
Introduction
Dehydroepiandrosterone (DHEA), an important precursor of activate steroid hormone, is produced abundantly by the adrenal cortex with an age-dependent pattern [1]. Age-dependent decrease of DHEA production has attracted extensive attention due to its possible benefit to psychological well-being in the elderly [2]. The literature evidences reveal the changes in DHEA level are associated with multiple pathologies, whereas a replacement therapy might alleviate age-associated declines in physiological functions [3-7]. This results in a large-scale self-administration of DHEA as an “anti-ageing” drug in dietary supplement. Many studies had reported that DHEA had antioxidant effects in various acute and chronic oxidative stress experiments [7-10]. However, the effects of DHEA administration can be antioxidant [11] or pro-oxidant [12, 13], depending on the administered dose and specific tissue [14]. Gallo et al. reported that slightly higher concentrations of DHEA protected cells against lipid peroxidation induced by oxidative stress while pharmacological doses of DHEA displayed a pro-oxidant activity [15]. In addition, microsomes isolated from DHEA-treated rats are resistant to iron-dependent lipid peroxidation, whereas microsomes pre-incubated with DHEA in vitro are not resistant, suggesting that a cell or tissue dependent signaling or transformation is required for DHEA’s antioxidant action [16].
Aging results in the progressive deterioration of physiological function, and free radical theory is the most accepted ageing theories [17]. In aging process, reactive oxygen species (ROS), can be generated; reactive oxygen species is toxic at high concentrations [17]. ROS can interact with many molecules which will result in DNA mutation, protein denaturation, lipid peroxidation, membrane destruction and so on [18]. Furthermore, excess ROS can activate apoptotic pathways [19]; a biochemical hallmark of apoptosis is DNA damage [20]. Administration of antioxidants attenuates free radical-mediated oxidative damage in several organs including the testis [21]. Oxidative stress plays a key role in cell damage [22] and the risk of oxidative damage is especially high for steroid synthesizing tissues, which use molecular oxygen for steroids biosynthesis [23]. The study shows that DHEA exerts its effects by rapidly transforming into biologically active steroids in target tissues [24]. It had proposed that increasing serum DHEA (60-79 years old) concentration to the levels found in young people may have anti-ageing effects [1]. Our previous study demonstrated that administration of DHEA markedly increased serum testosterone concentration in rats [25]. In males, ~95% of androgen biosynthesis and secretion occurs in Leydig cells, and it had been certified that functional changes in Leydig cells is account for the observed reduction in serum testosterone level [2].
Taken these points together, we presumed that DHEA protects cell from oxidative damage, which might be a major reason for the anti-ageing action of DHEA. In addition, the effect of DHEA on the antioxidant function of Leydig cells, a major target cell of DHEA convert to active steroids, is unknown. Thus, the present study aimed to investigate the effect of DHEA on ROS generation, antioxidant enzymes activity, DNA damage, cell apoptosis and apoptosis-related factors in H2O2-treated rat Leydig cells, and this information is important to understand the molecular mechanism of anti-ageing action of DHEA.
Results
Protective effect of DHEA on cell viability
Testosterone content was significantly increased in primary Leydig cells after DHEA treatment (P < 0.01) (Figure 1A). Treated with H2O2 reduced cell viability in a dose-dependent manner, and 300μM H2O2 treatment significantly decreased cell viability relative to that in H2O2-free group (Figure 1B). Pre-treatment with 1-50μM DHEA significantly enhanced cell viability (P < 0.05) (Figure 1C). Meanwhile, 50-100μM DHEA significantly increased testosterone content when compared to H2O2-treated group (P < 0.01) (Figure 1D).
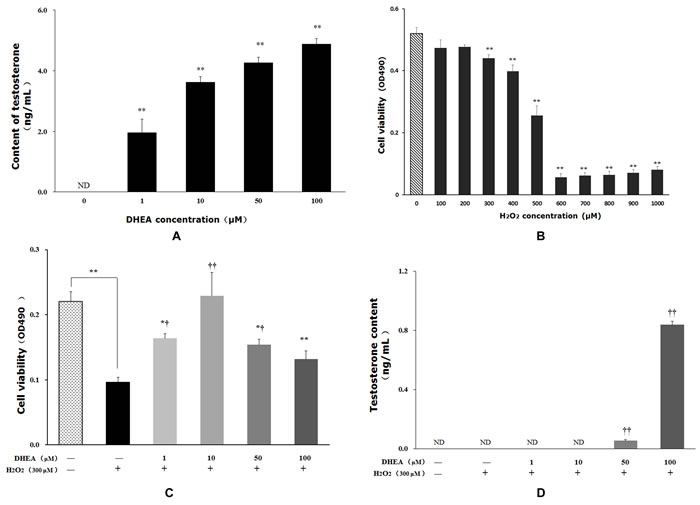
Figure 1: Impact of DHEA on testosterone content and cell viability in Leydig cells. Data are presented as means ± SD from three independent experiments, with six samples taken at each treatment group (n = 6). A. Testosterone content in Leydig cell with DHEA-treated; B. Cell viability in Leydig cell with H2O2-induced; C. Effect of DHEA-pretreated on cell viability in H2O2-induced Leydig cells; D. Effect of DHEA-pretreated on testosterone content in H2O2-induced Leydig cells. ND = none detected. *P < 0.05 and ** P < 0.01, relative to DHEA and H2O2-free control; † P < 0.05 and †† P < 0.01, relative to H2O2-induced control.
