
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous group of hematological malignancies with variable responses to therapy. Age and karyotype have been recognized as the most prominent prognostic factors in AML patients. Over the last decade, it has been reported that mutations of some genes of interest may have prognostic significance, especially in patients with cytogenetically normal (CN) AML. Prognostic impact within CN-AML has been well established for FLT3 internal tandem duplication (FLT3–ITD), NPM1 and CEBPA mutations. Mutations of these three genes allowed a refined prognostic classification of CN-AML and improved risk stratification in this subset of AML patients [1, 2]. With recent progress in genomic technologies, a large number of recurrent somatic mutations have been discovered in AML, including mutations in ASXL1 [3], TET2 [4], IDH1/2 [5], DNMT3A [6, 7], PFH6 [8], and BCOR [9], thus providing new insights into the mechanisms of leukemogenesis and further evidence of the genetic complexity of AML. Retrospective studies have suggested that these novel mutations may have prognostic significance in AML, but these findings need to be confirmed and validated prospectively in clinical trial cohorts [10–12].
For years, standard induction chemotherapy in AML patients has relied on the combination of cytarabine and anthracycline, the so-called 7 + 3 regimen. We recently published the results of the ALFA-0701 Phase 3 trial, showing that the addition of fractionated doses of gemtuzumab ozogamicin (GO) during induction and consolidation may significantly improve outcome of AML patients aged 50–70 years [13]. GO is a humanized immunoconjugate targeting the CD33 antigen, a myeloid antigen expressed on the majority of AML leukemic cells. Similar positive results have been reported in two Medical Research Council (MRC) trials [14, 15], and, as in the ALFA trial, it appeared that patients with favorable or intermediate cytogenetics, including those with CN-AML, may benefit from GO, while not those with adverse karyotype. In this context, there is a growing interest in identifying molecular determinants of response to this targeted agent.
Refining conventional cytogenetics (CC) represents another promising approach to improve prognostic classification of AML, as CC detects an abnormal karyotype in only half of AML patients. Single-nucleotide polymorphism array (SNP-A)–based karyotyping has revealed previously unrecognized acquired copy number abnormalities (CNA) in AML genome [16, 17]. In addition to a high level of resolution, SNP-A allows the detection of copy-neutral loss of heterozygosity (CN-LOH), also referred to as uniparental disomy (UPD) [17]. Moreover, some studies reported that novel lesions detected by SNP-A-based karyotyping may complement CC and improve AML outcome prediction [18–20].
In the present study, we thus performed SNP-A-based karyotyping and extensive mutational analysis in the 278 AML patients enrolled on the ALFA-0701 trial. Our objectives were first to evaluate the prognostic impact of these genetic alterations on clinical outcome, particularly within the subset of CN-AML, and second to analyze potential interactions between these genetic alterations and GO treatment.
RESULTS
Patient characteristics at diagnosis
Baseline patient and AML characteristics are provided in Tables S1 and S2 in the Supplementary Data, for all patients and CN-AML patients, respectively. Median age was 62 years in the whole cohort and in CN-AML. CC results were available for 254 patients, of which 146 had a CN-AML. SNP-A data were available for 248 patients. Among these 248 patients, 228 were also analyzed by CC, of which 132 had a CN-AML.
Conventional cytogenetics and SNP-A karyotyping
Among the 248 patients with available SNP-A data, we found 450 genomic aberrations in 135 patients. CNA (245 losses and 117 gains) concerned all chromosomes and UPD (n = 88) were located over all chromosomes except chromosomes 7, 14 and 18. Details of SNP-A lesions are provided in Table S3 in the Supplementary Data. Types and genomic distribution of SNP-A abnormalities were highly distinct between normal and abnormal karyotypes as defined by CC. Indeed, in AML with abnormal karyotype (n = 96), SNP-A lesions were more often CNA (222 losses and 102 gains) than UPD (n = 32). The median size was 33 Mb (range, 0.16–243) for deletions, 87 Mb (range, 0.4–249) for gains, and 24 Mb (range, 3–141) for UPDs. Most deletions were identified at chromosomes 5 (n = 28) and 7 (n = 27), gains at chromosome 8 (n = 23) and UPD at the short arm of chromosome 17 (n = 5). Nineteen patients with abnormal conventional karyotype had no detectable SNP-A lesions, including 5 with balanced chromosomal translocations or inversions, 13 with subclonal chromosomal abnormalities, and 1 with complex karyotype and several chromosome markers that may result from complex balanced translocations. In CN-AML, a total of 72 genomic aberrations were detected by SNP-A analysis in 50/132 (38%) of the patients, with a mean of 1.44 lesions per patient (range, 1–5). We detected 13 deletions (median size, 2 Mb; range, 0.15–64), 11 gains (median size, 0.5 Mb; range, 0.12–191), and 48 regions of UPDs (median size, 38 Mb; range, 2–136). Most UPDs were identified at chromosomes 13 and 1p in 7 and 6 patients, respectively (Table S3 in the Supplementary Data).
Overall, in the 228 patients analyzed by both techniques, the combination of CC and SNP-A karyotyping leads to a higher proportion of abnormal karyotypes (64%) compared with CC alone (42%) or SNP-A alone (56%) (Figure S1 in the Supplementary Data).
Molecular findings
The incidence of gene mutations and EVI1 overexpression in the whole cohort and in the subset of CN-AML are respectively indicated in Table 2, and Tables S1 and S2 in the Supplementary Data. At least one molecular abnormality was identified in 89% of the patients with CN-AML and 60% showed mutations in more than one of the studied genes (not shown). The description of all gene mutations identified by Sanger sequencing is provided in Table S4 in the Supplementary Data.
Co-occurrence of genetic alterations in CN-AML
Integrated SNP-A karyotyping and molecular analysis in CN-AML allowed us to identify positive and negative associations between those different genetic alterations. We found that FLT3–ITD and DNMT3A mutations were significantly associated with NPM1 mutations (P < 0.001 for both comparisons). Overall, NPM1 mutations co-occurred with at least 1 other mutation in 90% of cases. Frequent associations between FLT3–ITD and DNMT3A mutations (P = 0.001), RUNX1 and ASXL1 mutations (P = 0.033), RUNX1 and MLL partial tandem duplication (MLL-PTD) (P = 0.049) were also observed. In contrast, RUNX1 (P = 0.009) and CEBPA (P = 0.033) mutations rarely co-existed with NPM1 mutations. NPM1 and ASXL1 mutations were mutually exclusive (P = 0.003) (Figure 1A and 1B). EVI1 overexpression was not found concomitantly with any gene mutation (P < 0.001) (Figure 1B).
