
INTRODUCTION
Gliomas are the most common type of primary brain tumor and are often fast growing with a poor prognosis for patient [1]. The World Health Organization (WHO) classified gliomas in 2016 using molecular parameters in addition to their histological and immunohistochemical resemblance to presumed cells of origin and graded them by increasing degrees of undifferentiation, anaplasia, and aggressiveness (i.e., mitotic figures, necrosis, and vascular endothelial hyperplasia) [2, 3]. High-grade gliomas represent 60-75% of all cases and include grade III anaplastic astrocytoma, anaplastic oligodendroglioma, mixed anaplastic oligoastrocytoma, and grade IV glioblastoma [4]. Glioblastoma (GBM) is the most common malignant primary brain tumor and is one of the most lethal human cancers [2, 5]. Recently, GBM was classified in three groups: (1) GBM, IDH-wildtype (about 90% of cases, generally corresponds to the clinical definition of primary GBM and is prevalent in patients over 55 years); (2) GBM, IDH-mutant (about 10 % of cases, corresponds closely to so-called secondary GBM and preferentially arises in younger patients); and (3) GBM, NOS (a diagnosis that is reserved for those tumors for which full IDH evaluation cannot be performed) [3]. In United States, GBM accounts for 15.1% of all primary brain tumors, 46.1% of primary malignant brain tumors, and its annual incidence is 3.2 per 100,000 people (or 10,787 new cases diagnosed per year) [6]. Even though GBM may develop at any age, it is more common in elderly with a higher incidence rate in ages between 75 to 84 years (15.24 new cases per 100,000 people between these ages per year). In addition, GBM is 1.6 times more common in males and its incidence rate is 2 times higher among caucasians [6].
Regarding histology, GBM is characterized by considerable cellularity and mitotic activity, vascular proliferation, and necrosis [7]. Because GBM cells vary in size and shape, i.e., they are pleomorphic, this glioma was frequently called glioblastoma multiforme, a term no longer in use. From a molecular point of view, GBM is a highly heterogeneous tumor [8]. Genome-wide expression studies have revealed 4 transcriptional subclasses of GBM, displaying features reminiscent of distinct cell types: classical, mesenchymal, proneural, and neural [9, 10]. Classical subclass typically displays chromosome 7 amplifications, chromosome 10 deletions, EGFR amplification, EGFR mutations, and Ink4a/ARF locus deletion. Mesenchymal subclass displays a high frequency of NF1 mutation/deletion, high expression of CHI3L1, MET, and genes involved in tumor necrosis factor and nuclear factor-κB (NF-κB) pathways. Proneural GBM is characterized by alterations of PDGFRA and mutations in IDH1 and TP53, sharing gene expression features with low-grade gliomas and secondary GBM (i.e., low-grade gliomas later recurred as GBM). Neural subclass is characterized by expression of neuronal markers. Many molecular abnormalities and mutations overlap across transcriptional subclasses, for example PTEN loss, and a large number of very rare mutations have been described [11, 12].
Although GBM is typically confined to Central Nervous System (CNS) and rarely performing metastases in distant organs, this and other malignant gliomas are highly invasive, infiltrating surrounding brain parenchyma [5]. After initial diagnosis, standard treatment for GBM consists of maximal surgical resection [13, 14]. This practice aims to relieve mass effect, achieve cytoreduction, and provide adequate tissue for histologic and molecular tumor characterization. Although surgical resection can greatly reduce tumor bulk, complete tumor excision is frequently not reached due to infiltrative nature of GBM cells [15]. After surgical resection, adjuvant radiotherapy combined with chemotherapy should be considered for all patients. A radiotherapy dose of 60 Gy is frequently used [13]. In addition, the DNA alkylating agent named temozolomide (TMZ) is orally administered as first-line chemotherapy [5, 16]. This regimen is supported by a randomized phase III study [17], which demonstrated TMZ increased median survival to 15 months versus 12 months with radiotherapy alone (hazard ratio - HR = 0.63; P < .001). Two-year survival rate was also increased: 27% for chemotherapy plus radiotherapy versus 10% for radiotherapy alone [17]. Alternatively, biodegradable polymers containing the alkylating agent carmustine (BCNU) can be implanted into tumor bed after surgical resection. Nevertheless, a phase III trial has indicated a modest survival benefit of this regimen [18]. A humanized vascular endothelial growth factor (VEGF) monoclonal antibody named bevacizumab had been recently introduced as first-line monotherapy for progressive GBM [19]. Approval of bevacizumab by U.S. Food and Drug Administration was based on improvement of radiologic response rates observed in two single-arm or noncomparative phase II trials [20, 21]. However, two recent multicenter, phase III, randomized, double-blind, placebo-controlled trials [22, 23], have demonstrated bevacizumab increased median progression-free survival (10.6 vs. 6.2 months, HR: 0.64, p < 0.0001 [22]; 10.7 vs. 7.3 months, HR: 0.79, p = 0.004 [23]) but not overall survival of patients (16-17 months).
Although radiotherapy and chemotherapy improve patient’s survival, GBM remains among the most lethal and resistant malignant tumor [2, 24], and recurrence is nearly universal after a median progression-free survival of 7 to 10 months [25]. Thus, development of new therapies targeting surface molecules or signaling pathways that specifically regulate GBM proliferation or differentiation seems necessary.
In this context, in the present review we summarized the recent evidences demonstrating the participation of mGluR-mediated signaling pathways in GBM proliferation and differentiation, highlighting the putative role of these receptors as new molecular target for management and treatment of this neoplasia.
GLUTAMATE AS A GROWTH FACTOR FOR GLIOBLASTOMA
Several in vitro and in vivo studies have demonstrated GBM cells can release high levels of glutamate (L-Glu) to extracellular fluid. Released L-Glu may act as a neurotrophic factor, promoting proliferation and migration of glioma cells as well as contributing to tumor malignancy [26-28]. L-Glu autocrine secretion occurs mainly by cystine-glutamate antiporter (xCT), which exchanges extracellular cystine (Cys) for intracellular L-Glu at a 1:1 stoichiometric ratio [27, 29] (Figure 1, step 1). Moreover, due to loss of excitatory amino acid transporter 2 (EAAT2), GBM cells possess a low re-uptake rate of L-Glu from extracellular fluid, which keeps this aminoacid at a high concentration in extracellular fluid and increases tumor malignancy [27, 30] (Figure 1, step 2). Furthermore, higher levels of L-Glu can trigger a mechanism of neuronal cell death called excitotoxicity [31], which facilitates tumor bulk expansion [27, 32-34] (Figure 1, step 3).
Extracellular L-Glu activates two classes of membrane receptors: ionotropic glutamate receptors (iGluR: AMPA, NMDA and Kainate receptors), which are ligand-gated ion channels, and metabotropic glutamate receptors (mGluR), which are coupled to G proteins [35, 36]. mGluR family comprises eight subtypes subdivided in three groups according to their sequence homology, pharmacology, and associated-signaling pathway. Group I mGluR are coupled to Gq proteins and their activation stimulates phospholipase C (PLC) and phosphatidylinositol 4,5-biphosphate (PIP2) hydrolysis. PIP2 hydrolysis generates inositol (1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG), which stimulates intracellular Ca2+ release from endoplasmic reticulum and activates protein kinase C (PKC), respectively [33]. In contrast, mGluR of group II and III are coupled predominantly to Gi/o proteins, inhibiting adenylyl cyclase (AC) and, thus, decreasing ion channel activity and other downstream signaling pathways [37, 38] (Figure 2). Beyond the well-established role of glutamate receptors in glutamatergic neurotransmission, several evidences are emerging regarding the role of these receptors in cancer biology, especially in malignant brain tumors [39-41].
Activation of a mutated Ca+2-permeable form of AMPA receptors (AMPAR) enhanced migration and proliferation of high-grade gliomas (Figure 1, step 4). Blockage of AMPAR by NBQX led to inhibition of glioma growth and induced apoptosis of remaining cells [42, 43]. AMPAR-mediated tumor proliferation seemed to involve a Ca2+-dependent activation of Akt/PKB signaling pathway [44], since both NBQX (AMPAR antagonist) and Wortmannin (specific inhibitor of PI3K) reduced the Akt/PKB phosphorylation and decreased the number of tumoral viable cells in culture [45].
Parallel to iGluR, several evidences have demonstrated that mGluR are also functionally important for proliferation and differentiation of distinct types of cancer, including GBM [46]. Group III mGluR is involved in malignancy of a lot of cancers [47]. The implication of mGluR7 in tumor formation has yet to be characterized [46]. mGluR6 gene expression was shown to correlate with higher-grade pediatric CNS tumors [48]. Increased expression of mGluR4 and mGluR8 was reported in human lung adenocarcinoma samples and lung carcinoma cell line and treatment with mGluR8 agonist reduced cell growth and increased apoptosis in this lineage [49]. mGluR4 is overexpressed in more than 40% of malignant melanomas, laryngeal squamous cell carcinomas, and breast carcinomas and its overexpression was correlated with increased mortality in colorectal carcinoma [50]. mGluR4 inhibition suppressed proliferation of mGluR4-expressing colon cancer cell lines [50] whereas mGluR4 activation reduced cell proliferation in medulloblastoma cell lines and inhibits medulloblastoma cell xenografts progression in nude mice [51]. In addition, approximately 77% of human medulloblastoma samples expressed mGluR4, which was inversely correlated with tumor severity, spread, and recurrence [51].
Among Group I mGluR, increased mGluR5 immunoreactivity in human oral squamous cell carcinomas is associated with improved overall survival [52]. mGluR5 antagonist reduced tumor cell migration, invasion, and adhesion in human tongue cancer cells [52] and inhibits cell proliferation of laryngeal cancer [53]. Additionally, mGluR5 overexpression has been shown to induce melanoma development in transgenic mice [54]. Ectopic expression of mGluR1 in normal melanocytes induced melanocyte hyperproliferation in vitro and promote melanoma tumor development in vivo [55-59]. mGluR1 expression has been widely explored in breast cancer, supporting angiogenesis in these tumors, and silencing of this receptor (GRM1 shRNA) resulted in inhibition of cell proliferation [60]. In addition, mutations and single nucleotide polymorphisms (SNPs) of GRM1 were described in prostate cancer [61] and eight somatic variations of GRM1 were identified in cancers, including lung adenocarcinoma [62]. Group II mGluR is also implicated in a variety of cancer types [47], including melanoma [63]. Both group I and II mGluR are involved in glioma progression and the action in vitro and in vivo of agonists and antagonists of these receptors will be reported in detail in this review.
