Two out of four patients harboring MLL-AF9 fusion protein (Supplementary Table 3) resulted in successful engraftment into NSG mice. The kinetics of such engraftment, measured by percentage of human CD45+ cells in the peripheral blood varying between 22.3% to 42.2%, ranged from 47 to 67 days and led to expansion of leukemic cells with the same immunophenotype and cytogenetic features of the original patient sample (data not shown). Ex vivo experiments demonstrated that ZNF521 depletion strongly impaired viability and colony formation of mononuclear cells obtained from two primary MLL-AF9 AML patient-derived xenografts (Figure 4B, 4C). Most importantly, an increased expression of myeloid differentiation markers CD11b and CD14 (Figure 4D) and morphological features of mature monocytes/macrophage blast-like was observed (Figure 4E). These findings suggest that ZNF521 overexpression is critical to maintain an immature phenotype consistent with the MLL-rearranged cell lines results.
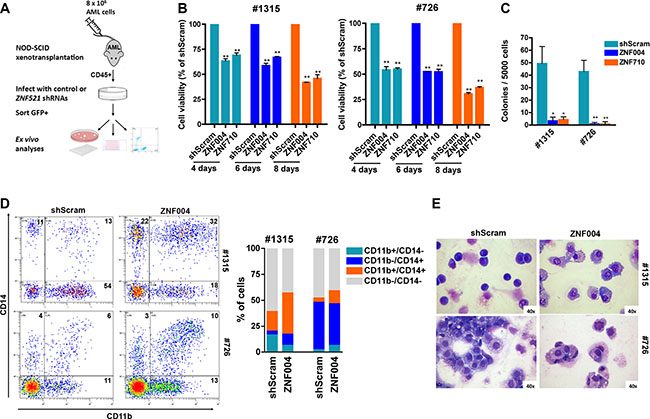
Figure 4: ZNF521 depletion impairs cell growth and induces differentiation on primary MLL-AF9 AML patient-derived xenograft cells. (A) Flow chart of experimental procedure for analyzing the role of ZNF521 in ex vivo cells obtained from patient-derived xenografts. Leukemic cells from patient #726 or patient #1315 were isolated from primary AML mice and infected with lentivirus encoding an shRNA to ZNF521 or shScram. Four days after transduction cells were FACS-sorted for GFP expression and cultured. (B) MTT cell viability assay in ex vivo cells. Data are represented as mean ± SD of three independent experiments. **P < 0.001, t-test. (C) Clonogenic growth of transduced ex vivo GFP+ cells following 14 days in methylcellulose culture. Data are shown as the means ± SD for triplicate analyses. *P < 0.05, **P < 0.001, t-test. (D) Representative flow cytometry dot plots showing expression of CD11b and CD14 in human CD45+ cells in the GFP+ cells population. Numbers indicate percentage of the four populations. The mean percentage of CD11b+/CD14−, CD11b−/CD14+, CD11b+/CD14+ and CD11b−/CD14− populations of three biological replicates are shown in the right panel. (E) Representative Wright-Giemsa-stained cytospins of ex vivo GFP+ cells at day 4 post transduction with ZNF521 shRNA (ZNF004) or shScram. Original magnification, × 40.
Gene expression changes after ZNF521 depletion in THP-1 cells
To investigate the gene expression pattern in MLL-AF9 AML cells expressing high levels of ZNF521, we performed microarray analysis of shZNF521- or shScram-transduced THP-1 cells. Since that the differentiation was overt after 7 days of transduction as reported above, we performed gene expression profiling at day 4 after transduction. A total of 158 genes showed a significant change of expression (>1.5-fold change, FDR< 0.05), 58 were upregulated while 100 were downregulated (Figure 5A and Supplementary Table 4). Gene Set Enrichment Analysis (GSEA) confirmed that ZNF521 depletion affected cell cycle progression and cell fate differentiation related genes [25] (Figure 5B, 5C). These results showed also positive enrichment of genes downregulated in CD133+ HSCs when compared with the CD133- cell [26], and negative enrichment of embryonic stem cells (ESC) associated genes [27] (Figure 5D, 5E). The enrichment of stemness-related genes found by our analysis is in line with proposed role of ZNF521 in the regulation of hematopoietic stem cell homeostasis [28]. Furthermore, GSEA revealed a negative enrichment with genes that are upregulated in MLL-rearranged pediatric AML compared with non-MLL-rearranged AML [29] (Figure 5F, 5G). Interestingly, the ZNF521 depletion gene set revealed positive enrichment with genes that are upregulated in hematopoietic precursors conditionally expressing HOXA9 and MEIS1, including HOXA9 target genes upregulated in hematopoietic stem cells [8, 30] (Figure 5H, 5I). In addition, genes up-regulated or downregulated upon knockdown of HOXA9 [11] were also similarly regulated in ZNF521-transduced THP-1 cells (Figure 5J, 5K). Considering individual genes, we found deregulated genes with known relevance in MLL-fusion-mediated AML (TET1, CDK6 and Musashi2) [31–33] and in myeloid progenitors differentiation (CD14 and MEF2A) [34] (Supplementary Figure 5). Taken together, these results indicate that ZNF521 expression negatively modulates genes involved in myeloid differentiation, and is required to maintain expression programs associated with MLL-induced transformation.