DHEA inhibit reactive oxygen species generation
ROS, •OH and MDA contents were significantly increased in H2O2-treated group when compared to control group (P < 0.05) (Figure 2). Pre-treatment with 10μM DHEA reduced intracellular ROS levels relative to that in H2O2-treated group (P < 0.05) (Figure 2A). Pre-treatment with 1-100μM DHEA significantly decreased •OH content (P < 0.01) (Figure 2B), while no significant differences were observed on the O2- content (Figure 2C). Compared to H2O2-treated group, MAD contents were decreased in the cells pre-treated with 10 and100μM DHEA (P < 0.05) (Figure 2D).
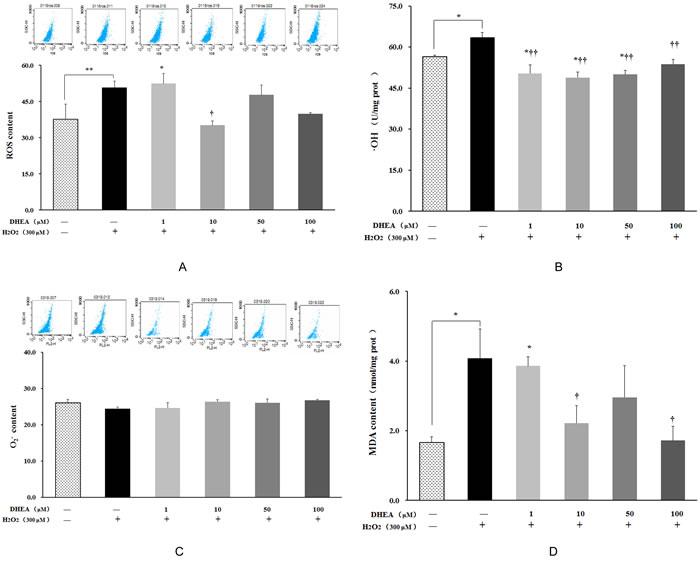
Figure 2: Impact of DHEA on oxidant generation and malondialdehyde content in H2O2-induced Leydig cells. Data are presented as means ± SD from three independent experiments, with six samples taken at each treatment group (n = 6). A. Reactive oxygen species (ROS) content; B. Superoxide anion (O2-); content C. Hydroxyl Radical (•OH) content; D. Malondialdehyde (MDA) content. *P < 0.05 and ** P < 0.01, relative to DHEA and H2O2-free control; † P < 0.05 and †† P < 0.01, relative to H2O2-induced control.
Impact of DHEA on antioxidant enzyme activity
As shown in Figure 3, no changes were observed on SOD, GSH-Px, CAT or POD activities in H2O2-treated group (P>0.05). Pre-treatment with 50μM and 100μM DHEA significantly increased POD activity when compared to H2O2-treated group (P < 0.05) (Figure 3C), while 1μM and 10μM DHEA significantly decreased GSH-Px activity relative to that in H2O2-treated group (P < 0.05) (Figure 3D). Compared to H2O2-treated group, pre-treatment with different dose DHEA have no significant effect on SOD and CAT activities (P>0.05) (Figure 3A and 3B).
Table 1: Prime sequence of β-actin and targeted gene
Gene |
Genebank accession Number |
Primer sequences(5’-3’ ) |
Orientation |
Product size(bp) |
β-actin |
NM_031144 |
CCCTGTGCTGCTCACCGA ACAGTGTGGGTGACCCCGTC |
Forward Reverse |
186 |
Bax |
NM_007527 |
GCAGGGAGGATGGCTGGGGAGA TCCAGACAAGCAGCCGCTCACG |
Forward Reverse |
352 |
Bcl-2 |
NM_016993 |
CGACTTTGCAGAGATGTCCA CATCCACAGAGCGATGTTGT |
Forward Reverse |
202 |
Caspase-9 |
NM_031632 |
GCCTCATCATCATCAACAACG TCTACGACAGGGTGGTC |
Forward Reverse |
283 |
Caspase-3 |
NM_012922 |
CTGGACTGCGGTATTGAG CGAATGAGATGGCGTGGG |
Forward Reverse |
342 |
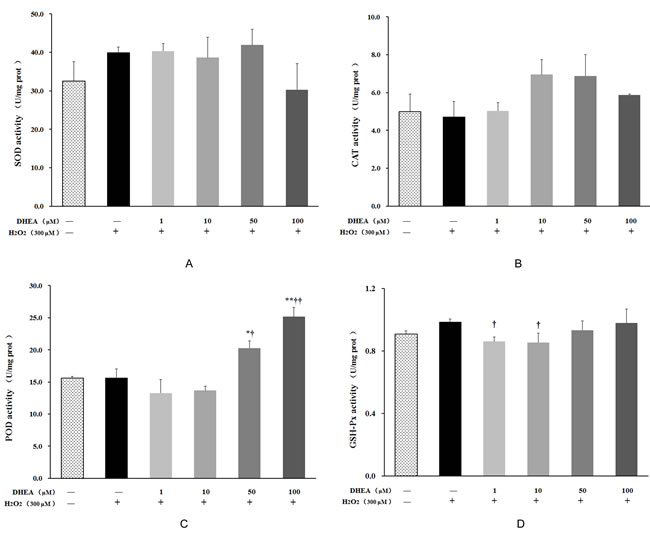
Figure 3: Effect of DHEA on antioxidant enzyme activity in H2O2-induced Leydig cells. Data are presented as means ± SD from three independent experiments, with six samples taken at each treatment group (n = 6). *P < 0.05 and ** P < 0.01, relative to DHEA and H2O2-free control; † P < 0.05 and †† P < 0.01, relative to H2O2-induced control. SOD = superoxide dismutase; CAT = catalase; POD = peroxidase; GSH-Px = glutathione peroxidase.