When correlating mutational profile with SNP-A data, we found a higher incidence of RUNX1 mutations (P = 0.01) and a lower incidence of IDH2R140 mutations (P = 0.03) in patients with SNP-A lesions compared to patients without SNP-A lesions (Table S5 in the Supplementary Data). We also observed that mutations affecting CEBPA, RUNX1, DNMT3A, TET2 or WT1 were found in 8/14 patients with UPDs (n = 12) or mono-allelic losses (n = 2) encompassing the corresponding gene locus. In addition, all 7 patients with UPD(13q) harbored a FLT3–ITD (Figure 1).
Response to induction therapy
Overall, complete remission (CR) or CRp were achieved in 217 patients (202 CR and 15 CRp), including 104 patients in the control arm and 113 patients in the GO arm. In the whole cohort, the presence of unfavorable karyotype (P < 0.001), SNP-A lesion(s) (P = 0.014) were associated with a lower CR/CRp rate, in contrast to FLT3–ITD and NPM1 mutations, which conferred a higher CR/CRp rate (P = 0.035 and P = 0.022, respectively). However, neither SNP-A lesion(s) nor gene mutations had an impact on response to induction therapy in the subset of CN-AML patients (Table S6 in the Supplementary Data). In multivariate analysis of the whole cohort, unfavorable karyotype remained the only factor significantly associated with a lower probability of achieving CR/CRp (P < 0.001). Randomization in the GO arm was associated with a trend (P = 0.053) towards a higher probability of CR/CRp achievement (Table S7 in the Supplementary Data).
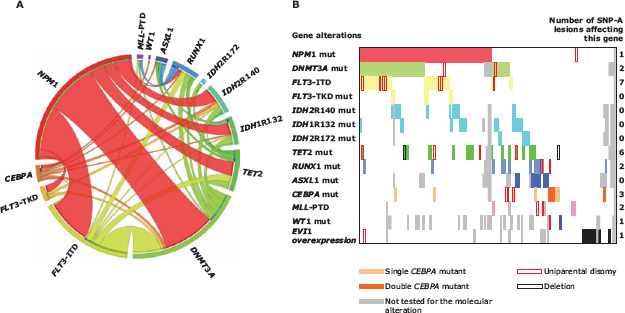
Figure 1: (A) Circos plot diagram illustrating the pairwise co-occurrence of gene mutations in cytogenetically normal acute myeloid leukemia. (B) Bar coding representing the co-occurrence of molecular alterations and SNP-A lesions in cytogenetically normal acute myeloid leukemia. Each patient is represented by a virtual column. Colored cells indicate the presence of a mutation in the gene(s) described in that row on the left. Abbreviations: SNP-A, single-nucleotide polymorphism array; mut, mutation; ITD, internal tandem duplication; TKD, tyrosine kinase domain; PTD, partial tandem duplication.
Prognostic analyses for relapse risk and survival
The median duration of follow-up from time to randomization was 25 months (interquartile range, 16–33). In the whole cohort, the presence of unfavorable karyotype and SNP-A lesions were predictive of a higher cumulative incidence of relapse (CIR) (P < 0.001 and P = 0.017, respectively). None of the molecular markers studied significantly influenced CIR. In the subset of CN-AML, MLL-PTD (P = 0.028) and DNMT3A mutations (P = 0.004) were both associated with an increased risk of relapse (Table 1).
Karyotype had a strong influence on overall survival (OS), as expected (Table 2). Interestingly, we found that OS was also significantly reduced in patients with SNP-A lesion(s), either in the whole cohort (Figure 2A) or in the subset of CN-AML (Figure 2B). In the whole cohort, NPM1 mutations were associated with a longer OS (P = 0.022), whereas EVI1 overexpression was associated with a shorter OS (P = 0.010). In the CN-AML subset, DNMT3A mutated patients had markedly reduced OS compared to DNMT3A wild-type patients (Figure 3C). Other molecular abnormalities studied did not significantly affect OS of CN-AML patients (Table 2).