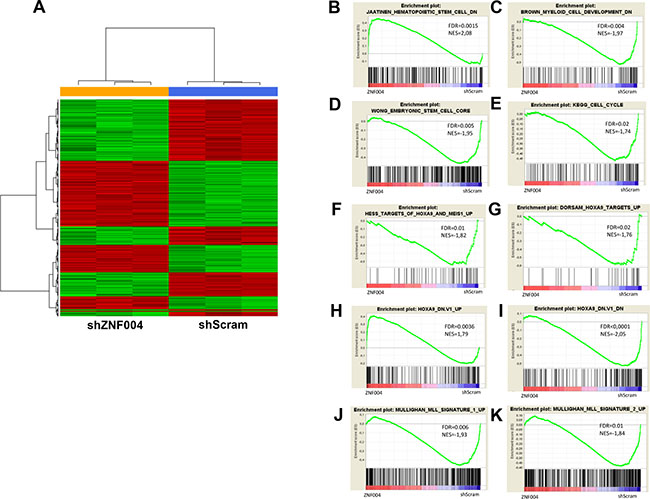
Figure 5: Microarray results of ZNF521 depletion in THP-1 cells. (A) Hierarchical clustering analysis of differently gene expression profiles associated with transduced THP-1 cells with ZNF521 shRNA (ZNF004) or control shScram after 4 days of transduction. Each column represents a sample and each row represent a gene. Relative levels of gene expression are depicted with a color scale where red represents the highest level of expression and green represents the lowest level. (B–E) GSEA plot showing gene expression signature of (B) negative enrichment of cell cycle signature, (C) negative enrichment of downregulated genes in myeloid cell development signature, (D) positive enrichment of downregulated genes in HSCs signature and (E) negative enrichment of embryonic stem cell core signature. (F, G) GSEA plot showing negative enrichment of MLL signature up-regulated genes in pediatric AML. (H–K) GSEA showing enrichment of upregulated genes in HOXA9 up-regulated (H) and down-regulated (I) signatures in HOXA9 knockdown cells, (J) positive enrichment of HOXA9 targets up-regulated and (K) negative enrichment of HOXA9 targets down-regulated in hematopoietic stem cells. The normalization enrichment score (NES) and the false discovery rate (FDR) values are indicated in each panel. Red and blue color bars indicated the positive and negative enrichment, respectively.
ZNF521 gene promoter is activates by MLL fusion proteins
Finally, to investigate the molecular mechanism that upregulates ZNF521 in MLL-rearranged AML, we performed luciferase reporter and ChIP assays using the Flag-tagged MLL-AF9 expression plasmid. To this end, we generated a series of constructs in which 5.0 kb of the genomic region upstream of the ZNF521 transcription start site (TSS) was subdivided in 4 fragments (ZNF521P1, ZNF521P2, ZNF521P3 and ZNF521P4) and inserted into a pGL4-basic reporter plasmid (Figure 6A). Luciferase assays in 293T cells showed that MLL-AF9 strongly activated the promoter region that lay between -1.3 to -3 kb (ZNF521P3) of the TSS (Figure 6A). To further confirm the region of ZNF521 activated by MLL-AF9, we generated 3 constructs (ZNF521P3.1, ZNF521P3.2 and ZNF521P3.3) spanning the ZNF521P3 fragment (Figure 6B). We found that the pGL4-ZNF521P3.3 construct showed the highest luciferase activity (Figure 6B), indicating that the MLL-AF9 responsive elements likely reside between −1.0 and −1.6 kb upstream of the ZNF521 TSS. Furthermore, to determine whether ZNF521 activation was MLL fusion-dependent, we performed ZNF521-driven luciferase reporter assay in another MLL fusion gene (MLL-ENL) and in two non-MLL-associated fusion genes such as AML1-ETO and PML-RARAα. We observed that both AML1-ETO and PML-RARAα yielded only a minimal luciferase activity compared with MLL-ENL that showed even a higher promoter binding affinity than MLL-AF9 (> 2.5 fold) (Figure 6C). Besides, wild-type (WT) MLL did not affect luciferase activity under the same settings, providing evidence that only MLL-fusion proteins likely activate ZNF521 expression (Figure 6C). Consistent with these results, ChIP analyses showed that both MLL-AF9 and MLL-ENL bind to ZNF521 promoter region in transfected 293T cells (Figure 6D). In order to validate MLL-AF9 binding to the ZNF521 promoter in AML cells, we performed ChIP with lysate from NOMO-1 and HL60 cell lines that endogenously expressing MLL-AF9 and WT MLL, respectively. Since that MLL-AF9 lacks the MLL-C portion of WT MLL, an anti-MLL N-terminal (MLLN) and an anti-MLL C-terminal (MLLC) antibodies were used for this experiment. ChIP assays showed that MLLN bound specifically to the ZNF521 promoter region in NOMO-1 but not in HL60 (Figure 6E; upper panel). By contrast, there was not apparently association with MLLC and ZNF521 in both NOMO-1 and HL60 cells (Figure 6E; lower panel). Together, these findings demonstrated that ZNF521 promoter is specifically bound by MLL-AF9, and provide further evidence that MLL fusion oncoproteins may drive aberrant expression of ZNF521, which may in turn lead to a block in differentiation.