Protective effect of DHEA on H2O2 -induced oxidation damage
The comet assay on DNA damage in H2O2-induced Leydig cells was shown in Figure 4 and Table 2. Exposure of Leydig cells to H2O2 caused a significant increase in DNA damage, as indicated by the greater migration of DNA fragments on the agarose gel (Figure 4). Comet length, tail moment and olive tail moment were significantly higher in cells exposed to 300μM H2O2 when compared to the H2O2-free control (P < 0.05) (Table 2). Pre-treatment with DHEA prevented H2O2-induced DNA damage in a dose-dependent pattern (Figure 4). As shown in Table 2, pre-treatment with 50-100μM DHEA significantly decreased comet length relative to that in H2O2-treated group (P < 0.01). When compared to H2O2-treated group, tail moment and olive tail moment were significant decreased in 10-100μM DHEA-pretreated group (P < 0.01).
Table 2: Effect of DHEA on the H2O2-induced DNA damage reflecting as different comet parameters in Leydig cell
Comet parameters |
Comet length (μm) |
Tail moment (μm) |
Olive tail moment (μm) |
Head DNA (%) |
Tail DNA (%) |
control |
69.25±5.08 |
13.18±1.89 |
3.42±0.80 |
72.43±4.31 |
27.57±4.31 |
300μM H2O2 |
102.79±8.06 * |
50.16±7.08 ** |
18.61±4.30** |
59.43±3.77* |
40.57±3.77* |
1μM DHEA+ 300μM H2O2 |
118.11±15.18** |
49.88±9.88** |
15.63±3.92* |
65.45±2.97 |
34.55±2.97 |
10μM DHEA+ 300μM H2O2 |
98.00±8.85 |
21.59±3.64†† |
6.81±1.59†† |
63.02±3.60 |
36.99±3.60 |
50μM DHEA+ 300μM H2O2 |
70.48±4.68 †† |
22.67±3.00†† |
4.32±0.47†† |
65.63±4.54 |
34.37±4.54 |
100μM DHEA+ 300μM H2O2 |
64.00±2.56†† |
6.16±1.08†† |
0.68±0.14†† |
90.34±1.18†† |
29.66±1.18†† |
Data are presented as means±SD from three individual experiments, with six samples taken at each time point (n = 6). *P < 0.05 and **P < 0.01, relative to DHEA and-H2O2-free control; †P < 0.05 and ††P < 0.01, relative to H2O2-induced control.

Figure 4: Typical DNA comet images of Leydig cells treated with DHEA and H2O2. A. control group; B. H2O2-induced control; C.-F. 1-100μM DHEA treatment group with 300μM H2O2-induced oxidation damage.
DHEA reduced H2O2-induced cells apoptosis
No differences were observed on late apoptosis ratio, while total apoptosis ratio and early apoptosis ratio were significantly increased in H2O2-treated group when compared to the H2O2-free control (P < 0.05) (Figure 5). Pre-treatment with 1-10μM DHEA significantly decreased total apoptosis ratio (P < 0.05) and 1-100μM DHEA significantly decreased early apoptosis ratio when compared to H2O2-treated group (P < 0.01), while no differences were observed on the late apoptosis ratio (P>0.05) (Figure 5).
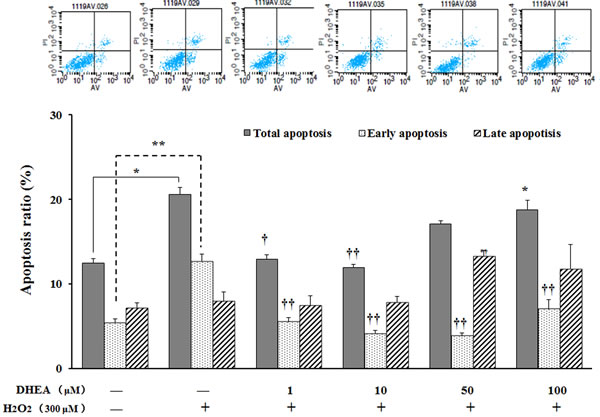
Figure 5: Effect of DHEA on cells apoptosis in H2O2-induced Leydig cells. Data are presented as means ± SD from three independent experiments, with six samples taken at each treatment group (n = 6). *P < 0.05 and ** P < 0.01, relative to DHEA and H2O2-free control; † P < 0.05 and †† P < 0.01, relative to H2O2-induced control.
Effect of DHEA on the mRNA expression levels of apoptosis related factors
As shown in Figure 6, Bax and caspase-3 mRNA levels were significantly increased (P < 0.01), while Bcl-2 mRNA level was significantly decreased in H2O2-treated group when compared to the H2O2-free control (P < 0.05). Pre-treatment with DHEA has no effect on Bcl-2 mRNA level (P>0.05) (Figure 6B), while Bax and capase-3 mRNA levels were significantly decreased in 10-100μM DHEA-pretreated group relative to that in H2O2-treated group (P < 0.05) (Figure 6A and 6D). Compared to H2O2-treated group, pre-treatment with 10μM and 100μM DHEA significantly decreased capase-9 mRNA level (P < 0.05) (Figure 6C).