Table 1: Incidence of genetic abnormalities and univariate prognostic analysis for cumulative incidence of relapse
All patients |
CN-AML patients |
|||||||
Covariate |
n (%) |
HR |
IC95% |
P-value |
n (%) |
HR |
IC95% |
P-value |
Karyotype |
||||||||
Favorable |
8/217 (3.7) |
0.74 |
0.27–2.02 |
0.56 |
NA |
NA |
NA |
NA |
Intermediate |
161/217 (74.2) |
0.76 |
0.49–1.16 |
0.20 |
NA |
NA |
NA |
NA |
Normal karyotype |
131/217 (60.4) |
0.79 |
0.53–1.17 |
0.24 |
NA |
NA |
NA |
NA |
Unfavorable |
30/217 (13.8) |
2.46 |
1.50–4.03 |
<0.001 |
NA |
NA |
NA |
NA |
Not available |
18/217 (8.3) |
0.58 |
0.25–1.32 |
0.20 |
NA |
NA |
NA |
NA |
SNP-array karyotyping |
||||||||
Presence of SNP-A lesion(s) |
97/193 (50.3) |
1.70 |
1.10–2.63 |
0.017 |
43/118 (36.4) |
1.46 |
0.82–2.61 |
0.20 |
Molecular findings |
||||||||
MLL-PTD |
4/210 (1.9) |
2.40 |
0.58–9.85 |
0.23 |
3/127 (2.4) |
5.11 |
1.20–21.86 |
0.028 |
NPM1 mutation |
81/216 (37.5) |
1.00 |
0.67–1.51 |
0.99 |
68/130 (52.3) |
1.37 |
0.80–2.36 |
0.25 |
FLT3–TKD mutation |
10/216 (4.6) |
0.80 |
0.32–1.97 |
0.62 |
4/130 (3.1) |
0.97 |
0.24–4.00 |
0.97 |
FLT3–ITD |
44/216 (20.4) |
1.37 |
0.84–2.25 |
0.21 |
33/130 (25.4) |
1.54 |
0.85–2.79 |
0.15 |
CEBPA sm + dm |
15/211 (7.1) |
0.57 |
0.23–1.41 |
0.23 |
11/128 (8.6) |
0.29 |
0.07–1.19 |
0.086 |
CEBPA dm |
9/211 (4.3) |
0.55 |
0.18–1.75 |
0.31 |
5/128 (3.9) |
NC |
NC |
NC |
EVI1 overexpression |
15/184 (8.1) |
1.32 |
0.64–2.74 |
0.45 |
10/123 (8.1) |
1.37 |
0.49–3.81 |
0.55 |
RUNX1 mutation |
20/208 (9.6) |
1.50 |
0.82–2.76 |
0.19 |
11/125 (8.8) |
1.70 |
0.72–4.02 |
0.22 |
WT1 mutation |
7/180 (3.9) |
1.10 |
0.35–3.51 |
0.87 |
2/106 (1.9) |
NC |
NC |
NC |
ASXL1 mutation |
17/191 (8.9) |
0.83 |
0.40–1.73 |
0.63 |
7/113 (6.2) |
0.58 |
0.14–2.37 |
0.44 |
DNMT3A mutation |
NA |
NA |
NA |
NA |
42/124 (33.9) |
2.24 |
1.29–3.87 |
0.004 |
TET2 mutation |
NA |
NA |
NA |
NA |
15/125 (12.0) |
0.80 |
0.34–1.87 |
0.60 |
IDH1R132 mutation |
NA |
NA |
NA |
NA |
13/123 (10.6) |
1.64 |
0.80–3.36 |
0.18 |
IDH2R140 mutation |
NA |
NA |
NA |
NA |
12/123 (9.8) |
0.47 |
0.17–1.31 |
0.15 |
IDH2R172 mutation |
NA |
NA |
NA |
NA |
5/123 (4.1) |
1.00 |
0.31–3.22 |
0.99 |
Abbreviations: CN-AML: cytogenetically normal acute myeloid leukemia; HR, hazard ratio; CI, confidence interval; SNP-A, single-nucleotide polymorphism array; NA, not applicable; NC, non-convergent; PTD, partial tandem duplication; ITD, internal tandem duplication; sm, single-mutation; dm, double-mutation.
Table 2: Incidence of genetic abnormalities and univariate prognostic analysis for overall survival
All patients |
CN-AML patients |
|||||||
Covariate |
n (%) |
HR |
IC95% |
P-value |
n (%) |
HR |
IC95% |
P-value |
Karyotype |
||||||||
Favorable |
9/278 (3.2) |
0.53 |
0.17–1.67 |
0.28 |
NA |
NA |
NA |
NA |
Intermediate |
186/278 (66.9) |
0.44 |
0.31–0.63 |
<0.001 |
NA |
NA |
NA |
NA |
Normal karyotype |
146/278 (52.5) |
0.47 |
0.32–0.67 |
<0.001 |
NA |
NA |
NA |
NA |
Unfavorable |
59/278 (21.2) |
3.69 |
2.51–5.42 |
<0.001 |
NA |
NA |
NA |
NA |
Not available |
24/278 (8.6) |
0.76 |
0.37–1.57 |
0.47 |
NA |
NA |
NA |
NA |
SNP-array karyotyping |
||||||||
Presence of SNP-A lesion(s) |
135/248 (54.4) |
2.62 |
1.71–4.03 |
<0.001 |
50/132 (37.9) |
2.47 |
1.34–4.56 |
0.004 |
Molecular findings |
||||||||
MLL-PTD |
6/268 (2.2) |
1.35 |
0.43–4.27 |
0.61 |
4/142 (2.8) |
1.88 |
0.45–7.78 |
0.38 |
NPM1 mutation |
94/276 (34.1) |
0.62 |
0.41–0.93 |
0.022 |
75/145 (51.7) |
0.89 |
0.50–1.59 |
0.70 |
FLT3–TKD mutation |
14/276 (5.1) |
0.73 |
0.32–1.67 |
0.46 |
5/145 (3.4) |
NC |
NC |
NC |
FLT3–ITD |
49/276 (17.8) |
0.94 |
0.57–1.55 |
0.80 |
36/145 (24.8) |
1.56 |
0.83–2.92 |
0.16 |
CEBPA sm + dm |
18/271 (6.6) |
0.60 |
0.26–1.37 |
0.23 |
12/143 (8.7) |
0.58 |
0.18–1.87 |
0.37 |
CEBPA dm |
10/271 (3.7) |
0.35 |
0.09–1.40 |
0.14 |
5/143 (3.6) |
NC |
NC |
NC |
EVI1 overexpression |
25/251 (10.0) |
2.10 |
1.19–3.70 |
0.010 |
10/135 (7.4) |
1.26 |
0.38–4.12 |
0.71 |
RUNX1 mutation |
26/266 (9.8) |
1.19 |
0.67–2.12 |
0.55 |
13/139 (9.4) |
1.26 |
0.50–3.22 |
0.62 |
WT1 mutation |
7/230 (3.0) |
0.64 |
0.16–2.61 |
0.54 |
2/119 (1.7) |
NC |
NC |
NC |
ASXL1 mutation |
21/247 (8.5) |
0.87 |
0.44–1.73 |
0.70 |
9/127 (7.1) |
1.87 |
0.66–5.26 |
0.24 |
DNMT3A mutation |
NA |
NA |
NA |
NA |
47/138 (34.1) |
2.29 |
1.27–4.11 |
0.006 |
TET2 mutation |
NA |
NA |
NA |
NA |
19/139 (13.7) |
1.55 |
0.72–3.33 |
0.27 |
IDH1R132 mutation |
NA |
NA |
NA |
NA |
15/137 (10.9) |
1.18 |
0.53–2.66 |
0.69 |
IDH2R140 mutation |
NA |
NA |
NA |
NA |
12/138 (8.7) |
0.29 |
0.07–1.19 |
0.085 |
IDH2R172 mutation |
NA |
NA |
NA |
NA |
6/137 (4.4) |
0.80 |
0.19–3.31 |
0.76 |
Abbreviations: CN-AML: cytogenetically normal acute myeloid leukemia; HR, hazard ratio; CI, confidence interval; SNP-A, single-nucleotide polymorphism array; NA, not applicable; NC, noCN-convergent; PTD, partial tandem duplication; ITD, internal tandem duplication; sm, single-mutation; dm, double-mutation.