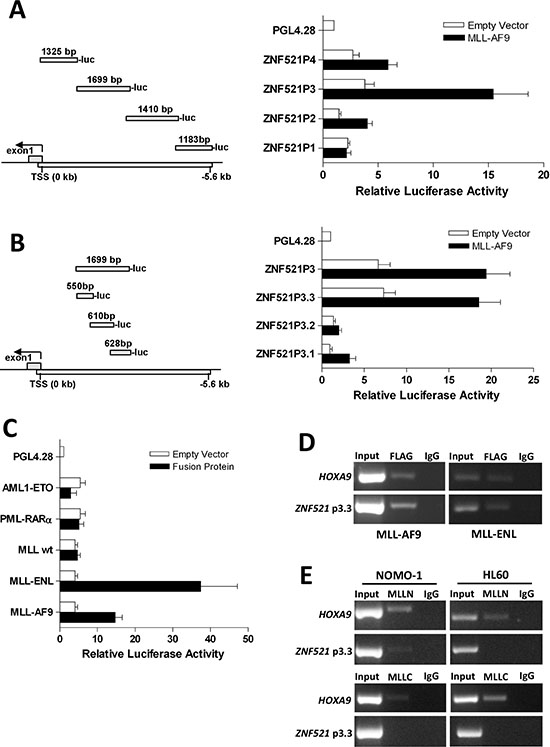
Figure 6: MLL-AF9 and MLL-ENL fusion oncoproteins bind to ZNF521 promoter. (A) An illustration of the 4 fragments representing 5058 bp of ZNF521 promoter and their positions are indicated in the left panel. The numbers above each part are referred to the length (bp) of the genomic fragment that was PCR amplified and then cloned upstream of the luciferase coding sequence (luc) of PGL42.8 plasmid. In the right panel, horizontal bars represent the luciferase activity generate by each construct following transient transfection in 293T cells with MLL-AF9 plasmid. (B) An illustration of the 3 fragments of ZNF521P3 and their respective positions are shown in the left panel. The numbers above each part are referred to the length (bp) of the genomic fragment cloned into PGL42.8 plasmid. In the right panel, horizontal bars represent the luciferase activity generate by each construct as described in A. (C) Luciferase activity of the ZNF521P3.3 fragment after transient transfection in 293T cells with AML1-ETO, PML-RARα, wild-type (WT) MLL, MLL-ENL or MLL-AF9 is shown. For each panel (A, B and C), luciferase activity is expressed relative to the empty vector of each expression plasmid (white bars) and normalizes to Firefly/Renilla luciferase activities considering the empty pGL42.8 vector as 1. Data are represented as mean ± SD of three independent experiments. (D) Both MLL-AF9 and MLL-ENL fusion oncogenes associates with the ZNF521P3.3 promoter region. ChIP assays were performed with the crossed-linked genomic DNA isolated from 293T cells transfected with either Flag-MLL-AF9 or Flag-MLL-ENL and using anti-Flag and anti-IgG antibodies. Normal IgG was used as a negative control. Input DNA from sonicated chromatin and immunoprecipitated DNA were subjected to PCR amplification with primers spanning the ZNF521P3.3 promoter region. PCR amplification with primers specific to the HOXA9 promoter region was used as positive control. Data from a representative of three replicate experiments are shown. (E) ChIP analysis of the ZNF521P3.3 promoter in NOMO-1 cells, which express MLL-AF9 and HL60 cells, which express WT MLL but not MLL-AF9, using antibodies directed to the N-terminus of MLL (MLLN; upper panel), C-terminus of MLL (MLLC; lower panel) or IgG. Immunoprecipitated chromatin samples were analyzed by PCR using primers corresponding to promoter ZNF521P3.3 region. Note that no PCR product for ZNF521 promoter was obtained when anti-MLLC, which recognizes only the WT MLL but not MLL-AF9, was used for immunoprecipitation.
DISCUSSION
We present data showing that pediatric AML patients carrying MLL translocations have a significantly upregulation of ZNF521 expression independently of the fusion partner involved in the translocation with MLL. The overexpression of ZNF521 is a robust transcriptional feature of MLL-rearranged AML, consistent across independent adult and pediatric microarray datasets [19–21]. From these data, we started our study with the aim to decrypt the ZNF521 function as transcription factor in MLL-rearranged AML, and understand if it might deserve attention as potential therapeutic target.