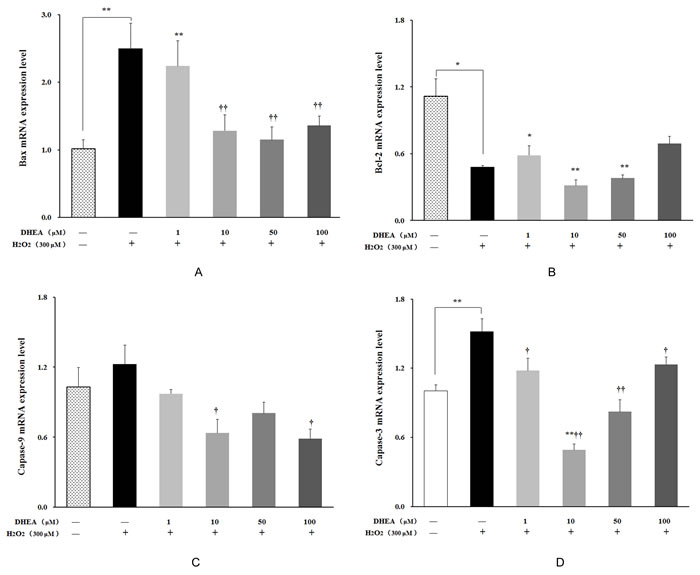
Figure 6: Effect of DHEA on Bax, Bcl-2, Capase-9 and Capase-3 mRNA levels in H2O2-induced Leydig cells. Data are presented as means ± SD from three independent experiments, with six samples taken at each treatment group (n = 6). A. Bax mRNA level; B. Bcl-2 mRNA level; C. Capase-9 mRNA level; D. Capase-3 mRNA level. *P < 0.05 and ** P < 0.01, relative to DHEA and H2O2-free control; † P < 0.05 and †† P < 0.01, relative to H2O2-induced control.
Discussion
In aging individuals, lipid peroxidation, oxidative stress and damage of cellular macromolecules caused by the excessive reactive oxygen species has been hypothesized to be one of the major contributors to the aging process [23]. As one of the most important ROS, hydrogen peroxide (H2O2) has been extensively used to induce oxidative stress in vitro models [28]. Our results showed that the cell viability significantly decreased in 300-1000μM H2O2-treated groups than that in H2O2-free group. Previous study had reported that exposure to low doses of H2O2 induces apoptosis in a variety of cell types, thereby directly establishing oxidative stress as a mediator of cell death [29, 30]. The high doses of H2O2 have been shown to be cytotoxic by triggering a disorderly form of cell death, or necrosis [30, 31]. Thus, we used 300μM H2O2 treatment in the subsequent experiments, and the results showed that H2O2 significantly increased ROS content and total apoptosis ratio in Leydig cells. Meanwhile, the comet assay showed that Leydig cells exposed to 300μM H2O2 exhibited a significant increase in comet length, tail moment and olive tail moment. These results indicated that H2O2 caused oxidative stress, providing a model for further investigation the protective effect of DHEA on H2O2-induced oxidative damage in Leydig cells.
Previous studies had reported that administration of DHEA produce a number of beneficial effects in elderly [1-4, 6]. This study found that pre-treatment with DHEA decreased the ROS content by inhibiting •OH generation and prevented cell death induced by H2O2. This is consistent with the results of Gabriel et al. [10], who reported that DHEA reduced ROS content in ovariectomized rats. Oxidative stress defined as excessive production of ROS include O2-, H2O2 and •OH, and excessive ROS is toxic to human body [17]. Many recent evidences indicate that endogenous free radicals contribute to spontaneous mutagenesis by directly inducing DNA damage [32]. Pre-treatment with DHEA prevented H2O2-induced DNA damage in a dose-dependent pattern in Leydig cells, as suggested by the decrease of comet length, tail moment and olive tail moment in DHEA-treated groups. Based on the data above, we presumed that DHEA might prevent DNA oxidative damage in H2O2-induced Leydig cells by inhibiting •OH generation.
It is well-known that body utilizes non-enzymatic and enzymatic antioxidants to avoid or to retard cellular damage arising from oxidative stress [10]. The antioxidant enzyme activity maintain the balance between the formation and scavenging of ROS [33, 34]. No changes were observed on the SOD and CAT activities, while pre-treatment with 50μM and 100μM DHEA significantly increased POD activity in H2O2-treated Leydig cells. DHEA supplementation demonstrated the antioxidant effects in various acute and chronic oxidative stress experiments in rodents [36]. It had reported that DHEA exhibits antioxidant properties by suppressing superoxide anion production [35]. Another antioxidant factor is the reduced glutathione (GSH), which act as a substrate in the detoxification of xenobiotic and in control of hydrogen peroxide and other peroxides concentrations [9]. Our results showed that 1μM and 10μM DHEA treatment decreased GSH-Px activity in H2O2-treated Leydig cells. NADPH is a cofactor required for the conversion of GSSG to GSH by glutathione reductase, and glucose-6-phosphate dehydrogenase (G6PDH) is an important enzyme involved in the production of NADPH [21]. In vivo and in vitro studies had shown that DHEA inhibits G6PDH activity which in turn reduced the production of NADPH [9, 21]. Thus, we speculated that DHEA decreased NADPH level through inhibiting G6PDH activity and finally decreased GSH-Px activity in H2O2-induced Leydig cells. These result indicated that pre-treatment with DHEA increased POD activity which results in the decrease of ROS contents in H2O2-treated Leydig cells.