The above-mentioned evidences indicate that group I and III mGluR may be considered a potential therapeutic target for both gliomas and other forms of cancer. Moreover, from physiological and pharmacological point of view, there is a growing number of evidences suggesting mGluR are better drug targets than iGluR [64-67]. Compared to iGluR, mGluR play a ‘modulatory’ rather than ‘mediatory’ role in glutamatergic excitatory synaptic transmission [68-71]. Consequently, mGluR ligands (such as agonists or antagonists) might lead to more subtle effects on fast excitatory transmission than iGluR antagonists, which indicates their therapeutic use may be more tolerable for patients [64, 65].
IN VITRO STUDIES EVALUATING THE ROLE OF mGluR ON GLIOMA PROLIFERATION
Glioma cell cultures have been widely used to elucidate the role of mGluR in cancer malignancy. Some studies have used GBM lineages as cellular model [68, 72-75] while others have used primary cultures from human GBM resections [68, 76-79]. Table 1 (A, B and C) and Table 2 summarizes current literature evaluating mRNA expression and protein immunocontent of mGluR, respectively, in glioma cultures and human glioma resections. Group II mGluR (mGluR2/3) was the most investigated (expressed - mRNA - and immunodetected - protein) in the majority of human glioma biopsy samples, primary cultures, and glioma lineages. In order to clarify the role of mGluR on proliferation, invasiveness, and migration, several assays were performed treating GBM cells with antagonists and/or agonists of these receptors.
Table 1A: RNA expression of group I mGluR subtypes in cellular malignant glioma models.
Metabotropic glutamate receptors |
mRNA |
||||
Sample |
Expression |
Reference |
|||
Group I |
mGluR1 |
Human Glioma Lineage |
U-87 MG |
Yes |
[72] |
No |
[73] |
||||
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
Resection |
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
||
AZ8 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ3 (Low grade astrocytoma) |
No |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB4 (Glioblastoma) |
Yes |
[48] |
|||
GB3 (Glioblastoma) |
Yes |
[48] |
|||
GB2 (Glioblastoma) |
Yes |
[48] |
|||
GB1 (Glioblastoma) |
No |
[48] |
|||
mGluR5 |
Human Glioma Lineage |
U-87 MG |
Yes |
[72] |
|
No |
|||||
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
U-178 MG |
Yes |
[74] |
|||
U-251 |
No |
[68] |
|||
Primary culture from human glioma |
FCN-9 |
No |
[68] |
||
MZC-12 |
No |
[68] |
|||
CDR-97 |
No |
[68] |
|||
Resection |
Anaplastic astrocytoma (52 years old) |
No |
[79] |
||
Astrocytoma grade II (12 years old) |
Yes |
[79] |
|||
Glioblastoma (53 years old) |
No |
[79] |
|||
Glioblastoma (68 years old) |
No |
[79] |
|||
Glioblastoma (82 years old) |
No |
[79] |
|||
Glioblastoma (58 years old) |
No |
[79] |
|||
Glioblastoma (55 years old) |
No |
[79] |
|||
Astrocytoma grade I (38 years old) |
No |
[79] |
|||
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ8 (Low grade astrocytoma) |
No |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
No |
[48] |
|||
AZ3 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB4 (Glioblastoma) |
Yes |
[48] |
|||
GB3 (Glioblastoma) |
Yes |
[48] |
|||
GB2 (Glioblastoma) |
Yes |
[48] |
|||
GB1 (Glioblastoma) |
Yes |
[48] |
|||
Table 1B: RNA expression of group II mGluR subtypes in cellular malignant glioma models.
Metabotropic glutamate receptors |
mRNA |
||||
Sample |
Expression |
Reference |
|||
Group II |
mGluR2 |
Human Glioma Lineage |
U-87 MG |
Yes |
|
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
U-251 |
Yes |
[68] |
|||
Primary culture from human glioma |
FCN-9 |
No |
[68] |
||
MZC-12 and MSS-5 |
Yes |
[68] |
|||
FLS-10 |
No |
[68] |
|||
LTN-12 |
No |
[68] |
|||
BRT-3 |
No |
[68] |
|||
CRL-8 |
No |
[68] |
|||
CDR-97 |
No |
[68] |
|||
Glioma Stem Cells |
No |
||||
Resection |
Glioblastoma |
Yes |
[78] |
||
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ8 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ3 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB1, GB2, GB3 and GB4 (Glioblastomas) |
Yes |
[48] |
|||
mGluR3 |
Human Glioma Lineage |
U-87 MG |
Yes |
||
U-343 |
No |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
U-251 |
Yes |
[68] |
|||
Primary culture from human glioma |
FCN-9 |
Yes |
[68] |
||
MZC-12 |
Yes |
[68] |
|||
FLS-10, LTN-12 and CDR-97 |
No |
[68] |
|||
MSS-5 |
Yes |
[68] |
|||
BRT-3 |
Yes |
[68] |
|||
CRL-8 |
Yes |
[68] |
|||
Glioma Stem Cells |
Yes |
||||
Resection |
Glioblastoma |
Yes |
[78] |
||
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ8 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ3 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB1, GB3 and GB4 (Glioblastomas) |
Yes |
[48] |
|||
GB2 (Glioblastoma) |
No |
[48] |
|||
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
Resection |
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
||
AZ8 (Low grade astrocytoma) |
No |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ3 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB1, GB3 and GB4 (Glioblastomas) |
Yes |
[48] |
|||
GB2 (Glioblastoma) |
No |
[48] |
|||
Table 1C: RNA expression of group III mGluR subtypes in cellular malignant glioma models.
Metabotropic glutamate receptors |
mRNA |
||||
Sample |
Expression |
Reference |
|||
Group III |
mGluR4 |
Human Glioma Lineage |
U-87 MG |
No |
|
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
Resection |
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
||
AZ8 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
No |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ3 (Low grade astrocytoma) |
No |
[48] |
|||
AZ2 (Low grade astrocytoma) |
No |
[48] |
|||
GB1, GB2, GB3 and GB4 (Glioblastoma) |
Yes |
[48] |
|||
mGluR6 |
Human Glioma Lineage |
U-87 MG |
Yes |
[72] |
|
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
Resection |
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
||
AZ8 (Low grade astrocytoma) |
No |
[48] |
|||
AZ7 (Low grade astrocytoma) |
No |
[48] |
|||
AZ6 (Low grade astrocytoma) |
No |
[48] |
|||
AZ5 (Low grade astrocytoma) |
No |
[48] |
|||
AZ4 (Low grade astrocytoma) |
No |
[48] |
|||
AZ3 (Low grade astrocytoma) |
No |
[48] |
|||
AZ2 (Low grade astrocytoma) |
No |
[48] |
|||
GB4 (Glioblastoma) |
Yes |
[48] |
|||
GB3 (Glioblastoma) |
Yes |
[48] |
|||
GB2 (Glioblastoma) |
Yes |
[48] |
|||
GB1 (Glioblastoma) |
No |
[48] |
|||
mGluR7 |
Human Glioma Lineage |
U-87 MG |
Yes |
[72] |
|
No |
[73] |
||||
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
Resection |
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
||
AZ8 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
No |
[48] |
|||
AZ3 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB1, GB2, GB3 and GB4 (Glioblastoma) |
Yes |
[48] |
|||
mGluR8 |
Human Glioma Lineage |
U-87 MG |
Yes |
[72] |
|
U-343 |
Yes |
[72] |
|||
MOGGCCM |
Yes |
[72] |
|||
Resection |
AZ21 (Low grade astrocytoma) |
Yes |
[48] |
||
AZ8 (Low grade astrocytoma) |
No |
[48] |
|||
AZ7 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ6 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ5 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ4 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ3 (Low grade astrocytoma) |
Yes |
[48] |
|||
AZ2 (Low grade astrocytoma) |
Yes |
[48] |
|||
GB1, GB3 and GB4 (Glioblastoma) |
Yes |
[48] |
|||
GB2 (Glioblastoma) |
No |
[48] |
|||
Arcella et al. (2005) showed pharmacological blockade of mGluR2/3 induced antiproliferative effects in U-87 MG glioma cell line. Daily addition (four days) of mGluR2/3 antagonists (LY 341495 - 1 µM; MTPG - 100 µM; or EGLU - 100 µM) to U-87 MG cultures reduced cell proliferation in a time-dependent manner, while did not induce apoptosis [73] (Figure 1, step 5). However, this treatment altered cell cycle, since FACS analysis showed antagonist reduced percentage of cells in S and G2M phases after two but not after four days of exposure. Furthermore, in cultures deprived of serum by 72 h, LY 341495 treatment reduced EGF-induced cyclin D1/D2 protein expression (early marker of the G1/S phase transition [79]).
In another study, using primary cultures from human GBM biopsies, D’Onofrio et al. (2003) showed pharmacological blockade of mGluR3 reduced cell proliferation [68]. This effect was observed in all selected cultures mGluR3+. Application of antagonist LY 341495 (1 µM) to growing medium once a day (for four days) reduced linear phase of growth in mGluR3+ cultures, with cell number being substantially reduced at 4th day of treatment. Cell growth was restored two days after washing out LY 341495, indicating that antiproliferative effect was reversible (i.e., antagonist was cytostatic, not cytotoxic). For excluding involvement of other mGluR, authors performed an additional experiment using lower concentrations of LY 341495 and this antagonist was able to reduce linearly cell growth at concentrations of 1 and 10 nM, indicating that inhibition of mGluR3 was responsible for antiproliferative effect of LY 341495. This result was further corroborated by evidence that EGLU (100 µM), another antagonist of mGluR3, mimicked LY 341495 action on cell growth.