A major hallmark of leukemia and a consequence of MLL fusion proteins expression is a block in hematopoietic differentiation [35]. On this way, our data show that the most relevant effect of ZNF521 depletion was to enhance myeloid differentiation of leukemia cells as evidenced by changes in cell morphology, immunophenotype and increase of a myeloid-specific gene expression in MLL-rearranged cell lines and primary cells. The requirement of ZNF521 in the maintenance of an undifferentiated status associated with MLL-rearranged AML was also supported by the fact that ZNF521 expression drastically decreased upon treatment with specific differentiation-induced agents, such as ATRA. The observed growth defect, cell cycle arrest and reduced colony formation upon ZNF521 depletion were secondary to cells entering into a differentiation program. Thus, MLL fusion proteins might promote leukemogenesis not only by HOXA9 and MEIS1 upregulation, but also by keeping the ZNF521 overexpressed, which in turn contributes to a block of differentiation or to the maintenance of an undifferentiated state of leukemia cells.
Consistent with this finding, others have reported that loss of ZNF521 enhanced erythroid differentiation and increased B-lineage maturation in cell lines and primary hematopoietic progenitor cell, respectively [15, 16]. Moreover, it is well established that Zfp521, the mouse counterpart of human ZNF521, in other cellular contexts including, embryonic stem cells (ESCs), neural cells, osteoblasts and chondrocytes mainly function to control cell differentiation of primitive or mature cells by modulating the activity of specific transcription factors [36–39]. Consistent with these findings, our GSEAs in THP-1 cells depleted for ZNF521 showed enrichment of hematopoietic stem cells (HSCs)- and ESC-associated downregulated genes and sets of genes associated with differentiation program [25, 26]. Based on these findings, we assumed that ZNF521 has not only a role in promoting self-renewal and maintenance of HSC but it also acts in MLL rearranged AML. Furthermore, our results support the direct activation of ZNF521 by MLL fusion proteins increasing the importance of this transcription factor in the transformation of the leukemia cells. In fact, we showed enrichment of set of genes related to MLL fusion-dependent transformation signatures as well as to HOXA9-mediated gene expression program [11, 30, 40]. Thus, the events documented after ZNF521 depletion, which in part resemble what has been previously observed in MLL-rearranged cells after loss of HOXA9, gave a further support that ZNF521 plays a critical role in MLL-fusion-mediated leukemia. Interestingly, the expression of either HOXA9, a canonical downstream target for MLL-rearranged leukemia [10, 41] or ZNF521 have been shown to be restricted in CD34+ progenitor cells [28, 30].
Nevertheless, in our gene-expression analysis, loss of ZNF521 does not affect HOXA9 expression, implying that both are MLL-dependent but might act in a non-mutually exclusive and additive manner.
Supporting the idea that ZNF521 is particularly required for MLL-mediated leukemia, our data of luciferase reporter and ChIP assays revealed that ZNF521 is a direct target of both MLL-AF9 and MLL-ENL fusion proteins. We defined a genomic region of 555 bp in 5′ ZNF521 promoter that is thought to be crucial for ZNF521 activation by MLL fusion proteins. This finding is consistent with prior observations that showed how the modulation of MLL-AF9 levels resulted in concordant changes in ZNF521 expression in different human in vitro models [42, 43]. Surprisingly, the inspection of ChIP-seq data from Bernt et al. [44] did not show peak in the vicinity of the Zfp521 gene in an MLL-AF9 mouse leukemia model. Of note, this is also observed for other well-known targets of MLL fusion proteins such as EVI1 and PLZF [45, 46] About ZNF521, this can be explained by the different approaches used, and the fact that in mouse BM Zfp521 is primarily expressed in the HSC fraction and significantly reduced in granulocyte-monocyte-progenitor cells (GMPs) (http://servers.binf.ku.dk/bloodspot/?gene=ZFP521&dataset=nl_mouse_data), in which the analysis has been done. Future ChIP-seq experiments on human transformed HSC will likely shed further light on ZNF521-MLL-AF9 target gene specificity.
In summary, this study unravels the anti-differentiation function of ZNF521 in MLL-rearranged cells and showed the mechanism by which ZNF521 participates in MLL-fusion mediated transformation. This data also indicate that ZNF521 is highly expressed in the majority of MLL-rearranged AML pediatric patients, and thus ZNF521 could be a potential molecular target for this subtype of aggressive leukemia.
MATERIALS AND METHODS
Patient samples and cell lines
All of the pediatric AML patient samples were obtained at the time of diagnosis from the University-Hospital of Padua and stratified according to the AIEOP AML 2002/01 protocol AML 2002/01 [47]. Patient characteristics are listed in Supplementary Table 1. Seven BM samples from healthy donors were obtained as control. All human myeloid cell lines (THP-1, NOMO-1, OCI-AML4, ML2, HL60, K562, Kasumi-1, NB4, U-937, SEM and REH) were obtained from DSMZ (Braunschweig, Germany) and 293T cells were obtained from ATCC (Manassas, VA, USA). All cell lines were maintained under standard conditions suggested by the manufacturer.