Present study showed that pre-treatment with1μM and 10μM DHEA significantly decreased the total apoptosis ratio and early apoptosis ratio, but not the late apoptosis ratio. This result indicated that DHEA prevented H2O2-induced Leydig cells apoptosis and this action was achieved mainly through inhibiting early apoptosis. Permeabilization of the mitochondrial outer membrane allows the release of pro-apoptotic factors from intermembrane space into cytosol in early apoptosis [37]. Cytochrome c release can promotes and amplify the apoptotic cascade, which is considered as the commitment step of programmed cell death [37-39]. The Bcl-2 family proteins, including Bax and Bcl-2, are key regulators in the early stages of apoptotic [40-44]. Our results showed that pre-treatment with 10-100μM DHEA decreased the pro-apoptotic protein Bax mRNA level in H2O2-treated Leydig cells, while there is no effect on anti-apoptotic protein Bcl-2 mRNA level. It well known that depending on the Bcl-2/Bax ratio, sequential activation of caspases-9 and caspases-3 plays a central role in the execution of cell apoptosis [45-47]. Our results also found that pre-treatment with 1-100μM DHEA significantly decreased the caspases-3 mRNA levels in H2O2-treated Leydig cells. In addition, pre-treatment with10μM and 100μM DHEA treatment decreased the caspases-9 mRNA levels in H2O2-treated Leydig cells. These results indicated that DHEA prevented H2O2-induced Leydig cells apoptosis mainly through inhibiting pro-apoptotic protein Bax mRNA levels which resulted in the decreasing of caspases-9 and caspases-3 mRNA levels.
In conclusion (as shown in Figure 7), our results demonstrated that pre-treatment with DHEA prevented H2O2-induced Leydig cells oxidative damage by increasing the POD activity which result in the inhibition of •OH generation, and it decreased H2O2-induced Leydig cells apoptosis through decrease the pro-apoptotic protein Bax and caspases-9, caspases-3 mRNA levels. This information shed light on the potential application for DHEA in elderly and treatment of oxidative stress induced related diseases. Certainly, further studies are warranted to better understand the underlying mechanisms on how DHEA prevents oxidative damage in the cells.
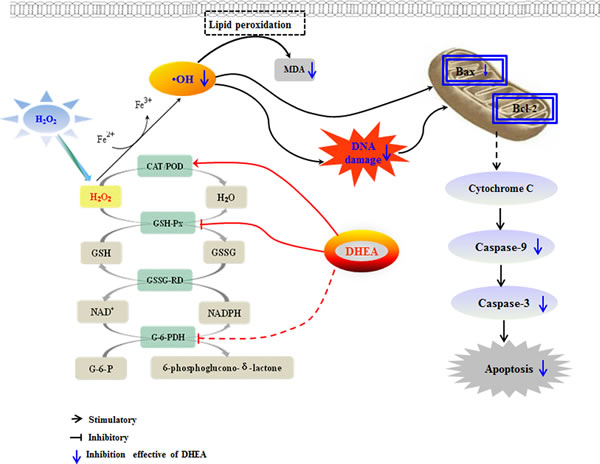
Figure 7: Mechanism of DHEA protective oxidation damage in H2O2-induced Leydig cells. DHEA prevented H2O2-induced Leydig cells oxidative damage by increasing POD activity which resulted in the inhibiting •OH generation. Meanwhile, DHEA decreased H2O2-induced cells apoptosis which mainly achieved through decrease the pro-apoptotic protein Bax, caspases-9 and caspases-3 mRNA levels.
Materials and methods
Isolation of primary Leydig cells
Male Sprague-Dawley (SD) rats (200±20g) were purchased from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China). Animals were housed under 25 °C and 50% humidity with a 12-h light/dark cycle. The food and water were available ad libitum. All animal handling procedures were performed in strict accordance with guide for the Care and Use of Laboratory Animals central of the Nanjing Agricultural University. The protocol was approved by the Institutional Animal Care and Use Committee of Nanjing Agricultural University. All experiments were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Leydig cells were isolated by enzymatic digestion and purified by a discontinuous Percoll gradient, as described by Murugesan et al. [23]. Briefly, testes were decapsulated with fine forceps without breaking the seminiferous tubules and digested in DMEM-F12 containing collagenase (0.5%) at 37°C for 15min in shaking water bath. After incubation, collagenase-free DMEM-F12 was added to each reaction tube which was allowed to stand for 3min at room temperature. Supernatants were combined and centrifuged at 2500×g for 5min at 4°C. The pellet resuspended in 2mL of DMEM-F12 was used as a crude testicular interstitial cell suspension, which was subject to discontinuous Percoll gradients for further purification. Two millilitre of 75% Percoll gradient was added to the centrifuge tube. Above this layer, 70%, 58%, 30% and 5% gradients of Percoll were laid gently one over the other. 2mL of crude Leydig cell suspension was then applied on top of this discontinuous gradient and centrifuged at 3000×g for 30min at 4°C. After centrifugation, most of Leydig cells were observed in 30% Percoll gradients. Leydig cells were transferred to centrifuge tube containing DMEM-F12 medium. After mixing, the tube was centrifuged at 2500×g for 10min at 4°C. Cell purity was assessed by histochemical localization analysis of 3β-HSD according to Aldred & Cooke [26]. Leydig cells were incubated with to DMEM-F12 supplemented with 10% fetal bovine serum (FBS), 5mg/mL transferrin, 2mM L-glutamine, 1.75mM HEPES, 100IU/mL penicillin and 100mg/mL streptomycin, and cultured in an atmosphere of 95% air and 5% CO2 at 37°C.
Detection of testosterone content by radioimmunoassay (RIA)
The concentration of testosterone in primary Leydig cells under basal or stimulated conditions was determined with RIA kit. After culturing for 24 h in DMEM-F12 medium at 37°C, cells were incubated with DHEA (0μM, 1μM, 50μM or 100μM) for 24 h, partly cells was collected and other cells was exposed to 300μM H2O2 for 8h. After incubation, cells were disrupted ultrasonically on ice and centrifuged at 2500×g for 10min at 4°C. The testosterone concentrations in the supernatants were determined (intra-variation coefficients < 10%, inter-variation coefficients < 15%).