Glioma stem cells (GSC) are brain tumor-initiating undifferentiated cells [80], which have multipotential differentiation capacity, high tumorigenic potential and low proliferation rate [81, 82]. Normally, GSC are obtained from adult human GBM biopsy samples and form in culture classical floating aggregates, named tumor spheres. These cells are very chemoresistant and radioresistant and therefore probably responsible for tumor progression and recurrence after conventional GBM resection [82]. Ciceroni et al. (2008) and Zhou et al. (2014) observed GSC express mGluR3 protein (but not mGluR2) and its pharmacological blockade promoted an astroglial differentiation of GSC [76, 78]. Zhou et al. (2014) showed mGluR3 blockade by LY 341495 (100 nM) for 48 h resulted in a decreased proliferation combined with an increase in GSC GFAP+ cells (classical marker for mature astrocytes) and a decrease of GSC nestin+ cells (classical marker for neural stem cells) [78]. In addition, stimulation of mGluR3 for 48 h by specific agonist LY 379268 (100 nM) has no effect on GSC proliferation and differentiation, which suggests mGluR3 activity is necessary to maintain proliferation but is incapable of stimulating it per se (Figure 1, step 5).
Ciceroni et al. (2008) showed treatment of GSC with antagonist LY 341495 (100 nM) promoted astrocytic-like differentiation of cells (increasing GFAP+ cells) and nestin+ cells were virtually absent after 14 days of treatment [76]. Treatment with agonist LY 379268 (100 nM) did not affect differentiation of cells. In another set of experiments, these authors cultured GSC under differentiating conditions (medium deprived of mitogens and containing 10% of fetal calf serum) for 8 days and received treatments with mGluR3 ligants. After, cells were enzymatically dissociated, transferred to uncoated 96-well plates, and regrown under proliferating conditions without any mGluR3 ligants (medium containing epidermal growth factor - EGF - plus basic fibroblast growth factor - bFGF - and lacking serum) for plus 12 days. Treatment of cells with agonist LY 379268 during differentiation phase led to formation of tumor spheres in proliferative phase. In contrast, cells treated with antagonist LY 341495 originated cultures containing exclusively adherent astrocyte-like cells. This indicated that treatment with mGluR3 antagonist was sufficient to maintain GSC towards astroglial differentiation even under conditions in which they normally proliferate and maintain their undifferentiated state (Figure 1, step 5).
Table 2: Protein immunocontent of mGluR subtypes in cellular malignant glioma models.
Metabotropic glutamate receptors |
Protein (Western Blot) |
||||
Sample |
Expression |
Reference |
|||
Group I |
mGluR1 |
Human Glioma Lineage |
U-87 MG |
Yes |
[41] |
U-373 |
No |
[75] |
|||
Resection |
Astrocytoma II |
No |
[75] |
||
Astrocytoma Anaplastic |
No |
[75] |
|||
Glioblastoma |
No |
[75] |
|||
mGluR5 |
Human Glioma Lineage |
U-87 MG |
Yes |
||
U-373 |
Yes |
[75] |
|||
U-118 |
Yes |
[75] |
|||
Resection |
Astrocytoma II |
Yes |
[75] |
||
Astrocytoma Anaplastic |
Yes |
[75] |
|||
Glioblastoma |
Yes |
[75] |
|||
Group II |
mGluR2/3 |
Human Glioma Lineage |
U-87 MG |
Yes |
[68] |
U-373 |
No |
[68] |
|||
A172 |
Yes |
[68] |
|||
Primary culture from human glioma |
FCN-9 |
Yes |
[68] |
||
MZC-12 |
Yes |
[68] |
|||
MSS-5 |
Yes |
[68] |
|||
BRT-3 |
Yes |
[68] |
|||
CRL-8 |
Yes |
[68] |
|||
GSS 98 |
Yes |
[68] |
|||
DMD 126 |
Yes |
[68] |
|||
MTR4 |
Yes |
[68] |
|||
Glioma Stem Cells |
Yes |
||||
Resection |
Astrocytoma II |
Yes |
[75] |
||
Astrocytoma Anaplastic |
Yes |
[75] |
|||
Glioblastoma |
Yes |
[75] |
|||
mGluR2 |
Primary culture from human glioma |
Glioma Stem Cells |
No |
[78] |
|
Resection |
Glioblastoma |
Yes |
[78] |
||
mGluR3 |
Primary culture from human glioma |
Glioma Stem Cells |
Yes |
[78] |
|
Resection |
Glioblastoma |
Yes |
[78] |
||
For both above-mentioned studies [76, 78], GSC did not respond to agonist LY 379268 probably because mGluR3 were constantly stimulated by L-Glu already present in culture medium. Concentrations of extracellular L-Glu in GSC cultured for 8 and 14 days under differentiating conditions (medium deprived of mitogens and containing serum) were about 60 µM and 70 µM, respectively [76]. These concentrations exceed the reported EC50 value for L-Glu and recombinant mGluR3 by 5-10 fold [83], which is sufficient to saturate all mGluR3.These results point to the notion that activation of mGluR3 by endogenous L-Glu allows proliferation of GSC by limiting astroglial differentiation and that receptor blockage eliminates this constraint, thereby promoting cell differentiation. This hypothesis is supported by work performed by Yelskaya et al. (2013), in which U-87 MG cultures where treated with Riluzole (1-100 µM), a drug that blocks the secretion of L-Glu and enhances its uptake from extracellular space [47]. Riluzole inhibited proliferation of cells in a dose-dependent manner, suggesting that absence of L-Glu in extracellular medium prevents glutamate-dependent proliferation and putative activation of mGluR3 in U-87 MG cells [84].
Group II mGluR are known to be able to activate the MAPK and PI3K pathways [37, 85-89], which are usually activated in response to proliferating agents [90, 91]. Arcella et al. (2005) [73] showed treatment of U-87 MG cultures with antagonist LY 341495 (1 µM) reduced activation of MAPK (assessed by WB analysis of p-Erk1/2) and PI3K (assessed by WB analysis of p-Akt/PKB) pathways. All of these effects were reversed by addition of agonist LY 379268 (1 µM), which was inactive per se. WB analysis also showed exposure to LY 341495 did not alter mGluR2/3 immunocontent (at least up to four days of treatment). Another study indicated that activation of mGluR3 could have a permissive role on stimulation of MAPK and PI3K pathways in GSCs dissociated cultures [77]. GSCs dissociated from tumor spheres were starved from mitogens and then treated with agonist LY 379268 (100 nM), which inhibited forskolin-stimulated cyclic adenosine monophosphate (cAMP) formation and increased p-Erk1/2 and p-Akt/PKB levels. All these effects were reversed by antagonist LY341495 (100 nM). In addition, treatment with LY 341495 also reversed EGF- and bFGF-induced increase in p-Erk1/2 and p-Akt/PKB levels. LY 341495 alone did not affect EGF receptor (EGFR) autophosphorylation in response to EGF, which suggests that this drug had no direct effects on EGFR (Figure 1, step 6, 7, 9 and 10).
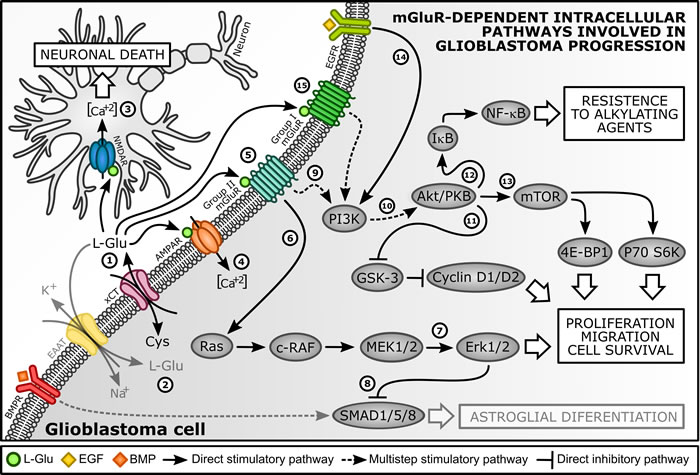
Figure 1: Regulation of GBM proliferative pathways by metabotropic glutamate receptors (mGluR). (1) GBM cells can release low levels of L-Glu mainly by xCT antiporter [27, 28]. (2) Due to loss of EAAT2, GBM cells possessed a low re-uptake rate of L-Glu, maintaining high concentrations of this amino acid in tumor environment [30]. (3) High levels of L-Glu can activate specific NMDAR, which causes neuronal death by excitotoxicity and facilitates tumor bulk expansion [27]. (4) GBM cells expressing mutated Ca+2-permeable AMPAR exhibited enhanced migration and proliferation and its blockade led to inhibition of growth and induction of apoptosis [42]. (5) mGluR3 activation by L-Glu induced GBM proliferation and kept these cells under undifferentiated state. In contrast, mGluR3 inhibition eliminated this constraint and promoted astroglial differentiation [68, 73, 76, 78]. (6) Accordingly to Arcella et al. (2005) [73] and Ciceroni et al. (2013) [77], MAPK axis supported mGluR3-induced GBM proliferation, (7) since mGluR3 stimulation increased Erk1/2 phosphorylation and its blockade reduced p-Erk1/2 levels. mGluR3 inhibition plus MEK1/2 blockade showed an additive antiproliferative effect on GBM cells [68]. (8) Moreover, mGluR3-dependet activation of MAPK pathway limited SMAD1/5/8-induced astroglial differentiation, which kept GBM cells under undifferentiated state. Exogenous SMAD1/5/8 stimulation or MEK inhibition prevented this effect [76]. (9) mGluR3 activation stimulated (10) phosphorylation of Akt/PKB via PI3K activation and this effect was reversed by receptor inhibition [73, 77]. (11) mGluR3-PI3K axis activation presented a permissive role on GBM cell cycle progression, since mGluR3 inhibition by LY341495 decreased cyclin D1/D2 immunocontent [68, 73], an early marker of G1/S phase transition [79]. (12) mGluR3-PI3K axis was also related to GBM chemoresistance. GSC become sensitive to TMZ, an alkylating agent, only if mGluR3 was inhibited or silenced [77], which was also mimicked by PI3K blockade. NF-κB activation by Akt/PKB limits pro-apoptotic activity of alkylating agents in GBM cells [100]. TMZ increased levels of p-IκB and this effect was reversed by mGluR3 or PI3K blockers. NF-κB bockade enabled TMZ toxicity, occluding permissive action of mGluR3 inhibition, indicating NF-κB lies downstream of Akt/PKB in pathway that restrains TMZ toxicity. (13) Akt/PKB also regulates mTOR, which promotes mRNA translation and protein synthesis through p70 S6K and 4E-BP1 phosphorylation [103]. This signaling pathway was showed to support GBM cells survival [106]. (14) Concomitant activation of EGFR and mGluR3 could act synergistically in GBM aggressiveness, since simultaneous inhibition of these receptors caused maximum apoptosis in GBM cells, as well as reduced their migration. (15) mGluR1 stimulation promoted GBM cells survival through PI3K-Akt/PKB-mTOR pathway activation [41]. mGluR1 inhibition markedly decreased cell viability and inhibited PI3K and Akt/PKB phosphorylation. mGluR1 inhibition also decreased levels of p-mTOR and P70 S6K
D’Onofrio et al (2003) examined the immunocontent of cyclin D1 and D2 and the activation of MAPK pathway in primary GBM cell cultures in serum-deprived conditions and in proliferative conditions (cells incubated with EGF for 8 h, for assessment of cyclin D1/D2, or for 10 min, for assessment of MAPK pathway) [68]. Addition of 1 µM of mGluR3 antagonist LY 341495 (in combination with EGF) reduced EGF-induced increase in immunocontent of both cyclin D1/D2 and p-Erk1/2. Effect on cyclin D1/D2 protein expression was partially reversed by agonist LY 379268 (1 µM) (Figure 1, step 11). To assess whether inhibition of MAPK pathway was the only mechanism responsible for antiproliferative effect of LY 341495, authors studied association of this drug with MAPK kinase (MEK) inhibitor, PD 98059. Both LY 341495 (1 µM) and PD 98059 (30 µM) reduced cell number in about 45%. Ability of LY 341495 and PD 98059 to reduce Erk1/2 phosphorylation suggests mGluR2/3-MAPK axis supports proliferation rate of human GBM cells. Since anti-proliferative effect of LY 341495 was not totally obliterated by PD 98059, other proliferation pathways may be also being controlled by mGluR2/3 in GBM cells (Figure 1, step 6 and 7).