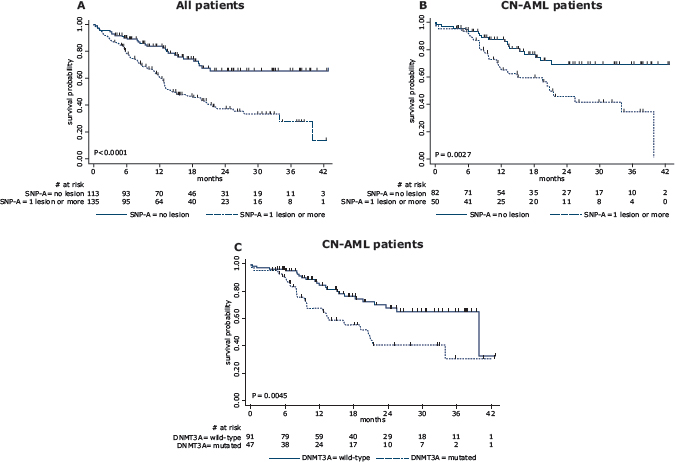
Figure 2: Kaplan-Meier estimates of overall survival according to SNP-array profile and gene mutational status. (A) in the whole patient cohort analyzed by SNP-A, 2–year OS was estimated at 68% (95%CI, 54–75) in those without SNP-A lesion versus 37% (95%CI, 27.5–47) in those with SNP-A lesion (P < 0.0001 by the log-rank test); (B) in CN-AML patients analyzed by SNP-A, 2–year OS was estimated at 70% (95%CI, 56–80) in those without SNP-A lesion versus 46% (95%CI, 29–61) in those with SNP-A lesion (P = 0.0027 by the log-rank test); (C) in CN-AML patients tested for DNMT3A mutation, 2–year OS was estimated at 68% (95%CI, 55–78) in those without mutation versus 41% (95%CI, 24–57) in those with mutation (P = 0.0045 by the log-rank test).
We then performed a multivariate analysis on CIR and OS considering the covariates previously selected as associated with clinical outcome at the 10% level, together with the treatment arm. The complete list of covariates that entered the multivariate models for CIR and OS is provided in Tables 3 and 4, respectively. The presence of unfavorable karyotype (P = 0.013) and randomization in the control arm (P = 0.007) were retained as significantly associated with higher CIR in the whole cohort, while, only the presence of MLL-PTD (P = 0.014) and DNMT3A mutations (P = 0.010) independently predicted higher CIR in CN-AML patients (Table 3). The presence of unfavorable karyotype (P < 0.001) and SNP-A lesion(s) (P = 0.001) independently predicted shorter OS in the whole cohort, while the presence of SNP-A lesion(s) (P = 0.006), DNMT3A mutations (P = 0.042) and randomization in the control arm (P = 0.043) independently predicted shorter OS in CN-AML patients (Table 4).
Interactions between treatment effect and baseline patient characteristics
Interactions between the treatment effect on CR/CRp achievement, CIR and OS, respectively, and the following baseline patient characteristics were studied: cytogenetics, SNP-A karyotyping, FLT3–ITD, and NPM1 mutations in the whole cohort and in the CN-AML subset, additionally with DNMT3A mutations in the CN-AML subset. When testing these potential interactions by the Gail and Simon statistics, no evidence of any statistically significant interaction was found regarding CR/CRp achievement (data not shown), CIR (Figure 3), and the variables SNP-A lesions, DNMT3A mutations, and NPM1 mutations, irrespective of the endpoint.
Interestingly, we observed that the survival benefit associated with GO addition was not apparent in patients with unfavorable cytogenetics, conversely to those with favorable/intermediate cytogenetics (HR, 1.44 versus 0.62; test for interaction, P = 0.04; Figure 3). More specifically, CN-AML patients benefited preferentially more from GO treatment as compared to AML patients with abnormal CC (HR, 0.52 versus 1.14; test for interaction, P = 0.04; Figure 3).
Moreover, the OS benefit associated with GO treatment appeared more pronounced in FLT3–ITD positive than in negative patients, in the whole cohort as well as in CN-AML (Figure 4), although the Gail and Simon test did not reach statistical significance (Figure 3). NPM1 mutated/FLT3–ITD negative genotype is considered in the ELN prognostic classification as a strong prognostic factor for favorable outcome in CN-AML [1]. In the ALFA-0701 trial, the NPM1 mutated/FLT3–ITD negative genotype showed no significant influence on outcome in CN-AML (data not shown). However, when comparing the outcome of CN-AML patients with NPM1 mutated/FLT3–ITD negative genotype to those with other genotypes, and according to the randomization arm, we observed that the NPM1 mutated/FLT3–ITD negative genotype was associated with favorable outcome in the control arm (P = 0.04), but not in the GO arm (P = 0.89) (Figure S4).
Table 3: Multivariate analysis for cumulative incidence of relapse.
All patients |
CN-AML patients |
|||||
Final model |
HR |
95% CI |
P-value |
HR |
95% CI |
P-value |
Presence of unfavorable karyotype |
2.06 |
1.17–3.64 |
0.013 |
NA |
NA |
NA |
Presence of SNP-A lesion(s) |
1.55 |
0.97–2.46 |
0.067 |
NA |
NA |
NA |
Presence of MLL-PTD |
NA |
NA |
NA |
6.30 |
1.44–27.51 |
0.014 |
Presence of DNMT3A mutation |
NA |
NA |
NA |
2.12 |
1.20–3.75 |
0.010 |
Randomization in the GO arm |
0.55 |
0.35–0.85 |
0.007 |
0.70 |
0.40–1.24 |
0.22 |
Abbreviations: HR, hazard ratio; CI, confidence interval; SNP-A, single-nucleotide polymorphism array; CN-AML: cytogenetically normal acute myeloid leukemia; NA, not applicable.