Cell viability assay
Cells were seeded on a 96-well plate (1×105 cells/well) and treated with 0, 1, 10, 50 or 100μM DHEA for 24h, then exposed to 300μM H2O2 for another 8h before addition of MTT solution. H2O2-free control cultures received an equal volume of dimethyl sulfoxide (DMSO, not exceeding 0.1%). 20μL of 5mg/mL MTT (3- [4, 5-dimethylthiazol-2-yl]-2, 5 diphenyl tetrazolium bromide) were added to each well. After 4h of culture, the culture medium was removed and the blue formazan crystals that had formed were dissolved in 50μL DMSO. The optical density of the formazan generated from MTT was measured at 490nm using a Microplate reader (Bio-Rad, USA).
Measurement of antioxidant parameters
Cells were grown in 6-well plates (1×105 cells/well) and treated with 0, 1, 10, 50 or 100μM DHEA for 24h, and then exposed to 300μM H2O2 for 8h. After incubation, the cells were then harvested, disrupted ultrasonically in ice and centrifuged at 2500×g for 10min at 4°C. The supernatants were collected and stored -20°C for subsequent analysis. The superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), catalase (CAT), peroxidase (POD) activities and malondialdehyde (MDA) contents were determined using commercially available assay kits (Jiancheng Bioengineering Institute, China) following the manufacturers’ protocols.
Reactive oxygen species content detected
ROS and O2- content were measured with the fluoroprobe 2’,7’-dichlorodihydrofluorescein diacetate (H2DCF-DA) and dihydroethidium (DHE), as previously described [27]. Briefly, cells were incubated with DHEA (0, 1, 10, 50 or 100μM) for 24h and then exposed to 300μM H2O2 for 8h. After incubation, cells were washed and incubated with 5mM H2DCF-DA or 2mM DHE for 30min at 37°C. The cells were washed and collected by centrifugation and suspended in PBS. Fluorescent intensity was measured by a FACSCalibur™ flow cytometry (Becton Dickinson, USA). Hydroxyl radical (•OH) contents were determined spectrophotometrically using commercially available assay kit (Biyuntian Bioengineering Institute, China) following the manufacturer’s protocols.
Assessment of DNA damage by the alkaline comet assay
DNA damage in Leydig cells was evaluated using a Trevigen Comet AssayTM kit (Trevigen Inc., USA) according to the manufacturer’s instructions. Briefly, cells were incubated with DHEA (0, 1, 10, 50 or 100μM) for 24h and then exposed to 300μM H2O2 for 8h. After incubation, cells were washed and suspended in cold PBS at 1×105 cells/mL. 50μL cells suspension and 500μL low-melting agarose (1%) were mixed and spread onto the comet slide, another layer of 1% agarose gel was then added. The slide was immersed in lysis buffer (2.5M NaCl, 100mM EDTA, 10mM Tris-base, 1% sodium lauryl sarcosinate, 1% Triton X-100, pH10) for at least 60 min at 4°C. Denaturation was performed in an alkali solution (300mM NaOH, 1mM EDTA, pH13.5) for 20min in the dark. The slide was then transferred to fresh alkaline (300mM NaOH, 1mM EDTA, pH13.5) and subjected to electrophoresis at 1V/cm, 300mA for 40min in darkness at 4°C. Thereafter, the slides were washed with neutralization buffer (0.4M Tris-HCl, pH7.4) and immersed in ice cold 100% ethanol for 5min and air-dried. DNA was stained with 50mL of SYBR dye for 20min in the refrigerator and immediately analyzed using a Nikon epifluorescence microscope. For each slide, 50 randomly chosen comets were analyzed using a Nikon epifluorescence microscope. Fluorescent images of single cells were captured and computed for comet parameters: comet length, tail moment, olive tail moment, head DNA and tail DNA, using the Biolab Comet v1.0 image analysis software.
Annexin V/propidium iondine staining assay
Cells undergoing apoptosis were determined using Annexin V-FITC and propidium iodine (PI) dual staining and measured by flow cytometry. Briefly, cells were incubated with DHEA (0, 1, 10, 50 or 100μM) for 24h and then exposed to 300μM H2O2 for 8h. After incubation, the cells were harvested and washed with cold phosphate buffered saline. 200μL of Annexin V-FITC stock solution was added to the cells and incubation continued for 30min at 4°C in the dark. This was followed by a further incubation with the propidium iodide solution (10μL, containing RNase). The cells were then immediately detected by FACSCalibur™ flow cytometry (Becton Dickinson, USA) to measure the cell apoptosis. Approximately 10,000 cells were analyzed in each sample.
Assay of apoptosis factors mRNA level by Real-time quantitative PCR
After treatment, total RNA was extracted from cells using the Trizol reagent (TaKaRa, Japan), according to the manufacturer’s protocol. The RNA concentration was determined by measuring the absorbance at 260nm (Eppendorf Biophotometer, Germany). 2μg of total RNA were reverse transcribed by incubation for 1h at 37◦C in a 25μL mixture comprising of 100U M-KGV reverse transcriptase, 8U RNase inhibitor, 0.5μg of oligo (dT), 50mM Tris-HCl (pH 8.3), 3mM MgCl2, 75mM KCl, 10mM DDT and 0.8mM dNTP. An aliquot of cDNA sample was mixed with 25μL SYBR Green PCR Master Mix (TaKaRa, Japan), in the presence of 10pmol of forward and reverse primer for β-actin (use as an internal control), Bax, Bcl-2, capase-9 and capase-3 (Table 1). All samples were analyzed in duplicate using an ABI Prism 7300 Sequence Detection System (Applied Biosystems, Sweden) using a program of 95°C for 1min, followed by 35 cycles 95°C for 30s, 60°C for 30s and 72°C for 20s. Fold changes were calculated using the 2-ΔΔCT method. The primers were designed using Primes Premier 5 and synthesized by Takara Biotechnology Co. Ltd (Dalian, China).