Ciceroni et al. (2008) focused on interaction between mGluR3 and bone morphogenetic proteins (BMP), which are known to promote astroglial differentiation of GSC [92]. BMP bind to membrane receptors of BMP/TGF-ß/activin family, which leads to phosphorylation and translocation of Smad1/5/8 proteins to nucleus [93]. Addition of exogenous BMP4 (100 ng/mL) plus mGluR3 agonist LY 379268 (100 nM) prevented BMP4-induced nuclear translocation and phosphorylation of Smad. Inhibitory action of LY 379268 was unaffected by non-metabolizable cAMP analogue, 8-Bromo-cAMP (1 mM), but was prevented by MAPK kinase (MEK) inhibitor, UO126 (30 µM), which was inactive per se. Moreover, cultures exposed to mGluR3 antagonist LY 341495 (100 nM) enhanced Smad1/5/8 phosphorylation to an extent similar to BMP4. These results suggested that activation of mGluR3 could inhibit BMP4 receptor signaling by activation of MAPK pathway. In addition, activation of mGluR3-MAPK pathway by endogenous L-Glu presented in medium may limit BMP4-induced differentiating activity, thus contributing to support undifferentiated state of GSCs, and eventually GBM growth and relapse [76] (Figure 1, step 8).
Yelskaya et al. (2013) reported that a combination of mGluR2/3 antagonist LY 341495 and Gefitinib, an EGFR inhibitor, works most efficiently to inhibit proliferation and migration of U-87 MG cells and induced apoptosis in this cell lineage [84]. They also investigated the efficacy of different classes of drugs (AMPAR antagonist - NBQX; mGluR2/3 antagonist - LY341495; EGFR inhibitor - Gefitinib; and PI3K inhibitors - Wortmannin and PI 828) in inhibiting proliferation of U-87 MG cells (24 h). A combination of Gefitinib (25 µM) with LY 341495 (1 µM) or PI 828 (2 µM) was more effective to inhibit proliferation of U-87 MG cells when compared to individual drugs alone. Using TUNEL assay, authors showed treatment with Gefitinib resulted in increased apoptosis compared to control group. Gefitinib in combination with PI 828 or Wortmannin (5 µM) did not increase apoptosis in cell cultures. However, treatment with Gefitinib plus NBQX (5 µM) or Gefitinib plus LY 341495 increased apoptosis compared to Gefitinib, NBQX or LY 341495 alone. Maximum percentage of apoptosis was observed in treatment with Gefitinib plus LY341495. Distance migrated by cells (wound healing assay) was significantly reduced with Gefitinib plus LY 341495 treatment when compared to treatments with Gefitinib or LY 341495 alone or control group (Figure 1, step 14).
An interesting question that arises from above-mentioned works is whether mGluR3 antagonists could interact with classical chemotherapies and whether activation of this receptor in malignant gliomas could control expression of proteins implicated in chemoresistance. In this context, Ciceroni et al. (2013) showed mGluR3 inhibition enables cytotoxic action of TMZ in GSC cultures [77]. TMZ (250 µM) did not affect cell viability when applied alone, but became toxic when combined with LY 341495 (100 nM) and LY 2389575 (100 nM), two mGluR3 antagonists. mGluR3 agonist LY 379268 (100 nM) was inactive per se, but reversed the permissive action of LY 341495 on TMZ toxicity. siRNA-induced knockdown of mGluR3 also enabled TMZ toxicity and antagonist LY 341495 did not further amplify TMZ toxicity in mGluR3 silenced cells [77]. This data suggests that a possible activation of mGluR3 by GSC-autocrine release of L-Glu could restrain toxic action of TMZ, increasing chemoresistance of these tumor cells (Figure 1, step 12). GSC were treated with other anticancer drugs (etoposide, irinotecan, irinotecan metabolite - SN 38, cisplatin or paclitaxel) alone or combined with mGluR3 antagonist LY341495 and these treatments had no significant effect on GSC viability, suggesting mGluR3 receptors selectively control responses of cells to TMZ and could not be extended to other chemotherapeutic agents.
In order to evaluate mechanisms underlying mGluR3-induced chemoresistance, Ciceroni et al. (2013) treated GSC with molecules that interfere in three major signaling pathways activated by mGluR3 [77]: inhibition of adenylyl cyclase (AC) activity, activation of MAPK pathway and activation of PI3K pathway [85, 88, 94, 95]. Cell permeable cAMP analog, 8-Bromo-cAMP (1 mM), did not affect synergism between mGluR3 blockade and TMZ. MAPK kinase (MEK) inhibitor, UO126 (30 µM), had mild effect on TMZ toxicity. In contrast, PI3K inhibitor LY 294002 (10 µM) had a permissive action on TMZ toxicity, mimicking the effect of mGluR3 blockade. Actions of LY 294002 and LY 341495 were less than addictive, suggesting mGluR3 inhibition facilitates cytotoxicity by limiting activation of PI3K pathway. This hypothesis was supported by the use of GSCs expressing a constitutively active form of t PI3K substrate, Akt/PKB [96]. In these cells, in which PI3K pathway was active in spite of mGluR3 blockade, synergism between LY 341495 and TMZ was largely attenuated (Figure 1, step 12).
The sensitivity of cancer cell lineages to various inhibitors and chemotherapeutic drugs is often associated with genetic mutations of key elements in the Ras-Raf-MEK-ERK and PI3K-PTEN-Akt/PKB-mTOR pathways [97, 98]. Akt/PKB is known to activate nuclear factor-κB (NF-κB) by phosphorylating IκB kinase [99], which limits the pro-apoptotic activity of DNA-alkylating agents in glioma cells [100]. In Ciceroni et al. (2013) study, treatment with TMZ activated NF-κB, showed by increased levels of IκB phosphorylation. This effect was reversed by mGluR3 antagonist, LY 341495, or by PI3K inhibitor, LY 294002. The specific NF-κB inhibitor JSH-23 (10 µM) [101] enabled TMZ toxicity and occluded permissive action of LY 341495 in GSCs. Similar effects were obtained with salicylic acid, which also inhibits NF-κB [102]. As opposed to LY 341495, JSH-23 could still enhance TMZ toxicity in GSCs expressing the constitutively active form of Akt/PKB, indicating NF-κB lies downstream to Akt/PKB in pathway that restrains TMZ toxicity (Figure 1, step 12). Akt/PKB also regulates mammalian target of rapamycin (mTOR), which promotes mRNA translation and protein synthesis by phosphorylating p70 S6K and 4E-BP1 [103]. mGluR stimulation activates mTOR pathway [104, 105] and inhibitors of Akt/PKB-mTOR pathway are under development for treatment of cancer, including malignant gliomas [106, 107]. In Ciceroni et al. (2013) work, it was shown selective mTOR inhibitor rapamycin (10 nM) did not mimic, but rather abolished permissive action of mGluR3 blockade on TMZ toxicity (Figure 1, step 13).
Clinical efficacy of TMZ is limited by DNA-repairing enzyme, O6-methylguanine-DNA methyltransferase (MGMT), which removes DNA adducts generated by alkylating agents [108]. In Ciceroni et al. (2013) [77] study, GSC clones constitutively expressed MGMT and treatment with TMZ alone increased MGMT mRNA levels 3 h after its application and slightly reduced MGMT protein levels at 24 and 48 h. When TMZ was combined with LY 341495, MGMT mRNA did not increase and MGMT protein levels were markedly reduced. Moreover, action of LY341495 was mimicked by siRNA-induced knockdown of mGluR3, or PI3K inhibitor (LY 294002), or by NF-κB inhibitor (JSH-23). Finally, the permissive action of LY 341495, LY 294002, or JSH-23 was no longer seen in GSC overexpressing MGMT, demonstrating that synergistic action of mGluR3 blockade and TMZ treatment was mediated by inhibition of MGMT expression. This hypothesis was supported by evidence that treatment with MGMT inhibitor, O6-benzylguanine (10 µM), enabled TMZ toxicity. These results suggest that mGluR3-dependent TMZ toxicity restrains is supported by induction of MGMT transcription via an intracellular signaling pathway that sequentially involves PI3K and NF-κB.