Quantitative real time PCR
Total RNA was extracted with Trizol reagent (Invitrogen) and reverse transcribed into cDNA using the Superscript III First-Strand Synthesis System (Life Technology). The mRNA levels of ZNF521, PU.1, CEBPa, HOXA9 and MEIS1 were measured by quantitative RT-PCR (qRT-PCR) with SYBR green on an AB 7900HT real time system (Applied Biosystem) using the comparative Ct method and the GAPDH gene expression as internal control [48]. The primer sequences for quantitative qRT-PCR are listed in Supplementary Table 2.
Lentiviral shRNA vector, transduction and FACS-sorting
For knockdown studies, two shRNAs against ZNF521 and a control scrambled shRNA (shScram) were used (Mission pLKO.1-puro-CMV-TurboGFP system, Cat Number TCRN0000229710 and TCRN0000229004, Sigma-Aldrich) (see Supplementary Table 2 for shRNA sequences). Lentiviral cell transduction was performed as described previously [49]. After culture in fresh medium, GFP-positive cells were sorted 96 hours after infection using a MoFlo XDP cell sorter (Beckman Coulter) and used for experiments. Alternatively, cells were gated for GFP expression and subjected to flow cytometry analyses. ZNF521 knockdown efficiency was measured by qRT-PCR and western blot analyses.
Plasmids constructs, transient transfection and luciferase assay
pMSCV-neo-Flag-MLL-AF9, MSCV-PML-RARA-IRES-GFP, MSCV-AML1-ETO-GFP, pCMVMLL-3xFlag and pCMVMLL-ENL-3xFlag have been previously described [42, 50–51]. Flag-tagged proteins were previously verified by Western blot with anti-Flag M2 antibody (Sigma), as well as the GFP-tagged proteins by expression of green fluorescence protein (GFP) in vitro. The procedure to generate the various constructs of the ZNF521 promoter into pGL4.28 plasmid (Promega) is described in Supplementary Materials and Methods. For luciferase assay, 293T cells were cotransfected with 0.5 μg of the reporter plasmid, 1 μg of expression plasmid or empty vector and 0.5 μg of Renilla luciferase reporter vector (Promega) as internal control for normalization of transfection efficiency, for a total of 2 μg of combined plasmids per well. The cells were then harvested at 48 hours after transfection using a Dual-Luciferase reporter assay system (Promega) and the Victor3 TM 1420 Multilabel Counter (PerkinElmer). Data are presented as the mean ratio for triplicate experiments.
Chromatin immunoprecipitation (ChIP) and PCR detection
ChIP assay was performed using the Imprint Chromatin Immunoprecipitation kit (Sigma), according to the manufacturer’s protocol with minor modifications. Briefly, 293T cells (3.5 × 106 cells) were transfected with 10 μg of Flag-MLL-AF9 or Flag-MLL-ENL expression plasmids. 48 hours post transfection, were cross-linked with 1% formaldehyde (Sigma) for 15 minute at room temperature. Subsequently, the lysed cells were isolated and sonicated on ice to shear DNA into fragments of 200 bp to 1 kb. Then, the chromatin complexes were incubated into pre-treated Stripwells (Sigma) with anti-Flag M2 monoclonal antibody (Sigma), or normal mouse IgG (Sigma) as indicated. The input DNA was isolated from sonicated lysates before immunoprecipitation as a positive control. Purified DNA was then resuspended in TE buffer (10 mM Tris-HCL and 1 mM EDTA, pH 8.0) for PCR. ChIP assay from 2 × 106 of MLL-AF9-expressing NOMO-1 cells or HL60 cells was performed as above reported using a N-terminal MLL monoclonal antibody (Santa Cruz Biotechnology) or a C-terminal MLL polyclonal antibody (Sigma) or a mouse IgG (Sigma) as indicated. Purified ChIP DNA was amplified by regular PCR. Primers amplifying the ZNF521 promoter region and the HOXA9 promoter region used for the ChIP PCR are listed in Supplementary Table 2.