Statistical analyses
Data were expressed as means ± SD and differences were considered significant at P < 0.05. The effect of DHEA on testosterone and the cell viability induced by H2O2 were analyzed by t-test. Other data were analyzed with one-way ANOVA and treatment differences were subjected to a Duncan’s multiple comparison tests. All statistical analyses were performed with SPSS 13.0 for Windows (StatSoft, Inc., Tulsa, OK, USA).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31572483), the Natural Science Foundation of Jiangsu Province (No. BK20151434) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflicts of interest
None of the authors declare competing financial interests.
Author contribution
Conceived and designed the experiments: Haitian Ma. Performed the experiments: Xiao Ding. Analyzed the data: Lei Yu. Contributed reagents/materials/analysis tools: Chongyang Ge. Wrote the paper: Xiao Ding and Lei Yu.
References
1. Genazzani AD, Lanzoni C and Genazzani AR. Might DHEA be considered a beneficial replacement therapy in the elderly? Drugs & Aging. 2007; 24(3):173-185.
2. Baulieu EE, Thomas G, Legrain S, Lahlou N, Roger M, Debuire B, Faucounau V, Girard L, Hervy MP, Latour F, Leaud MC, Mokrane A, Pitti-Ferrandi H, Trivalle C, de Lacharriere O, Nouveau S, et al. Dehydroepiandrosterone (DHEA), DHEA sulfate, and aging: Contribution of the DHEAge Study to a sociobiomedical issue. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97(8):4279-4284.
3. Kurita H, Maeshima H, Kida S, Matsuzaka H, Shimano T, Nakano Y, Baba H, Suzuki T and Arai H. Serum dehydroepiandrosterone (DHEA) and DHEA-sulfate (S) levels in medicated patients with major depressive disorder compared with controls. Journal of Affective Disorders. 2013; 146(2):205-212.
4. Perret M and Aujard F. Aging and season affect plasma dehydroepiandrosterone sulfate (DHEA-S) levels in a primate. Experimental Gerontology. 2005; 40(7):582-587.
5. Rice SPL, Zhang L, Grennan-Jones F, Agarwal N, Lewis MD, Rees DA and Ludgate M. Dehydroepiandrosterone (DHEA) treatment in vitro inhibits adipogenesis in human omental but not subcutaneous adipose tissue. Molecular and Cellular Endocrinology. 2010; 320(1-2):51-57.
6. Sato K, Iemitsu M, Aizawa K, Mesaki N, Ajisaka R and Fujita S. DHEA administration and exercise training improves insulin resistance in obese rats. Nutrition & Metabolism. 2012; 9.
7. Savineau JP, Marthan R and de la Roque ED. Role of DHEA in cardiovascular diseases. Biochem Pharmacol. 2013; 85(6):718-726.
8. Huerta-Garcia E, Luis Ventura-Gallegos J, Crescencio Victoriano ME, Montiel-Davalos A, Tinoco-Jaramillo G and Lopez-Marure R. Dehydroepiandrosterone inhibits the activation and dysfunction of endothelial cells induced by high glucose concentration. Steroids. 2012; 77(3):233-240.
9. Schwartz AG and Pashko LL. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Research Reviews. 2004; 3(2):171-187.
10. Vianna Metello Jacob MH, Janner DdR, da Rosa Araujo AS, Jahn MP, Rios Kucharski LC, Moraes TB, Dutra Filho CS, Marques Ribeiro MF and Bello-Klein A. Dehydroepiandrosterone improves hepatic antioxidant reserve and stimulates Akt signaling in young and old rats. Journal of Steroid Biochemistry and Molecular Biology. 2011; 127(3-5):331-336.
11. Jacob MHVM, Janner DDR, Belló-Klein A, Llesuy SF and Ribeiro MFM. Dehydroepiandrosterone modulates antioxidant enzymes and Akt signaling in healthy Wistar rat hearts. Journal of Steroid Biochemistry & Molecular Biology. 2008; 112(1-3):138-144.
12. Mastrocola R, Aragno M, Betteto S, Brignardello E, Catalano MG, Danni O and Boccuzzi G. Pro-oxidant effect of dehydroepiandrosterone in rats is mediated by PPAR activation. Life Sciences. 2003; 73(3):289-299.
13. Jacob MHVM, Janner DDR, Jahn MP, Kucharski LC, Belló-Klein A and Ribeiro MFM. Age-related effects of DHEA on peripheral markers of oxidative stress. Cell Biochemistry & Function. 2010; 28(1):52-57.
14. Pélissier MA, Muller C, Hill M and Morfin R. Protection against dextran sodium sulfate-induced colitis by dehydroepiandrosterone and 7α-hydroxy-dehydroepiandrosterone in the rat. Steroids. 2006; 71(3):240-248.
15. Gallo M, Aragno M, V, Tamagno E, Brignardello E, Manti R, Danni O and Boccuzzi G. Protective effect of dehydroepiandrosterone against lipid peroxidation in a human liver cell line. European Journal of Endocrinology. 1999; 141(1):35-39.
16. Tamagno E, Aragno M, Boccuzzi G, Gallo M, Parola S, Fubini B, Poli G and Danni O. Oxygen free radical scavenger properties of dehydroepiandrosterone. Cell Biochemistry & Function. 1998; 16(1):57-63.
17. Yoshioka J, Schreiter ER and Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxidants & Redox Signaling. 2006; 8(11-12):2143-2151.
18. Molassiotis AN, Sotiropoulos T, Tanou G, Kofidis G, Diamantidis G and Therios I. Antioxidant and anatomical responses in shoot culture of the apple rootstock MM 106 treated with NaCl, KCl, mannitol or sorbitol. Biologia Plantarum. 2006; 50(3):331-338.