A recent study described, for the first time, an anti-cancer role of mGluR1 in gliomas [41]. Zhang et al. (2015) showed treatment with selective mGluR1 antagonist Bay 36-7620 (50µM) or Riluzole (50µM), a glutamate release inhibitor approved for amyotrophic lateral sclerosis [47, 109], markedly decreased cell viability and increased LDH release in U-87 MG lineage. These treatments and mGluR1 knockdown (using siRNA technology) significantly increased apoptotic rate in these glioma cells. In addition, Riluzole or Bay 36-7620 increased immunocontent of cleaved PARP and caspase-3, whereas immunocontent of pro-PARP and pro-caspase-3 was not altered. Similar results were observed after mGluR1 knockdown. Authors also tested whether treatment with Riluzole or Bay 36-7620 and transfection with mGluR1 targeted siRNA could induce inhibition of PI3K activity in U87 cells. U-87 MG cells showed a significantly decreased phosphorylation of PI3K after mGluR1 blockade as well as after mGluR1 targeted siRNA. mGluR1-dependent inhibition of PI3K resulted in inhibition of Akt/PKB phosphorylation at both Ser473 and Thr308 residues. Moreover, mGluR1 inhibition decreased expression levels of p-mTOR and P70 S6K, one of the best-characterized targets of mTOR complex. In contrast, expression of PTEN was not changed by antagonists or siRNA transfection. All these data provided strong evidence that activation of PI3K-Akt/PKB-mTOR pathway was suppressed after mGluR1 inhibition (Figure 1, step 15).
The majority of above-mentioned in vitro works indicates that endogenous activation of mGluR1 and mGluR3 increase the proliferation of GBM cells and that MAPK and PI3K pathways may be involved in this process. All of these studies are particularly interesting from a therapeutic standpoint since ligands of these types of mGluR represent an expanding class of drugs endowed with high receptor affinity, elevated brain penetration, and good profile of safety and tolerability [65, 83]. These features make these drugs potential candidates for in vivo studies evaluating progression and aggressiveness of malignant brain tumors.
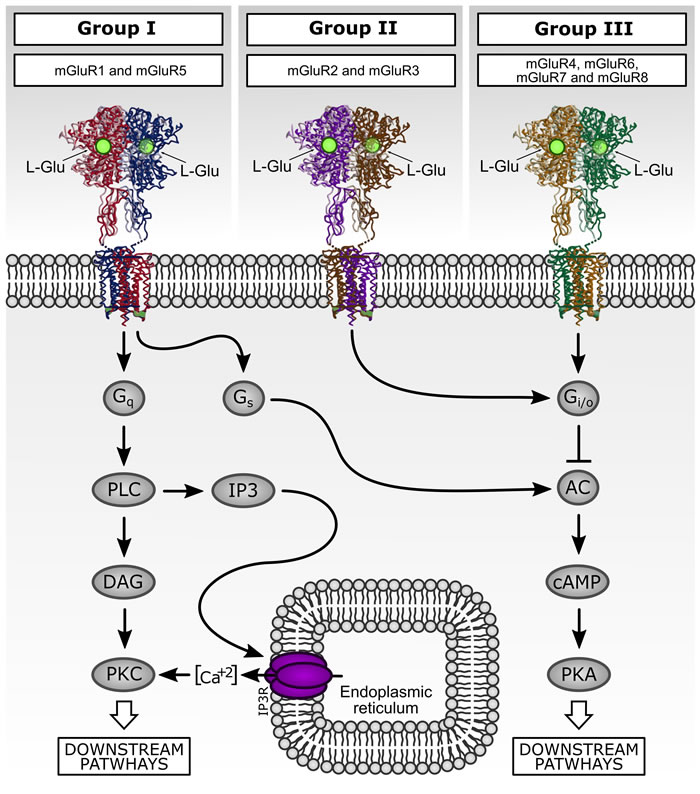
Figure 2: Downstream signaling pathways activated by metabotropic glutamate receptors (mGluR). mGluR family comprises eight subtypes subdivided in three groups according their sequence homology, pharmacology, and second messenger signaling pathway association. Group I mGluR are coupled to Gq proteins and their activation stimulates phospholipase C (PLC) and phosphatidylinositol 4,5-biphosphate (PIP2) hydrolysis. PIP2 hydrolysis generates inositol (1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG), which stimulates intracellular Ca2+ release from endoplasmic reticulum and activates protein kinase C (PKC), respectively. In contrast, mGluR of group II and III are coupled predominantly to Gi/o proteins and classically related to inhibition of adenylyl cyclase (AC) and directly regulate ion channel activity and other downstream signaling partners via liberation of Gβγ subunits.
IN VIVO STUDIES EVALUATING THE ROLE OF mGluR ON GLIOMA TUMOR PROGRESSION
Arcella et al. (2005) [73] have shown that a continuous systemic infusion of mGluR2/3 antagonist LY 341495 reduced growth of GBM cells in two independent in vivo models. U-87 MG cells were implanted under skin (2 x 106 cells/mL) of mice, which were subcutaneously implanted with osmotic minipumps releasing saline, LY 341495 (1 mg/kg per day), EGLU (1 mg/kg per day), LY 379268 (1 mg/kg per day), or LY 341495 plus LY 379268 during 28 days. Analysis of tumor weight showed chronic infusion of antagonists LY 341495 or EGLU reduced GBM growth. On the other hand, infusion of mGluR2/3 agonist LY 379268 did not affect tumor growth and failed to fully reverse LY341495 effect. These results corroborate with Zhou et al. (2014) in vitro study [78], in which activation of mGluR2/3-dependent signaling pathway is necessary to maintain tumor growth but is incapable of stimulating it per se.
In another set of experiments, Arcella et al. (2005) implanted U-87 MG cells into brain left caudate nucleus of nude mice and immunohistochemical analysis revealed that these cells showed a higher expression of mGluR2/3 and Ki-67 (a cellular marker for proliferation) [73]. After magnetic resonance imaging (MRI) analysis (7th day after cell implantation), selected mice with similar tumor sizes were subcutaneously implanted with osmotic minipumps releasing either saline or antagonist LY 341495 (10 mg/kg per day). Treatment during 7 days with LY 341495 reduced tumor size and drug effect was particularly evident during exponential phase of tumor growth (between the 21st and 28th days after cell implantation). Withdrawal of LY 341495 on 21st day allowed growing of tumor to the same extent as control group, suggesting effect of LY 341495 was reversible and cytostatic. Treatment with LY 341495 also reduced the number of Ki-67+ cells in tumor specimens.
These two xenograft models above-described may be complementary (Figure 3A and 3B). Growth of implanted cells in a soft tissue (i.e., subcutaneously) evaluates mainly proliferation rate of tumor on an adequate energy supply. On the other hand, growth of glioma cells in brain requires multiple processes, such as excitotoxic-mediated neuronal death, expression of enzymes that degrade extracellular matrix, and expression of ion channels that drive movement of water out of cell [110]. Unfortunately, work performed by Arcella et al. (2005) have no information regarding outcome of mice treated with LY 341495, since animals were not allowed to survive beyond the 4th week of tumor growth. Nevertheless, authors said none of the five mice died during the four weeks of observation in group treated with this antagonist [73].
Ciceroni et al. (2008) showed continuous pharmacological blockade of mGluR3 reduced growth of infiltrating brain tumors originating from GSC xenografts [76]. GSC spheres were suspended in their growing medium and then infused into left striatum of nude mice (3 x 105 cells/3 µL). Immediately after cell transplantation, mice were subcutaneously implanted with osmotic minipumps releasing mGluR3 agonist LY 379268 or antagonist LY 341495 (both at a rate of 1 mg/kg per day during 3 months), or filled with saline solution. MRI analysis carried out at 3 months showed signal alterations in brain parenchyma of control and LY 379268 treated mice, however, no changes were observed in mice treated with antagonist LY 341495. Histological analysis revealed presence of small and large aggregates of GBM cells in brain parenchyma, which were characterized by nuclear atypia and high mitotic activity. These features are consistent with primitive advanced-stage GBM, where tumors migrate and disseminate asymmetrically along blood vessels and fiber tracts and not grow uniformly [111]. Cell aggregates were consistently found in ipsilateral neostriatum, as well as along the intra- and inter-hemispheric white matter in both control and LY 379268 treated mice. On the other hand, aggregates were absent or present to a very low extent in animals treated with antagonist LY 341495, which indicates that mGluR3 blockage inhibited GSC-dependent generation of tumor cell progeny and/or reduced the growth of GBM malignant cells.
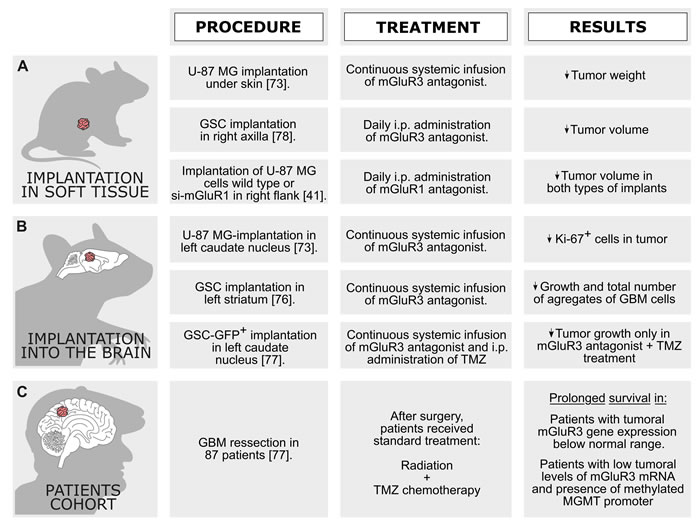
Figure 3: In vivo models employed to study the role of mGluR on brain tumor growth and aggressiveness. A. and B. Effects of mGluR ligands in two mice xenograft models. C. Relation between tumoral mGluR3 mRNA expression and GBM patients’ survival.