Table 4: Multivariate analysis for overall survival
All patients |
CN-AML patients |
|||||
Final model |
HR |
95% CI |
P-value |
HR |
95% CI |
P-value |
Presence of unfavorable karyotype |
2.54 |
1.56–4.12 |
<0.001 |
NA |
NA |
NA |
Presence of SNP-A lesion(s) |
2.25 |
1.39–3.65 |
0.001 |
2.38 |
1.28–4.43 |
0.006 |
Presence of NPM1 mutation |
1.12 |
0.67–1.87 |
0.68 |
NA |
NA |
NA |
Presence of EVI1 overexpression |
1.63 |
0.87–3.06 |
0.12 |
NA |
NA |
NA |
Presence of DNMT3A mutation |
NA |
NA |
NA |
1.91 |
1.02–3.56 |
0.042 |
Presence of IDH2R140 mutation |
NA |
NA |
NA |
0.38 |
0.09–1.63 |
0.19 |
Randomization in the GO arm |
0.75 |
0.49–1.13 |
0.17 |
0.51 |
0.27–0.98 |
0.043 |
Abbreviations: HR, hazard ratio; CI, confidence interval; SNP–A, single–nucleotide polymorphism array; CN-AML: cytogenetically normal acute myeloid leukemia; NA, not applicable.
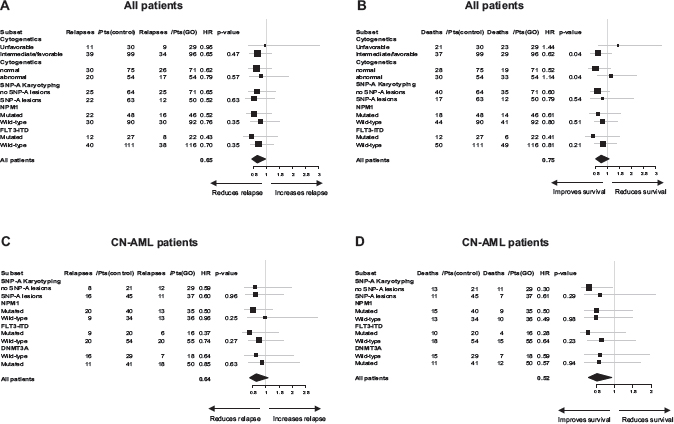
Figure 3: Forest plots of patient subsets and pooled data for (A) cumulative incidence of relapse and (B) overall survival in all patients, and (C) cumulative incidence of relapse and (D) overall survival in patients with cytogenetically normal acute myeloid leukemia. P-values obtained with the Gail and Simon interaction tests are also indicated.
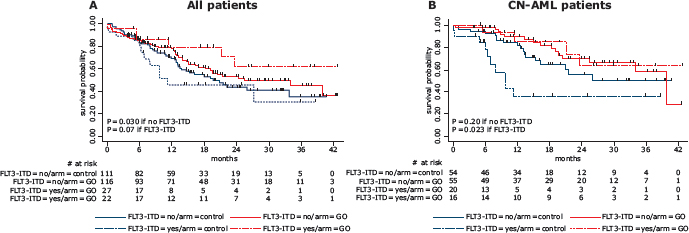
Figure 4: Kaplan-Meier estimates of overall survival according to FLT3–ITD status and treatment arm. (A) Overall survival according to FLT3–ITD status and treatment arm in all patients. In FLT3–ITD positive patients, 2–year OS was estimated at 46% (95%CI, 23–66) in the control arm versus 62% (95%CI, 32–82) in the GO arm (P = 0.07 by the log-rank test); In FLT3–ITD negative patients, 2–year OS was estimated at 44% (95%CI, 32–55) in the control arm versus 53% (95%CI, 42–63) in the GO arm (P = 0.30 by the log-rank test). (B) Overall survival according to FLT3–ITD status and treatment arm in patients with cytogenetically normal acute myeloid leukemia. In FLT3–ITD positive patients, 2-year OS was estimated at 36% (95%CI, 14–59) in the control arm versus 64% (95%CI, 29–85) in the GO arm (P = 0.023 by the log-rank test); in FLT3–ITD negative patients, 2-year OS was estimated at 55% (95%CI, 36–71) in the control arm versus 70% (95%CI, 53–82) in the GO arm (P = 0.20 by the log-rank test). Abbreviations: CN-AML: cytogenetically normal acute myeloid leukemia; ITD, internal tandem duplication.
DISCUSSION
Our study is the first to prospectively analyze SNP-A lesions along with a large panel of gene mutations in a homogeneous cohort of adult patients with primary AML. This combined analysis allowed us to evaluate the relationships between these genetic alterations and their prognostic significance in the context of treatment with GO. Furthermore, we studied the interactions between treatment with GO and the most frequent genetic markers in AML.
Several studies in AML [18–20] have shown that SNP-A analysis allows a higher detection rate of chromosomal defects than CC, mostly due to its ability to detect small CNA and UPDs. In our cohort, we showed that, whilst having a normal conventional karyotype, 38% of the patients had ≥ 1 SNP-A lesion, UPDs being the most common abnormalities. The spectrum of gene mutations we observed is in agreement with previous studies and underlines the particularly high incidence of NPM1 (52%), DNMT3A (34%), and FLT3–ITD (25%) mutations in older patients with CN-AML. Our study also confirms the previously reported patterns of cooperativity between gene mutations in CN-AML [11, 12, 21].