Microarray analysis
Total RNA from sorted THP-1 cells transduced with shRNAs was isolated using Trizol as above reported and processed for microarray analysis using the Affymetrix GeneChip 3′IVT express Kit (Affimetrix) after RNA quality control using Agilent 2100 Bioanalyzer (Agilent). Gene expression profile was performed using a Human Genome U133 2.0 Plus chip (Affymetrix), as previously described [52]. The data were RMA-normalized using R software (http://www.r-project.org/) with BioConductor package (www.bioconductor.org). Shrinkage t test was used to identify differentially expressed genes between shScram and shRNA ZNF521 THP-1 cells selected with a local FDR < 0.05 (FDR). Hierarchical clustering analyses were performed using Euclidian distance and Ward’s methods. Gene set enrichment analysis (GSEA) was performed using GSEA version 2.0 software (Broad Institute; http://www.broadinstitute.org/gsea) with genes ranked by difference of class and statistical significance by 1000 gene set permutations. Gene set permutation was used to enable direct comparisons between shScram and shRNA ZNF521 results (< 7 replicates). Median of probes was used to collapse multiple probe sets to a single value per gene for each sample. Gene sets with a FDR < 0.05 were declared to be statistically significant. The microarray gene expression data have been submitted in NCBI’s Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) under accession GSE79110
Western blot and immunofluorescence staining
Western blot and immunofluorescence were performed using standard procedures. The antibody against ZNF521 was from Novus Biologicals (72009). Gamma-Tubulin (T6557), Actin (A5316), Flag M2 (F3165) antibodies were from Sigma Aldrich. For immunofluorescence analysis, antibody against CDKN1A/p21 (2947S) was from Cell Signaling and antibody against CDKN1B/p27 (610241) was from BD Biosciences. Detail method for the immunofluorescence is described in Supplementary Materials and Methods.
Cell function analyses
Cell viability, clonogenicity, cell cycle, apoptosis, expression of CD11b and CD14, morphological analysis and cell differentiation induction were performed as detailed in Supplementary Materials and Methods.
Studies with AML patient-derived xenograft cells
Primary MLL-AF9-expressing cells were obtained from BM samples of diagnosed AML pediatric patients stored in the BioBank of the laboratory of Pediatric Hematology of the University Hospital of Padua, (Italy) according to the guidelines of the local ethics committee. Initial AML xenografts were established by tail vein injection with 8 × 106 primary cells suspended in 300 μl of PBS in 6- to 8-week-old NSG mice, which were purchased from Charles River (Wilmington, MA, USA). All animal experiments were performed in accordance with institutional guidelines and established protocols [53]. Engraftment was monitored by weekly blood collections and flow cytometry analysis with antihuman CD45 (BD Biosciences). The engraftment rate was defined by the number of days required for the transplanted human CD45+ cells to reach at least 20% in the peripheral blood. Human leukemic cells from the spleens of engrafted mice were collected and cultivated in RPMI supplemented with 10% Human serum (Euroclone), antibiotics, and cytokines SCF, FLT-3L and TPO (40 ng/ml for each), IL-3 and IL-6 (20 ng/ml for each). (All cytokines were obtained from Inalco, Milan, Italy). For ex vivo experiments, two independent biological replicates were performed.
Data analysis
Data are presented as mean ± SD. Each experiment was performed at least 3 times, except where stated otherwise. The differences were examined using 2-tailed t test, Mann–Whitney U-test or kruskal-Wallis one-way analysis of variance followed Dunn’s test as appropriate (GraphPad Prism; GraphPad). Results were considered significant at P < 0.05.
Authors’ contributions
Contribution: G.G., G.M., and S.A. conceived and designed the experiments; M.Pi., S.M., and S.I. performed the animal experiments; G.G., G.M., and S.A. analyzed the data; L.P., P.B., G.V., C.T., C.F., S.B., and M.Pig contributed reagents, materials, samples and analysis tools; G.G., M.Pa., and S.A wrote the paper; G.B. supervised the study.
ACKNOWLEDGMENTS
This work was supported by research funding from CARIPARO, Istituto di Ricerca Pediatrica Fondazione Città della Speranza, University of Padua, and Associazione Italiana Ricerca sul Cancro (AIRC) grant IG14295 (S.I.). We thank to Jay L. Hess, Nabeel R. Yaseen, James J-D. Hsieh for providing plasmids, Samuela Francescato and Elena Bortoloso for help in cell morphology, Pamela Scarparo, Barbara Michielotto and Barbara Buldini for flow cytometry assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006; 368:1894–1907.
2. McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007; 1:338–345.
3. Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007; 1:324–337.
4. Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, Pombo de Oliveira M, Renneville A, Villarese P, Macintyre E, Cave H, Clappier E, Mass-Malo K, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013; 27:2165–2176.
5. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007; 7:823–833.
6. Balgobind BV, Zwaan CM, Pieters R, Van den Heuvel-Eibrink MM. The heterogeneity of pediatric MLL-rearranged acute myeloid leukemia. Leukemia. 2011; 25:1239–1248.
7. Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010; 115:453–474.
8. Hess JL, Bittner CB, Zeisig DT, Bach C, Fuchs U, Borkhardt A, Frampton J, Slany RK. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006; 108:297–304.
9. Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002; 30:41–47.
10. Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003; 17:2298–2307.
11. Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, Zwaan CM, Kung AL, Armstrong SA. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 2009; 113:2375–2385.
12. Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, Hess JL, Slany RK. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol. 2004; 24:617–628.