Zhou et al. (2014) used a model of subcutaneous implantation of GSC (1x103 cells) in right axilla of mice [78]. After 3 weeks of GSC implantation, tumors were large, ulcerative and had complete capsule. HE stained reveled cells with dark nuclei without evidences of necrosis. Intercellular heteromorphism was obvious and vascular structure was clear. Mice treated with LY 341495 intraperitoneally (1 mg/kg per day) presented a reduced tumor volume when compared to animals treated with agonist LY 379268 (1 mg/kg per day) and vehicle. This result was sustained during 10th, 15th and 20th days after cells implantation.
Ciceroni et al. (2013) have evaluated the effect of mGluR3 inhibition plus TMZ treatment on tumor growth in nude mice implanted with human GSCs in brain parenchyma [77]. All mice were subcutaneously implanted with osmotic minipumps releasing LY 341495 antagonist (3mg/kg per day for 28 days) or saline. At the same time, mice received three injections of TMZ (70mg/kg, intraperitoneally) or vehicle (every day) during the 1st week following minipump implantation. In one experiment, mice received drug treatments during 15 days after GSC implantation and were killed 30 days later (i.e. 45 days after cell implantation). In the other experiment, mice were treated with drugs during 45 days after GSC implantation and killed 30 days later (i.e. 75 days after cell implantation). Control mice (i.e., minipump releasing saline and vehicle injected intraperitoneally) showed presence of GSCs in brain with typical morphology of GBM. For mice killed 45 days after cell implantation, tumor cells were confined to medial portion of caudate nucleus close to wall of lateral ventricle. Mice killed at 75th day showed infiltrating mass tumors in ipsilateral caudate nucleus. Moreover, tumor cells spread to ipsi- and contralateral portion of corpus callosum. Treatment of animals with LY 341495 or TMZ alone did not alter tumor growth in all analyzed brain areas. However, a combined treatment with TMZ plus LY341495 significantly reduced tumor growth in analyzed brain areas. The authors did not observe any signs of systemic toxicity or motor impairment in mice treated with LY 341495 (3 mg/kg per day). In addition, in this work mice survived to acute experiments using doses higher than 300 mg/kg of LY 341495.
Zhang et al. (2015) also demonstrated an anti-tumor activity of mGluR1 inhibition in vivo using a U-87 MG xenograft GBM model in nude mice [41]. After implantation of U-87 MG, tumor-bearing mice were treated every day with Bay 36-7620 (mGluR1 antagonist - 10 mg/kg) intraperitoneally during 24 days. Treatment reduced significantly tumor volume from 22nd to 30th days when compared to control group. In other experiment, nude mice were injected with si-mGluR1 transfected U-87 MG cells or si-control transfected U-87 MG cells. As expected, tumor in si-mGluR1 group had a smaller volume when compared to si-control group.
Association between antitumoral features and apparently non-toxic effects of mGluR antagonists in vivo indicates these drugs are potential candidates for an adjuvant treatment for GBM in humans.
PATIENTS COHORT TO EVALUATE THE ROLE OF mGluR ON GLIOMA AGGRESSIVENESS
Only one study has evaluated the profile of mGluR mRNA expression in a cohort of patients with GBM (Figure 3C) [77]. It was analyzed a possible relationship between expression levels of mGluR3 and survival rate of patients with GBM undergoing surgery followed by radiotherapy and TMZ chemotherapy. The transcript of mGluR3 was measured by quantitative PCR in selected regions of tumor in a cohort of 87 patients. ‘Normal’ mGluR3 mRNA levels were defined as those measured in autoptic brain samples with no histological abnormalities. Levels of mGluR3 mRNA below normal range were detected in 42 GBM biopsies (48.3%), whereas 45 tumor samples (51.7%) presented higher mRNA levels. Kaplan-Meier (KM) survival analysis showed a prolonged survival rate for patients with tumoral mGluR3 RNA expression below normal range. Interestingly, five patients who survived longer than 36 months showed tumoral mGluR3 mRNA expression below normal range. On multivariate analysis, Karnofsky performance score and mGluR3 mRNA emerged as independent predictors for survival. Authors also stratified patients for mGluR3 expression and methylation of MGMT promoter. Group with low tumoral mGluR3 mRNA levels and methylated MGMT promoter showed a significantly higher survival rate as compared to low tumoral mGluR3 mRNA and unmethylated MGMT promoter group.
In summary, low levels of mGluR3 mRNA in tumor specimens may be a predictor for long survival rate in patients with GBM. In addition, methylation state of MGMT gene promoter influenced survival only in those patients whose GBM biopsies presented low expression of mGluR3 RNA. These data may encourage the use of mGluR3 antagonists as adjuvant drugs for treatment of GBM and suggest transcript levels of mGluR3 should be a potential predictor of GBM patients’ survival.
CONCLUSIONs
A large number of preclinical studies have suggested that metabotropic glutamate receptors could be considered a prospective molecular target for treatment of several brain disorders, including depression [112], anxiety disorders [113], Alzheimer’s disease [114], Parkinson’s disease [115], and more recently malignant brain tumors. Several in vitro and in vivo studies have supported the putative involvement of mGluR-mediated signaling on progression, aggressiveness, and recurrence of malignant gliomas, which points to the notion that specific subtype-selective mGluR ligands may be considered as potential adjuvant chemotherapy for glioma treatment. Several academic groups [116-118], Pfizer [119], Roche [120, 121], Novartis [122] and Merck [123] have employed receptor structure-based design of mGluR selective negative allosteric modulators (NAMs) in their studies and ligand-receptor binding models were refined using mutagenesis and structure-activity data. Even though pharmaceutical and toxicological properties of all mGluR ligands are not yet entirely determined for humans (such as effective and maximum tolerable doses), several clinical studies on Phase I, II and III are being performed using mGluR modulators for treatment of distinct brain disorders and cancer. Addex Pharmaceuticals (Geneva, Switzerland) are recently performing a Phase I clinical trials on its mGluR5 ligand ADX48621 for the treatment of depression and anxiety (ClinicalTrials.gov Identifier: NCT02447640). Similarly, a compound LY 2140023, a prodrug of the group II metabotropic glutamate receptor agonist LY 404039, has been tested in a Phase III for the treatment of patients with schizophrenia (ClinicalTrials.gov Identifier: NCT01328093). Regarding cancer treatment, Barbara Ann Karmanos Cancer Institute in collaboration with National Cancer Institute (NCI) are performing a Phase I clinical trial to evaluated the efficacy of riluzole, a mGluR 1 blocker, into reduce the breast cancer growth (NCT00903214). If any of these compounds eventually obtain approval by the Food and Drug Administration (FDA), there will be a strong scientifically-based rationale for testing distinct group I and II mGluR antagonists in the treatment of malignant brain tumors through prospective, large-scale and randomized clinical trials.
Abbreviations
8-Bromo-cAMP: Non-metabolisable cAMP analogue
AC: Adenylyl cyclase
AMPAR: Ionotropic glutamate receptor AMPA
Bay 36-7620: mGluR1 antagonist
BCNU: Carmustine
bFGF: Basic fibroblast growth factor
BMP: Bone morphogenetic proteins
cAMP: Cyclic adenosine monophosphate
CD133: Stem cell marker
CNS: Central nervous system
Cys: Cystine
DAG: Diacylglycerol
DCG-IV: Group II mGluR agonist
DHPG: Group I mGluR agonist
EAAT2: Excitatory amino acid transporter 2
EAAT3: Excitatory amino acid transporter 3
EGF: Epidermal growth factor
EGFR: EGF receptor
EGLU: Selective group II antagonist
FACS: Fluorescence-activated cell sorting
FCS: Fetal calf serum
GBM: Glioblastoma
GFAP: Glial fibrillary acidic protein
GluR: Glutamate receptors
GSC: Glioma stem cells
HR: Hazard ratio
iGluR: Ionotropic glutamate receptors
IP3: Inositol (1,4,5)-trisphosphate
i.p.: intraperitoneal injection
JSH-23: NF-κB inhibitor
L-Glu: L-glutamate
LY 2389575: Group II mGluR antagonist
LY 294002: PI3K inhibitor
LY 341495: Group II mGluR antagonist
LY 379268: Group II mGluR agonist
MAPK: Mitogen-activated protein kinase
MEK: MAPK kinase
mGluR: Metabotropic glutamate receptors
MGMT: O6-methylguanine-DNA methyltransferase
MPEP: Selective group I antagonist
MRI: Magnetic resonance imaging
mTOR: Mammalian target of rapamycin
NBQX: AMPAR antagonist
NF-κB: Nuclear factor-κB
NMDAR: Ionotropic glutamate receptor NMDA
P70 S6K: p70 ribosomal protein S6 kinase
PCR: Polymerase chain reaction
PD 98059: MEK inhibitor
PI 828: PI3K inhibitor
PI3K: Phosphatidylinositol-4,5-bisphosphate 3-kinase
PiP2: Phosphatidylinositol-4,5-bisphosphate
PKC: Protein kinase C
PLC: Phospholipase C
siRNA: Small interfering RNA
SN 38: Irinotecan metabolite
TMZ: Temozolomide
UO126: MEK inhibitor
VEGF: Vascular endothelial growth factor
WHO: World Health Organization
Wortmannin: PI3K inhibitor
xCT: Cystine-glutamate antiporter.
ConflictS of Interest
There is no conflict of interest.
REFERENCES
1. Westphal M and Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nature reviews Neuroscience. 2011; 12:495-508.
2. Cloughesy TF, Cavenee WK and Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014; 9:1-25.
3. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P and Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131:803-820.
4. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97-109.
5. Omuro A and DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013; 310:1842-1850.
6. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncol. 2015; 17:iv1-iv62.
7. Kleihues P, Soylemezoglu F, Schäuble B, Scheithauer BW and Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995; 15:211-221.
8. Theeler BJ, Yung WK, Fuller GN and De Groot JF. Moving toward molecular classification of diffuse gliomas in adults. Neurology. 2012; 79:1917-1926.
9. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98-110.
10. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9:157-173.
11. Bredel M, Scholtens DM, Harsh GR, Bredel C, Chandler JP, Renfrow JJ, Yadav AK, Vogel H, Scheck AC, Tibshirani R and Sikic BI. A network model of a cooperative genetic landscape in brain tumors. JAMA. 2009; 302:261-275.