As previously described, the prognostic impact of age and white blood cell count, two well-established risk factors in AML, was not statistically significant in our cohort [13]. The only independent predictors of OS we identified in the whole cohort were unfavorable cytogenetics and SNP-A lesions. Unfavorable cytogenetics was also the only independent prognostic factor for relapse risk in the whole cohort. The potential of genetic profiling to refine prognosis in CN-AML has been highlighted by many studies [10–12, 19, 20]. In our study, multivariate analysis in CN-AML showed that presence of SNP-A lesions, MLL-PTD, DNMT3A mutations, and randomization in the control arm were associated with poor clinical outcome. Thus, our study confirmed the markedly poor prognostic impact of SNP-A lesions, both in the whole cohort and in the CN-AML subset, in accordance with previous reports [18–20]. The adverse prognostic impact of DNMT3A mutations in CN-AML has been consistently reported by several groups, including our own [22]. However, the molecular subgroup in which DNMT3A mutations are found significantly associated to a worse outcome varies across studies, most reports showing that the poor prognostic impact of DNMT3A mutations was most obvious in CN-AML with NPM1/FLT3 non-favorable genotypes [11, 23–26]. These subgroup analyses were not relevant in the ALFA-0701 trial since the prognostic impact of NPM1 mutated/FLT3–ITD negative genotype was not significant in the whole CN-AML cohort, but only in CN-AML patients treated in the control arm. In our study, the lack of favorable prognostic value of the NPM1 mutated/FLT3–ITD negative genotype in CN-AML patients treated with GO can be at least partially explained by the fact that FLT3–ITD mutations confer a higher sensitivity to GO (see below), which is likely to compensate the benefit of being FLT3–ITD negative. Besides, the prognostic analysis for less common molecular alterations like CEBPA or IDH1/2 mutations was limited by the small number of mutated cases in our cohort.
We and others [13–15] previously reported that the benefit of GO addition was limited to patients with cytogenetically favorable or intermediate-risk AML. Presence of FLT3–ITD has been recognized as a major predictor of outcome in AML, especially in the CN-AML subset [11]. Nevertheless, in the ALFA-0701 trial, FLT3–ITD did not significantly impact on CIR and OS in univariate analysis, suggesting that treatment with GO may improve outcome of FLT3–ITD positive patients. Actually, we found that the OS benefit associated with GO treatment was more pronounced in FLT3–ITD positive than in negative patients. Interestingly, these findings are consistent with results from Jawad et al. who found that leukemic stem and progenitor cells carrying FLT3–ITD were more sensitive to GO and that FLT3–ITD status was an independent predictor of in vitro chemosensitivity to GO [27]. However, the mechanisms underlying these differential responses remain unknown. Because GO has been withdrawn from the market, very few data on the genetic basis of response to GO in patients are available. In the MRC AML16 trial, FLT3–ITD mutations were found independently prognostic in older AML patients, but not predictive of response to GO. However, the cumulative dose of GO in this study was much lower than in the ALFA-0701 trial, which may account for these discordant results [15].
Overall, the current information about GO supports the efficacy of this agent in newly diagnosed AML, with acceptable toxicity [28, 29]. In light of three published randomized trials [13–15], the most appropriate indications for reapproval of GO in AML would be patients with cytogenetically favorable or intermediate-risk AML [13–15, 28, 29]. Moreover, the present data suggest that the addition of GO to standard chemotherapy may be a promising treatment option especially for FLT3–ITD positive AML. Although patients with FLT3–ITD positive AML are usually considered as candidates for allogeneic stem cell transplantation (allo-SCT), the benefit of this high-risk procedure in first CR for those patients is still a matter of debate [30–32]. During the last decade, FLT3 tyrosine kinase has emerged as an attractive therapeutic target in FLT3 mutated AML. Several anti-FLT3 compounds are undergoing evaluation in different phases of clinical trials, as monotherapy or in combination with standard chemotherapy. Despite promising in vitro studies, clinical responses to single-agent FLT3 inhibitors in AML patients are generally incomplete and not sustained, and no significant improvement in OS has been demonstrated so far [33, 34]. Therefore, in FLT3–ITD positive AML patients, particularly in those not eligible for allo-SCT, the addition of GO to standard chemotherapy appears as an interesting treatment option, that would be worthy of further investigation in clinical trials.
In conclusion, our study emphasizes that SNP-A lesions and DNMT3A mutations represent adverse prognostic factors, particularly in CN-AML. Both markers add independent prognostic information and, therefore, could contribute to improve risk stratification in patients with CN-AML. Importantly, our results suggest that addition of GO to standard chemotherapy might overcome the poor prognosis of FLT3–ITD. However, further studies based on larger patient cohorts are required to formally identify the molecular determinants of response to GO.
PATIENTS AND METHODS
Patients and treatment
From January 2008 to November 2010, 278 patients aged 50–70 years with previously untreated primary AML were included in the randomized multicentric Phase 3 ALFA-0701 trial (NCT00927498), investigating the benefit of the addition of low fractionated doses of GO to standard front-line chemotherapy (Supplementary Data) [13]. The study was approved by the ethics committee of Saint-Germain en Laye, France, and by the institutional review board (IRB) of the French Regulatory Agency and undertaken in accordance with the Declaration of Helsinki. All patients provided written informed consent. Bone marrow or peripheral blood samples collected at AML diagnosis and after treatment were obtained from the tissue bank “Tumorothèque du Centre de Référence Régional en Cancérologie de Lille (C.R.R.C.)" and an approval of this study was obtained from the IRB of CHRU of Lille (CSTMT089).
Conventional cytogenetic analysis
Cytogenetic R-banding analysis was performed on diagnostic bone marrow samples using standard methods. The karyotypes were described according to the International System for Human Cytogenetic Nomenclature recommendations [35] and classified according to the revised MRC criteria within 3 groups (favorable, intermediate and unfavorable) [36].
Cell isolation, nucleic acid extraction, and cDNA synthesis
Mononuclear cells from pretreatment bone marrow or peripheral blood were isolated by Ficoll density gradient centrifugation (MSL, Eurobio, Courtaboeuf, France). The blast percentage following enrichment was above 60% in all samples. Genomic DNA was extracted from mononuclear cells using the QIAamp DNA Mini Kit® (Qiagen, Courtaboeuf, France) following the manufacturer’s instructions. Total RNA was extracted from the same specimen and reverse transcribed using the High-Capacity cDNA Archive Kit (Applied Biosystems, Courtaboeuf, France) and according to the standardized protocol developed within the Europe Against Cancer (EAC) program [37].
Molecular analysis
MLL-PTD [38], FLT3–ITD [22], mutations of FLT3 tyrosine kinase domain (FLT3–TKD) (FLT3D835/I836) [22], NPM1 (exon 12) [39], CEBPA [40], WT1 (exons 7 and 9) [41], IDH1R132 [42], IDH2R140 [43], IDH2R172 [42], RUNX1 (exons 3–8) [44], ASXL1 (exon 12) [3], TET2 (exons 3–11) [4], and DNMT3A (exons 8–9, 11–23) [22] and EVI1 overexpression [45] were assessed centrally as previously described. The screening for TET2, IDH1/2, and DNMT3A mutations was restricted to CN-AML. Further details regarding gene mutation analysis are available in the Supplementary Data.