13. Lin AC, Roche AE, Wilk J, Svensson EC. The N termini of Friend of GATA (FOG) proteins define a novel transcriptional repression motif and a superfamily of transcriptional repressors. J Biol Chem. 2004; 279:55017–55023.
14. Bond HM, Mesuraca M, Carbone E, Bonelli P, Agosti V, Amodio N, De Rosa G, Di Nicola M, Gianni AM, Moore MA, Hata A, Grieco M, Morrone G, et al. Early hematopoietic zinc finger protein (EHZF), the human homolog to mouse Evi3, is highly expressed in primitive human hematopoietic cells. Blood. 2004; 103:2062–2070.
15. Matsubara E, Sakai I, Yamanouchi J, Fujiwara H, Yakushijin Y, Hato T, Shigemoto K, Yasukawa M. The role of zinc finger protein 521/early hematopoietic zinc finger protein in erythroid cell differentiation. J Biol Chem. 2009; 284:3480–3487.
16. Mega T, Lupia M, Amodio N, Horton SJ, Mesuraca M, Pelaggi D, Agosti V, Grieco M, Chiarella E, Spina R, Moore MA, Schuringa JJ, Bond HM, et al. Zinc finger protein 521 antagonizes early B-cell factor 1 and modulates the B-lymphoid differentiation of primary hematopoietic progenitors. Cell Cycle. 2011; 10:2129–2139.
17. Spina R, Filocamo G, Iaccino E, Scicchitano S, Lupia M, Chiarella E, Mega T, Bernaudo F, Pelaggi D, Mesuraca M, Pazzaglia S, Semenkow S, Bar EE, et al. Critical role of zinc finger protein 521 in the control of growth, clonogenicity and tumorigenic potential of medulloblastoma cells. Oncotarget. 2013; 4:1280–1292. doi: 10.18632/oncotarget.1176.
18. Hiratsuka T, Takei Y, Ohmori R, Imai Y, Ozeki M, Tamaki K, Haga H, Nakamura T, Tsuruyama T. ZFP521 contributes to pre-B-cell lymphomagenesis through modulation of the pre-B-cell receptor signaling pathway. Oncogene. 2015.
19. Kohlmann A, Schoch C, Dugas M, Schnittger S, Hiddemann W, Kern W, Haferlach T. New insights into MLL gene rearranged acute leukemias using gene expression profiling: shared pathways, lineage commitment, and partner genes. Leukemia. 2005; 19:953–964.
20. Jo A, Tsukimoto I, Ishii E, Asou N, Mitani S, Shimada A, Igarashi T, Hayashi Y, Ichikawa H. Age-associated difference in gene expression of paediatric acute myelomonocytic lineage leukaemia (FAB M4 and M5 subtypes) and its correlation with prognosis. Br J Haematol. 2009; 144:917–929.
21. Pigazzi M, Masetti R, Bresolin S, Beghin A, Di Meglio A, Gelain S, Trentin L, Baron E, Giordan M, Zangrando A, Buldini B, Leszl A, Putti MC, et al. MLL partner genes drive distinct gene expression profiles and genomic alterations in pediatric acute myeloid leukemia: an AIEOP study. Leukemia. 2011; 25:560–563.
22. Roy A, Banerjee S. P27 and Leukemia: Cell Cycle and Beyond. J Cell Physiol. 2015; 230:504–509.
23. Niitsu N, Hayashi Y, Sugita K, Honma Y. Sensitization by 5-aza-2'-deoxycytidine of leukaemia cells with MLL abnormalities to induction of differentiation by all-trans retinoic acid and 1alpha,25-dihydroxyvitamin D3. Br J Haematol. 2001; 112:315–326.
24. Gupta K, Chakrabarti A, Rana S, Ramdeo R, Roth BL, Agarwal ML, Tse W, Agarwal MK, Wald DN. Securinine, a myeloid differentiation agent with therapeutic potential for AML. PLoS One. 2011; 6:e21203.
25. Brown AL, Wilkinson CR, Waterman SR, Kok CH, Salerno DG, Diakiw SM, Reynolds B, Scott HS, Tsykin A, Glonek GF, Goodall GJ, Solomon PJ, Gonda TJ, et al. Genetic regulators of myelopoiesis and leukemic signaling identified by gene profiling and linear modeling. J Leukoc Biol. 2006; 80:433–447.
26. Jaatinen T, Hemmoranta H, Hautaniemi S, Niemi J, Nicorici D, Laine J, Yli-Harja O, Partanen J. Global gene expression profile of human cord blood-derived CD133+ cells. Stem Cells. 2006; 24:631–641.
27. Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007; 21:2762–2774.
28. Bond HM, Mesuraca M, Amodio N, Mega T, Agosti V, Fanello D, Pelaggi D, Bullinger L, Grieco M, Moore MA, Venuta S, Morrone G. Early hematopoietic zinc finger protein-zinc finger protein 521: a candidate regulator of diverse immature cells. Int J Biochem Cell Biol. 2008; 40:848–854.
29. Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012; 122:3407–3415.
30. Dorsam ST, Ferrell CM, Dorsam GP, Derynck MK, Vijapurkar U, Khodabakhsh D, Pau B, Bernstein H, Haqq CM, Largman C, Lawrence HJ. The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood. 2004; 103:1676–1684.
31. Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, Hong GM, Elkahloun AG, Arnovitz S, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2013; 110:11994–11999.
32. Placke T, Faber K, Nonami A, Putwain SL, Salih HR, Heidel FH, Kramer A, Root DE, Barbie DA, Krivtsov AV, Armstrong SA, Hahn WC, Huntly BJ, et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood. 2014; 124:13–23.
33. Park SM, Gonen M, Vu L, Minuesa G, Tivnan P, Barlowe TS, Taggart J, Lu Y, Deering RP, Hacohen N, Figueroa ME, Paietta E, Fernandez HF, et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J Clin Invest. 2015; 125:1286–1298.
34. Zheng R, Wang X, Studzinski GP. 1,25-Dihydroxyvitamin D3 induces monocytic differentiation of human myeloid leukemia cells by regulating C/EBPbeta expression through MEF2C. J Steroid Biochem Mol Biol. 2015; 148:132–137.
35. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005; 5:311–321.
36. Kamiya D, Banno S, Sasai N, Ohgushi M, Inomata H, Watanabe K, Kawada M, Yakura R, Kiyonari H, Nakao K, Jakt LM, Nishikawa S, Sasai Y. Intrinsic transition of embryonic stem-cell differentiation into neural progenitors. Nature. 2011; 470:503–509.
37. Hesse E, Saito H, Kiviranta R, Correa D, Yamana K, Neff L, Toben D, Duda G, Atfi A, Geoffroy V, Horne WC, Baron R. Zfp521 controls bone mass by HDAC3-dependent attenuation of Runx2 activity. J Cell Biol. 2010; 191:1271–1283.
38. Correa D, Hesse E, Seriwatanachai D, Kiviranta R, Saito H, Yamana K, Neff L, Atfi A, Coillard L, Sitara D, Maeda Y, Warming S, Jenkins NA, et al. Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes. Dev Cell. 2010; 19:533–546.
39. Shen S, Pu J, Lang B, McCaig CD. A zinc finger protein Zfp521 directs neural differentiation and beyond. Stem Cell Res Ther. 2011; 2:20.
40. Mullighan CG, Kennedy A, Zhou X, Radtke I, Phillips LA, Shurtleff SA, Downing JR. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia. 2007; 21:2000–2009.
41. Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007; 26:6766–6776.
42. Abdul-Nabi AM, Yassin ER, Varghese N, Deshmukh H, Yaseen NR. In vitro transformation of primary human CD34+ cells by AML fusion oncogenes: early gene expression profiling reveals possible drug target in AML. PLoS One. 2010; 5:e12464.
43. Fleischmann KK, Pagel P, Schmid I, Roscher AA. RNAi-mediated silencing of MLL-AF9 reveals leukemia-associated downstream targets and processes. Mol Cancer. 2014; 13:27–4598–13–27.
44. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011; 20:66–78.
45. Arai S, Yoshimi A, Shimabe M, Ichikawa M, Nakagawa M, Imai Y, Goyama S, Kurokawa M. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011; 117:6304–6314.
46. Ono R, Masuya M, Nakajima H, Enomoto Y, Miyata E, Nakamura A, Ishii S, Suzuki K, Shibata-Minoshima F, Katayama N, Kitamura T, Nosaka T. Plzf drives MLL-fusion-mediated leukemogenesis specifically in long-term hematopoietic stem cells. Blood. 2013; 122:1271–1283.
47. Pession A, Masetti R, Rizzari C, Putti MC, Casale F, Fagioli F, Luciani M, Lo Nigro L, Menna G, Micalizzi C, Santoro N, Testi AM, Zecca M, et al. Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood. 2013; 122:170–178.
48. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3: 1101–1108.
49. Indraccolo S, Habeler W, Tisato V, Stievano L, Piovan E, Tosello V, Esposito G, Wagner R, Uberla K, Chieco-Bianchi L, Amadori A. Gene transfer in ovarian cancer cells: a comparison between retroviral and lentiviral vectors. Cancer Res. 2002; 62:6099–6107.
50. Liu H, Cheng EH, Hsieh JJ. Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution:a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 2007; 21:2385–2398.
51. Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, Hess JL. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell. 2011; 20:563–575.
52. Bresolin S, Zecca M, Flotho C, Trentin L, Zangrando A, Sainati L, Stary J, de Moerloose B, Hasle H, Niemeyer CM, Te Kronnie G, Locatelli F, Basso G. Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol. 2010; 28:1919–1927.
53. Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, Gurney A, Hoey T, Seganfreddo E, Basso G, Valtorta S, Moresco RM, Amadori A, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia. 2014; 28:278–288.