12. Yadav AK, Renfrow JJ, Scholtens DM, Xie H, Duran GE, Bredel C, Vogel H, Chandler JP, Chakravarti A, Robe PA, Das S, Scheck AC, Kessler JA, et al. Monosomy of chromosome 10 associated with dysregulation of epidermal growth factor signaling in glioblastomas. JAMA. 2009; 302:276-289.
13. Tsao MN, Mehta MP, Whelan TJ, Morris DE, Hayman JA, Flickinger JC, Mills M, Rogers CL and Souhami L. The American Society for Therapeutic Radiology and Oncology (ASTRO) evidence-based review of the role of radiosurgery for malignant glioma. Int J Radiat Oncol Biol Phys. 2005; 63:47-55.
14. DeAngelis LM. Brain tumors. N Engl J Med. 2001; 344:114-123.
15. Wang Y and Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013; 331:139-146.
16. Malmström A, Grønberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, Abacioglu U, Tavelin B, Lhermitte B, Hegi ME, Rosell J, Henriksson R and NCBTSG. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012; 13:916-926.
17. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987-996.
18. Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, Whittle IR, Jääskeläinen J and Ram Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003; 5:79-88.
19. Niyazi M, Harter PN, Hattingen E, Rottler M, von Baumgarten L, Proescholdt M, Belka C, Lauber K and Mittelbronn M. Bevacizumab and radiotherapy for the treatment of glioblastoma: brothers in arms or unholy alliance? Oncotarget. 2016; 7:2313-2328. doi: 10.18632/oncotarget.6320.
20. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009; 27:4733-4740.
21. Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, Park J, Albert PS and Fine HA. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009; 27:740-745.
22. Gilbert MR, Dignam J, Won M, Blumenthal DT, Vogelbaum MA, Aldape KD, Colman H, Chakravarti A, Jeraj R, Armstrong TS, Wefel JS, Brown PD, Jaeckle KA, et al. RTOG 0825: Phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM). J Clin Oncol. 2013.
23. Chinot O, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Hilton M, Abrey LE and Cloughesy T. OT-03. Phase III trial of bevacizumab added to standard radiotherapy and temozolomide for newly-diagnosed glioblastoma: Mmature progression-free survival and preliminary overall survival results in AVAGLIO. Neuro-Oncology. 2012; 14:vi101-vi105.
24. Masui K, Cloughesy TF and Mischel PS. Review: molecular pathology in adult high-grade gliomas: from molecular diagnostics to target therapies. Neuropathol Appl Neurobiol. 2012; 38:271-291.
25. Omuro AM, Faivre S and Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther. 2007; 6:1909-1919.
26. Behrens PF, Langemann H, Strohschein R, Draeger J and Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J Neurooncol. 2000; 47:11-22.
27. Takano T, Lin JH, Arcuino G, Gao Q, Yang J and Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001; 7:1010-1015.
28. Ye ZC and Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999; 59:4383-4391.
29. Noch E and Khalili K. Molecular mechanisms of necrosis in glioblastoma: the role of glutamate excitotoxicity. Cancer Biol Ther. 2009; 8:1791-1797.
30. Ye ZC, Rothstein JD and Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999; 19:10767-10777.
31. Olney JW. Excitotoxicity, apoptosis and neuropsychiatric disorders. Curr Opin Pharmacol. 2003; 3:101-109.
32. de Groot J and Sontheimer H. Glutamate and the biology of gliomas. Glia. 2011; 59:1181-1189.
33. Nicoletti F, Arcella A, Iacovelli L, Battaglia G, Giangaspero F and Melchiorri D. Metabotropic glutamate receptors: new targets for the control of tumor growth? Trends Pharmacol Sci. 2007; 28:206-213.
34. Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008; 105:287-295.
35. Simeone TA, Sanchez RM and Rho JM. Molecular biology and ontogeny of glutamate receptors in the mammalian central nervous system. J Child Neurol. 2004; 19:343-360; discussion 361.
36. Ozawa S, Kamiya H and Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998; 54:581-618.
37. Niswender CM and Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010; 50:295-322.
38. De Blasi A, Conn PJ, Pin J and Nicoletti F. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci. 2001; 22:114-120.
39. Cavalheiro EA and Olney JW. Glutamate antagonists: deadly liaisons with cancer. Proc Natl Acad Sci U S A. 2001; 98:5947-5948.
40. Kalariti N, Pissimissis N and Koutsilieris M. The glutamatergic system outside the CNS and in cancer biology. Expert Opin Investig Drugs. 2005; 14:1487-1496.
41. Zhang C, Yuan XR, Li HY, Zhao ZJ, Liao YW, Wang XY, Su J, Sang SS and Liu Q. Anti-cancer effect of metabotropic glutamate receptor 1 inhibition in human glioma U87 cells: involvement of PI3K/Akt/mTOR pathway. Cell Physiol Biochem. 2015; 35:419-432.
42. Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, Sasaki T and Ozawa S. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002; 8:971-978.
43. Rzeski W, Turski L and Ikonomidou C. Glutamate antagonists limit tumor growth. Proc Natl Acad Sci U S A. 2001; 98:6372-6377.
44. Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T, Saito N, Tsuzuki K, Okado H, Miwa A, Nakazato Y and Ozawa S. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007; 27:7987-8001.
45. Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS and Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001; 61:6674-6678.
46. Teh J and Chen S. mGlu Receptors and Cancerous Growth. Wiley Interdiscip Rev Membr Transp Signal. 2012; 1:211-220.
47. Yu LJ, Wall BA, Wangari-Talbot J and Chen S. Metabotropic glutamate receptors in cancer. Neuropharmacology. 2016.
48. Brocke KS, Staufner C, Luksch H, Geiger KD, Stepulak A, Marzahn J, Schackert G, Temme A and Ikonomidou C. Glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol Ther. 2010; 9:455-468.
49. Li L, Homan KT, Vishnivetskiy SA, Manglik A, Tesmer JJ, Gurevich VV and Gurevich EV. G Protein-coupled Receptor Kinases of the GRK4 Protein Subfamily Phosphorylate Inactive G Protein-coupled Receptors (GPCRs). J Biol Chem. 2015; 290:10775-10790.
50. Chang HJ, Yoo BC, Lim SB, Jeong SY, Kim WH and Park JG. Metabotropic glutamate receptor 4 expression in colorectal carcinoma and its prognostic significance. Clin Cancer Res. 2005; 11:3288-3295.
51. Iacovelli L, Arcella A, Battaglia G, Pazzaglia S, Aronica E, Spinsanti P, Caruso A, De Smaele E, Saran A, Gulino A, D’Onofrio M, Giangaspero F and Nicoletti F. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J Neurosci. 2006; 26:8388-8397.
52. Park SY, Lee SA, Han IH, Yoo BC, Lee SH, Park JY, Cha IH, Kim J and Choi SW. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol Rep. 2007; 17:81-87.
53. Stepulak A, Luksch H, Uckermann O, Sifringer M, Rzeski W, Polberg K, Kupisz K, Klatka J, Kielbus M, Grabarska A, Marzahn J, Turski L and Ikonomidou C. Glutamate receptors in laryngeal cancer cells. Anticancer Res. 2011; 31:565-573.
54. Choi KY, Chang K, Pickel JM, Badger JD and Roche KW. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc Natl Acad Sci U S A. 2011; 108:15219-15224.
55. Marín YE and Chen S. Involvement of metabotropic glutamate receptor 1, a G protein coupled receptor, in melanoma development. J Mol Med (Berl). 2004; 82:735-749.
56. Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, Zhu H, Robbins C, Makalowska I, Shin SS, Marin Y, Roberts KG, Yudt LM, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003; 34:108-112.
57. Chen S, Zhu H, Wetzel WJ and Philbert MA. Spontaneous melanocytosis in transgenic mice. J Invest Dermatol. 1996; 106:1145-1151.
58. Zhu H, Reuhl K, Zhang X, Botha R, Ryan K, Wei J and Chen S. Development of heritable melanoma in transgenic mice. J Invest Dermatol. 1998; 110:247-252.
59. Ohtani Y, Harada T, Funasaka Y, Nakao K, Takahara C, Abdel-Daim M, Sakai N, Saito N, Nishigori C and Aiba A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene. 2008; 27:7162-7170.
60. Speyer CL, Hachem AH, Assi AA, Johnson JS, DeVries JA and Gorski DH. Metabotropic glutamate receptor-1 as a novel target for the antiangiogenic treatment of breast cancer. PLoS One. 2014; 9:e88830.
61. Ali S, Shourideh M and Koochekpour S. Identification of novel GRM1 mutations and single nucleotide polymorphisms in prostate cancer cell lines and tissues. PLoS One. 2014; 9:e103204.
62. Esseltine JL and Ferguson SS. Regulation of G protein-coupled receptor trafficking and signaling by Rab GTPases. Small GTPases. 2013; 4:132-135.
63. Prickett TD, Wei X, Cardenas-Navia I, Teer JK, Lin JC, Walia V, Gartner J, Jiang J, Cherukuri PF, Molinolo A, Davies MA, Gershenwald JE, Stemke-Hale K, et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat Genet. 2011; 43:1119-1126.
64. Nicoletti F, Bruno V, Copani A, Casabona G and Knöpfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996; 19:267-271.
65. Bruno V, Battaglia G, Copani A, D’Onofrio M, Di Iorio P, De Blasi A, Melchiorri D, Flor PJ and Nicoletti F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J Cereb Blood Flow Metab. 2001; 21:1013-1033.
66. Récasens M, Guiramand J, Aimar R, Abdulkarim A and Barbanel G. Metabotropic glutamate receptors as drug targets. Curr Drug Targets. 2007; 8:651-681.
67. Niswender CM, Jones CK and Conn PJ. New therapeutic frontiers for metabotropic glutamate receptors. Curr Top Med Chem. 2005; 5:847-857.
68. D’Onofrio M, Arcella A, Bruno V, Ngomba RT, Battaglia G, Lombari V, Ragona G, Calogero A and Nicoletti F. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J Neurochem. 2003; 84:1288-1295.
69. Dietrich D, Kral T, Clusmann H, Friedl M and Schramm J. Presynaptic group II metabotropic glutamate receptors reduce stimulated and spontaneous transmitter release in human dentate gyrus. Neuropharmacology. 2002; 42:297-305.