SNP-A analysis
Patient genomic DNA was processed and hybridized to Genome-Wide Human SNP 6.0 arrays (Affymetrix, Santa Clara, CA) according to the manufacturer instructions. Raw data were analyzed using the Genotyping Console version 4.1 software (Affymetrix). In order to distinguish somatic from constitutional SNP array lesions, we adopted a stringent and conservative algorithm. Aberrations were excluded as known copy number variants if there was > 50% overlap with variants from the public Database of Genomic Variants (DGV Beta version 10). Interstitial UPD < 20 Mb were considered as constitutional and excluded from the analysis. Subsequently, all CNA and UPD were validated by visual inspection and annotated based on the hg19 human genome assembly.
Statistical analysis
Median with interquartile range and percentages were computed as summary statistics. Clinical endpoints considered for prognostic analyses were CR/CRp rate, CIR and OS. CIR and OS were calculated after censoring patients who received allo-SCT in first CR at allo-SCT time. We focused here on the prognostic influence of CC, SNP-A, and molecular alterations. First, univariate analyses for CR/CRp were based on logistic regression models with prognostic influence measured by odds ratio (OR) with 95% confidence interval (95%CI). Univariate predictive analyses for CIR and OS were based on Cox models with prognostic influence measured by hazards ratio (HR) with 95% CI. Estimation of survival curves used the Kaplan-Meier method, compared by the log-rank test [46, 47]. Stepwise multivariable Cox models allowed assessing the additional prognostic influence of those variables previously selected at the 10% level [48]. Secondly, prognostic analyses were limited to the subset of patients with CN-AML. Finally, to identify optimal treatment groups, interactions were tested using the Gail and Simon statistics [49]. This consisted of testing the heterogeneity in the OR of CR/CRp or in the HR of relapse or death according to cytogenetics, SNP-A karyotyping, and molecular findings in the two randomized treatment arms. Statistical analysis was performed with SAS (version 9.2, SAS Inc., Cary, NC) and R (version 2.13.1) softwares. All reported P-values are two-sided, with a nominal type I error of 0.05.
ACKNOWLEDGMENTS
This work was supported by the Association Laurette Fugain, the Fondation de France (Leukemia Committee), the Ligue Contre le Cancer (North Center), North-West Canceropole (Onco-Hematology axis), SIRIC Oncolille, and Institut National du Cancer (Translational Research Project: DREAM), France.
DISCLOSURES
SCa declares consultancy fees from Wyeth/Pfizer from 2006 to 2008. The remaining authors have no relevant conflict of interest to disclose.
REFERENCES
1. Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010; 115:453–474.
2. Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A and Dohner H. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008; 358:1909–1918.
3. Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, Mozziconacci MJ, Vey N and Birnbaum D. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009; 145:788–800.
4. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lecluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009; 360:2289–2301.
5. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009; 361:1058–1066.
6. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010; 363:2424–2433.
7. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, Shi JY, Zhu YM, Tang L, Zhang XW, Liang WX, Mi JQ, Song HD, Li KQ, Chen Z and Chen SJ. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011; 43:309–315.
8. Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, Nicolas C, Payer AR, Fernandez HF, Tallman MS, Paietta E, Melnick A, Vandenberghe P, Speleman F, Aifantis I, Cools J, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011; 25:130–134.
9. Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, Spanhol-Rosseto A, Klein HU, Dugas M, Schindela S, Trifonov V, Schnittger S, Haferlach C, Bassan R, Wells VA, Spinelli O, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011; 118:6153–6163.
10. Marcucci G, Haferlach T and Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011; 29:475–486.
11. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012; 366:1079–1089.
12. Sanders MA and Valk PJ. The evolving molecular genetic landscape in acute myeloid leukaemia. Curr Opin Hematol. 2013; 20:79–85.
13. Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, de Revel T, Gastaud L, de Gunzburg N, Contentin N, Henry E, Marolleau JP, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012; 379:1508–1516.
14. Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, Yin JA, Hunter A, Goldstone AH and Wheatley K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011; 29:369–377.
15. Burnett AK, Russell NH, Hills RK, Kell J, Freeman S, Kjeldsen L, Hunter AE, Yin J, Craddock CF, Dufva IH, Wheatley K and Milligan D. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012; 30:3924–3931.
16. Bullinger L, Kronke J, Schon C, Radtke I, Urlbauer K, Botzenhardt U, Gaidzik V, Cario A, Senger C, Schlenk RF, Downing JR, Holzmann K, Dohner K and Dohner H. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2009; 24:438–449.
17. Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, Baty J, Heath S, Westervelt P, Watson MA, Tomasson MH, Nagarajan R, O'Gara BP, Bloomfield CD, Mrozek K, Selzer RR, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci U S A. 2009; 106:12950–12955.
18. Parkin B, Erba H, Ouillette P, Roulston D, Purkayastha A, Karp J, Talpaz M, Kujawski L, Shakhan S, Li C, Shedden K and Malek SN. Acquired genomic copy number aberrations and survival in adult acute myelogenous leukemia. Blood. 2010; 116:4958–4967.
19. Tiu RV, Gondek LP, O'Keefe CL, Huh J, Sekeres MA, Elson P, McDevitt MA, Wang XF, Levis MJ, Karp JE, Advani AS and Maciejewski JP. New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J Clin Oncol. 2009; 27:5219–5226.
20. Yi JH, Huh J, Kim HJ, Kim SH, Kim YK, Sohn SK, Moon JH, Kim KH, Won JH, Mun YC, Kim H, Park J, Jung CW and Kim DH. Adverse prognostic impact of abnormal lesions detected by genome-wide single nucleotide polymorphism array-based karyotyping analysis in acute myeloid leukemia with normal karyotype. J Clin Oncol. 2011; 29:4702–4708.
21. Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol. 2011; 4:36.
22. Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, Cayuela JM, Hayette S, Reman O, Contentin N, Bordessoule D, Pautas C, Botton S, Revel T, Terre C, Fenaux P, et al. Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia. 2012; 26:1247–1254.
23. Ostronoff F, Othus M, Ho PA, Kutny M, Geraghty DE, Petersdorf SH, Godwin JE, Willman CL, Radich JP, Appelbaum FR, Stirewalt DL and Meshinchi S. Mutations in the DNMT3A exon 23 independently predict poor outcome in older patients with acute myeloid leukemia: a SWOG report. Leukemia. 2013; 27:238–241.
24. Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, Figueroa ME, Zeilemaker A, Melnick A, Lowenberg B, Valk PJ and Delwel R. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012; 119:5824–5831.
25. Thol F, Damm F, Ludeking A, Winschel C, Wagner K, Morgan M, Yun H, Gohring G, Schlegelberger B, Hoelzer D, Lubbert M, Kanz L, Fiedler W, Kirchner H, Heil G, Krauter J, et al. Incidence and Prognostic Influence of DNMT3A Mutations in Acute Myeloid Leukemia. J Clin Oncol. 2011; 29:2889–2896.
26. Gaidzik VI, Schlenk RF, Paschka P, Stolzle A, Spath D, Kuendgen A, von Lilienfeld-Toal M, Brugger W, Derigs HG, Kremers S, Greil R, Raghavachar A, Ringhoffer M, Salih HR, Wattad M, Kirchen HG, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG). Blood. 2013; 121:4769–4777.
27. Jawad M, Seedhouse C, Mony U, Grundy M, Russell NH and Pallis M. Analysis of factors that affect in vitro chemosensitivity of leukaemic stem and progenitor cells to gemtuzumab ozogamicin (Mylotarg) in acute myeloid leukaemia. Leukemia. 2010; 24:74–80.
28. Ravandi F, Estey EH, Appelbaum FR, Lo-Coco F, Schiffer CA, Larson RA, Burnett AK and Kantarjian HM. Gemtuzumab ozogamicin: time to resurrect? J Clin Oncol. 2012; 30:3921–3923.
29. Rowe JM and Lowenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood. 2013; 121: 4838–4841.
30. Brunet S, Martino R and Sierra J. Hematopoietic transplantation for acute myeloid leukemia with internal tandem duplication of FLT3 gene (FLT3/ITD). Curr Opin Oncol. 2013; 25:195–204.
31. Gale RE, Hills R, Kottaridis PD, Srirangan S, Wheatley K, Burnett AK and Linch DC. No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML10 and 12 trials. Blood. 2005; 106:3658–3665.
32. Sengsayadeth SM, Jagasia M, Engelhardt BG, Kassim A, Strickland SA, Goodman S, Lucid C, Vnencak-Jones CL, Greer JP and Savani BN. Allo-SCT for high-risk AML-CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplant. 2012; 47:1535–1537.
33. Beffinger M and Skwarska A. The role of FLT3 kinase as an AML therapy target. Curr Pharm Des. 2012; 18:2758–2765.
34. Kayser S and Levis MJ. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: clinical implications and limitations. Leuk Lymphoma. 2013; e-pub ahead of print 5 June 2013; doi: 10.3109/10428194.2013.800198.
35. International System for Cytogenetic Nomenclature: Guidelines for Cancer Cytogenetics. In: Mitelmann F, ed. Supplement to an International System for Human Cytogenetic Nomenclature (Basel, Switzerland: S. Karger), 1991, pp. 1–53.
36. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ and Burnett AK. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010; 116:354–365.
37. Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cave H, Pane F, Aerts JL, De Micheli D, Thirion X, Pradel V, Gonzalez M, et al. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – a Europe Against Cancer program. Leukemia. 2003; 17:2318–2357.
38. Schnittger S, Wormann B, Hiddemann W and Griesinger F. Partial tandem duplications of the MLL gene are detectable in peripheral blood and bone marrow of nearly all healthy donors. Blood. 1998; 92:1728–1734.
39. Boissel N, Renneville A, Biggio V, Philippe N, Thomas X, Cayuela JM, Terre C, Tigaud I, Castaigne S, Raffoux E, De Botton S, Fenaux P, Dombret H and Preudhomme C. Prevalence, clinical profile, and prognosis of NPM mutations in AML with normal karyotype. Blood. 2005; 106:3618–3620.
40. Preudhomme C, Sagot C, Boissel N, Cayuela JM, Tigaud I, de Botton S, Thomas X, Raffoux E, Lamandin C, Castaigne S, Fenaux P and Dombret H. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood. 2002; 100:2717–2723.
41. Renneville A, Boissel N, Zurawski V, Llopis L, Biggio V, Nibourel O, Philippe N, Thomas X, Dombret H and Preudhomme C. Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer. 2009; 115:3719–3727.
42. Boissel N, Nibourel O, Renneville A, Gardin C, Reman O, Contentin N, Bordessoule D, Pautas C, de Revel T, Quesnel B, Huchette P, Philippe N, Geffroy S, Terre C, Thomas X, Castaigne S, et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol. 2010; 28:3717–3723.
43. Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H and Preudhomme C. Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2011; 117:3696–3697.
44. Preudhomme C, Renneville A, Bourdon V, Philippe N, Roche-Lestienne C, Boissel N, Dhedin N, Andre JM, Cornillet-Lefebvre P, Baruchel A, Mozziconacci MJ and Sobol H. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood. 2009; 113:5583–5587.
45. Groschel S, Lugthart S, Schlenk RF, Valk PJ, Eiwen K, Goudswaard C, van Putten WJ, Kayser S, Verdonck LF, Lubbert M, Ossenkoppele GJ, Germing U, Schmidt-Wolf I, Schlegelberger B, Krauter J, Ganser A, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010; 28:2101–2107.
46. Kaplan EL and Meier P. Non-parametric estimations from incomplete observations. JASA. 1958; 53:457–481.
47. Peto R and Peto J. Asymptotically efficient rank invariant test procedures. J R Stat Soc. 1972; 135:185–206.
48. Cox DR. Regression models and life tables. J R Stat Soc B. 1972; 34:187–220.
49. Gail M and Simon R. Testing for qualitative interactions between treatment effects and patient subsets. Biometrics. 1985; 41:361–372.