19. Fico A, Manganelli G, Cigliano L, Bergamo P, Abrescia P, Franceschi C, Martini G and Filosa S. 2-deoxy-d-ribose induces apoptosis by inhibiting the synthesis and increasing the efflux of glutathione. Free Radical Biology and Medicine. 2008; 45(2):211-217.
20. Silva JP, Gomes AC and Coutinho OP. Oxidative DNA damage protection and repair by polyphenolic compounds in PC12 cells. European Journal of Pharmacology. 2008; 601(1-3):50-60.
21. Chen HL, Cangello D, Benson S, Folmer J, Zhu H, Trush MA and Zirkin BR. Age-related increase in mitochondrial superoxide generation in the testosterone-producing cells of Brown Norway rat testes: relationship to reduced steroidogenic function? Experimental Gerontology. 2001; 36(8):1361-1373.
22. Boots AW, Haenen GRMM and Bast A. Health effects of quercetin: From antioxidant to nutraceutical. European Journal of Pharmacology. 2008; 585(2-3):325-337.
23. Murugesan P, Muthusamy T, Balasubramanian K and Arunakaran J. Studies on the protective role of vitamin C and E against polychlorinated biphenyl (Aroclor 1254) - induced oxidative damage in Leydig cells. Free Radical Research. 2005; 39(11):1259-1272.
24. Labrie F, Belanger A, Belanger P, Berube R, Martel C, Cusan L, Gomez J, Candas B, Chaussade V, Castiel I, Deloche C and Leclaire J. Metabolism of DHEA in postmenopausal women following percutaneous administration. Journal of Steroid Biochemistry and Molecular Biology. 2007; 103(2):178-188.
25. Song L, Tang X, Kong Y, Ma H and Zou S. The expression of serum steroid sex hormones and steroidogenic enzymes following intraperitoneal administration of dehydroepiandrosterone (DHEA) in male rats. Steroids. 2010; 75(3):213-218.
26. Aldred LF and Cooke BA. The effect of cell damage on the density and steroidogenic capacity of rat testis Leydig cells, using an NADH exclusion test for determination of viability. Journal of steroid biochemistry. 1983; 18(4):411-414.
27. Lee KE, Kim EY, Kim CS, Choi JS, Bae EH, Ma SK, Park JS, Jung YD, Kim SH, Lee JU and Kim SW. Macrophage-stimulating protein attenuates hydrogen peroxide-induced apoptosis in human renal HK-2 cells. European Journal of Pharmacology. 2013; 715(1-3):304-311.
28. Yang B, Oo TN and Rizzo V. Lipid rafts mediate H2O2 prosurvival effects in cultured endothelial cells. Faseb Journal. 2006; 20(9):1501-1503.
29. Simon HU, Haj-Yehia A and Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000; 5(5):415-418.
30. Buttke TM and Sandstrom PA. Oxidative stress as a mediator of apoptosis. Immunology Today. 1994; 15(1):7-10.
31. Kehrer JP. Cause-effect of oxidative stress and apoptosis. Teratology. 2000; 62(4):235-236.
32. Simic MG, Bergtold DS and Karam LR. Generation of oxy radicals in biosystems. Mutation Research. 1989; 214(1):3-12.
33. Xu S, Hu B, He Z, Ma F, Feng J, Shen W and Yang J. Enhancement of Salinity Tolerance during Rice Seed Germination by Presoaking with Hemoglobin. International Journal of Molecular Sciences. 2011; 12(4):2488-2501.
34. Wang Y, He W, Huang H, An L, Wang D and Zhang F. Antioxidative responses to different altitudes in leaves of alpine plant Polygonum viviparum in summer. Acta Physiologiae Plantarum. 2009; 31(4):839-848.
35. Mohan PF and Jacobson MS. Inhibition of macrophage superoxide generation by dehydroepiandrosterone. American Journal of the Medical Sciences. 1993; 306(1):10-15.
36. Aragno M, Parola S, Brignardello E, Manti R, Betteto S, Tamagno E, Danni O and Boccuzzi G. Oxidative stress and eicosanoids in the kidneys of hyperglycemic rats treated with dehydroepiandrosterone. Free Radical Biology and Medicine. 2001; 31(8):935-942.
37. Danial NN. BCL-2 family proteins: Critical checkpoints of apoptotic cell death. Clinical Cancer Research. 2007; 13(24):7254-7263.
38. Chipuk JE and Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends in Cell Biology. 2008; 18(4):157-164.
39. Kinnally KW and Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007; 12(5):857-868.
40. Antignani A and Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Current Opinion in Cell Biology. 2006; 18(6):685-689.
41. Dejean LM, Martinez-Caballero S and Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death and Differentiation. 2006; 13(8):1387-1395.
42. Er E, Oliver L, Cartron P-F, Juin P, Manon S and Vallette FM. Mitochondria as the target of the pro-apoptotic protein Bax. Biochimica Et Biophysica Acta-Bioenergetics. 2006; 1757(9-10):1301-1311.
43. Kroemer G, Galluzzi L and Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological Reviews. 2007; 87(1):99-163.
44. Leber B, Lin J and Andrews DW. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007; 12(5):897-911.
45. Johnson CR and Jarvis WD. Caspase-9 regulation: An update. Apoptosis. 2004; 9(4):423-427.
46. Porter AG and Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death and Differentiation. 1999; 6(2):99-104.
47. Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ and Akey CW. The Holo-Apoptosome: Activation of Procaspase-9 and Interactions with Caspase-3. Structure. 2011; 19(8):1084-1096.