70. Dubé GR and Marshall KC. Modulation of excitatory synaptic transmission in locus coeruleus by multiple presynaptic metabotropic glutamate receptors. Neuroscience. 1997; 80:511-521.
71. Knöpfel T and Uusisaari M. (2007). Modulation of Excitation by Metabotropic Glutamate Receptors. Inhibitory Regulation of Excitatory Neurotransmission.
72. Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, Rzeski W, Staufner C, Brocke KS, Turski L and Ikonomidou C. Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol. 2009; 132:435-445.
73. Arcella A, Carpinelli G, Battaglia G, D’Onofrio M, Santoro F, Ngomba RT, Bruno V, Casolini P, Giangaspero F and Nicoletti F. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro Oncol. 2005; 7:236-245.
74. Corti C, Clarkson RW, Crepaldi L, Sala CF, Xuereb JH and Ferraguti F. Gene structure of the human metabotropic glutamate receptor 5 and functional analysis of its multiple promoters in neuroblastoma and astroglioma cells. J Biol Chem. 2003; 278:33105-33119.
75. Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S and Troost D. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. 2003; 17:2106-2118.
76. Ciceroni C, Arcella A, Mosillo P, Battaglia G, Mastrantoni E, Oliva MA, Carpinelli G, Santoro F, Sale P, Ricci-Vitiani L, De Maria R, Pallini R, Giangaspero F, et al. Type-3 metabotropic glutamate receptors negatively modulate bone morphogenetic protein receptor signaling and support the tumourigenic potential of glioma-initiating cells. Neuropharmacology. 2008; 55:568-576.
77. Ciceroni C, Bonelli M, Mastrantoni E, Niccolini C, Laurenza M, Larocca LM, Pallini R, Traficante A, Spinsanti P, Ricci-Vitiani L, Arcella A, De Maria R, Nicoletti F, et al. Type-3 metabotropic glutamate receptors regulate chemoresistance in glioma stem cells, and their levels are inversely related to survival in patients with malignant gliomas. Cell Death Differ. 2013; 20:396-407.
78. Zhou K, Song Y, Zhou W, Zhang C, Shu H, Yang H and Wang B. mGlu3 receptor blockade inhibits proliferation and promotes astrocytic phenotype in glioma stem cells. Cell Biol Int. 2014; 38:426-434.
79. Hamel PA and Hanley-Hyde J. G1 cyclins and control of the cell division cycle in normal and transformed cells. Cancer Invest. 1997; 15:143-152.
80. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004; 432:396-401.
81. Stupp R and Hegi ME. Targeting brain-tumor stem cells. Nat Biotechnol. 2007; 25:193-194.
82. Codrici E, Enciu AM, Popescu ID, Mihai S and Tanase C. Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Targets. Stem Cells Int. 2016; 2016:5728438.
83. Schoepp DD, Jane DE and Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999; 38:1431-1476.
84. Yelskaya Z, Carrillo V, Dubisz E, Gulzar H, Morgan D and Mahajan SS. Synergistic inhibition of survival, proliferation, and migration of U87 cells with a combination of LY341495 and Iressa. PLoS One. 2013; 8:e64588.
85. Ferraguti F, Baldani-Guerra B, Corsi M, Nakanishi S and Corti C. Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur J Neurosci. 1999; 11:2073-2082.
86. Sanderson TM, Hogg EL, Collingridge GL and Corrêa SA. Hippocampal mGluR-LTD in health and disease: focus on the p38 MAPK and ERK1/2 pathways. J Neurochem. 2016.
87. Willard SS and Koochekpour S. Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci. 2013; 9:948-959.
88. Chun-Jen Lin C, Summerville JB, Howlett E and Stern M. The metabotropic glutamate receptor activates the lipid kinase PI3K in Drosophila motor neurons through the calcium/calmodulin-dependent protein kinase II and the nonreceptor tyrosine protein kinase DFak. Genetics. 2011; 188:601-613.
89. Gerber U, Gee CE and Benquet P. Metabotropic glutamate receptors: intracellular signaling pathways. Curr Opin Pharmacol. 2007; 7:56-61.
90. Schaeffer HJ and Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999; 19:2435-2444.
91. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E and Wang L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016; 7:33440-33450. doi: 10.18632/oncotarget.7961.
92. Piccirillo SG and Vescovi AL. Bone morphogenetic proteins regulate tumorigenicity in human glioblastoma stem cells. Ernst Schering Found Symp Proc. 2006; 59-81.
93. Fukuda S and Taga T. [Roles of BMP in the development of the central nervous system]. [Article in Japanese]. Clin Calcium. 2006; 16:781-785.
94. Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT and Pin JP. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011; 60:1017-1041.
95. Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, Barletta E, De Blasi A and Nicoletti F. Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem. 2002; 82:216-223.
96. Fiory F, Oriente F, Miele C, Romano C, Trencia A, Alberobello AT, Esposito I, Valentino R, Beguinot F and Formisano P. Protein kinase C-zeta and protein kinase B regulate distinct steps of insulin endocytosis and intracellular sorting. J Biol Chem. 2004; 279:11137-11145.
97. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012; 3:1068-1111. doi: 10.18632/oncotarget.659.
98. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Bäsecke J, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3:954-987. doi: 10.18632/oncotarget.652.
99. Vivanco I and Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002; 2:489-501.
100. Weaver KD, Yeyeodu S, Cusack JC, Baldwin AS and Ewend MG. Potentiation of chemotherapeutic agents following antagonism of nuclear factor kappa B in human gliomas. J Neurooncol. 2003; 61:187-196.
101. Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR and Kim Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004; 571:50-54.
102. Kopp E and Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994; 265:956-959.
103. Inoki K, Li Y, Zhu T, Wu J and Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002; 4:648-657.
104. Hou L and Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004; 24:6352-6361.
105. Bernard PB, Castano AM, Bayer KU and Benke TA. Necessary, but not sufficient: insights into the mechanisms of mGluR mediated long-term depression from a rat model of early life seizures. Neuropharmacology. 2014; 84:1-12.
106. Argyriou AA and Kalofonos HP. Molecularly targeted therapies for malignant gliomas. Mol Med. 2009; 15:115-122.
107. Nicoletti F, Fagone P, Meroni P, McCubrey J and Bendtzen K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov Today. 2011; 16:715-721.
108. Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004; 4:296-307.
109. Willard SS and Koochekpour S. Glutamate signaling in benign and malignant disorders: current status, future perspectives, and therapeutic implications. Int J Biol Sci. 2013; 9:728-742.
110. Bowman CL, Yohe L and Lohr JW. Enzymatic modulation of cell volume in C6 glioma cells. Glia. 1999; 27:22-31.
111. Giese A. Glioma invasion—pattern of dissemination by mechanisms of invasion and surgical intervention, pattern of gene expression and its regulatory control by tumorsuppressor p53 and proto-oncogene ETS-1. Acta Neurochir Suppl. 2003; 88:153-162.
112. Pilc A, Chaki S, Nowak G and Witkin JM. Mood disorders: regulation by metabotropic glutamate receptors. Biochem Pharmacol. 2008; 75:997-1006.
113. Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA and Schoepp DD. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005; 4:131-144.
114. Lee HG, Zhu X, O’Neill MJ, Webber K, Casadesus G, Marlatt M, Raina AK, Perry G and Smith MA. The role of metabotropic glutamate receptors in Alzheimer’s disease. Acta Neurobiol Exp (Wars). 2004; 64:89-98.
115. Conn PJ, Battaglia G, Marino MJ and Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nature reviews Neuroscience. 2005; 6:787-798.
116. Mølck C, Harpsøe K, Gloriam DE, Clausen RP, Madsen U, Pedersen L, Jimenez HN, Nielsen SM, Mathiesen JM and Bräuner-Osborne H. Pharmacological characterization and modeling of the binding sites of novel 1,3-bis(pyridinylethynyl)benzenes as metabotropic glutamate receptor 5-selective negative allosteric modulators. Mol Pharmacol. 2012; 82:929-937.
117. Dalton JA, Gómez-Santacana X, Llebaria A and Giraldo J. Computational analysis of negative and positive allosteric modulator binding and function in metabotropic glutamate receptor 5 (in)activation. J Chem Inf Model. 2014; 54:1476-1487.
118. Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J and Conn PJ. Probing the metabotropic glutamate receptor 5 (mGlus) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol. 2013; 83:991-1006.
119. Zhang L, Balan G, Barreiro G, Boscoe BP, Chenard LK, Cianfrogna J, Claffey MM, Chen L, Coffman KJ, Drozda SE, Dunetz JR, Fonseca KR, Galatsis P, et al. Discovery and preclinical characterization of 1-methyl-3-(4-methylpyridin-3-yl)-6-(pyridin-2-ylmethoxy)-1H-pyrazolo- [3,4-b]pyrazine (PF470): a highly potent, selective, and efficacious metabotropic glutamate receptor 5 (mGluR5) negative allosteric modulator. J Med Chem. 2014; 57:861-877.
120. Malherbe P, Kratochwil N, Mühlemann A, Zenner MT, Fischer C, Stahl M, Gerber PR, Jaeschke G and Porter RH. Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. J Neurochem. 2006; 98:601-615.
121. Lundström L, Bissantz C, Beck J, Wettstein JG, Woltering TJ, Wichmann J and Gatti S. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br J Pharmacol. 2011; 164:521-537.
122. Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezèau L, Carroll F, Pin JP, Cambria A, Vranesic I, Flor PJ, et al. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan [b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem. 2000; 275:33750-33758.
123. Noeske T, Trifanova D, Kauss V, Renner S, Parsons CG, Schneider G and Weil T. Synergism of virtual screening and medicinal chemistry: identification and optimization of allosteric antagonists of metabotropic glutamate receptor 1. Bioorg Med Chem. 2009; 17:5708-5715.
124. Condorelli DF, Dell’Albani P, Corsaro M, Giuffrida R, Caruso A, Trovato Salinaro A, Spinella F, Nicoletti F, Albanese V and Giuffrida Stella AM. Metabotropic glutamate receptor expression in cultured rat astrocytes and human gliomas. Neurochem Res. 1997; 22:1127-1133.