
INTRODUCTION
Therapeutic targeting of oncogenic mutations in signal transduction pathways has opened a new era in oncology and created a need for efficient mutational analyses in routine pathology. Presently, the complexity of cancer tissues is not taken into account in clinical mutation diagnostics which is performed on crude tissue extracts. Therefore, all different cell types present in a tumor sample – normal parenchymal cells, stromal cells, inflammatory cells, pre-neoplastic and fully developed malignant cells – contribute their wild-type and mutated alleles to the analysis. Thanks to next generation sequencing technology, knowledge about the genetic heterogeneity within a single cancer lesion with regard to acquired aberrations is increasing rapidly. The data support the concept of clonal evolution where different mutations can be enriched in different sub-clones due to an array of selection mechanisms at work in different compartments of the tumor bulk – hypoxia, inflammation, necrosis, inflammation and organ specific environmental factors [1]. To characterize intratumor heterogeneity in a routine diagnostic setting, tumor cells can be enriched by manual microdissection, but more detailed analysis of specific tumor regions requires laborious laser-assisted microdissection. Hence, methods that offer in situ mutation detection directly on tissue sections are highly warranted.
Recently, we published a novel strategy for in situ detection and genotyping of individual mRNA molecules [2]. In this approach, target transcripts are first converted into cDNA molecules and thereafter detected using padlock probes and target primed rolling-circle amplification (RCA). Padlock probes are short linear oligonucleotides that become circular when the ends are brought together by hybridization to a target sequence, and joined by a DNA ligase if perfectly matched [3-6]. The padlock probes contain tag sequences that after amplification act as detection sites for fluorescently labeled oligonucleotides. The resulting rolling circle products (RCPs) appear as bright signals localized in the cytoplasm of the cells. Thus, this in situ technique offers single transcript analysis directly on slides and circumvents traditional DNA extraction from heterogeneous tumor tissues. In addition to point mutations and single-nucleotide polymorphisms (SNPs), the method can provide information on RNA-edited transcripts, tissue specific allele expression [2], alternative splicing, fused transcript variants and small insertions/deletions [7].
The aim of this study was to develop an in situ assay for mutation analysis in clinical oncology and diagnostic molecular pathology, especially with regard to use in routinely collected formalin–fixed, paraffin-embedded (FFPE) tissue. A primary goal of the present study was to design a multiplexed in situ mutation detection assay for point mutations in KRAS, one of the most frequently activated oncogenes in cancer. In colorectal cancer, the presence of mutations in the KRAS gene indicates that the tumor will not respond to EGFR antibody therapy [8]. There are seven point mutations in codon 12 and 13 that together account for approximately 95% of all KRAS mutations in colorectal cancer [9]. In lung adenocarcinoma KRAS mutations are associated with poor prognosis and non-responsiveness to EGFR inhibitors whereas KRAS wild-type tumors with EGFR mutations are linked to better prognosis and response to EGFR inhibitors [10]. The secondary goal of this study was to apply the technique to explore if specific mutations are present in separate cancer sub-clones, and if differences in the balance between expressed mutated and wild-type alleles can be linked to any geographical areas or histologic patterns in a cancer lesion. To this end, we designed individualized patient-specific in situ assays for tumors with multiple known oncogene mutations selected from a cohort [11] of lung cancer cases with characterized mutations in EGFR, KRAS and TP53 [12].
RESULTS
Assay design
We designed padlock probes for point mutations in KRAS codons 12, 13 (G12S, G12R, G12C, G12D, G12A, G12V and G13D) and 61 (Q61H), as well as for EGFR (G719A, G719C, S768I and L858R) and TP53 (S127F and P190S). Padlock probes for the wild-type forms of the different targets were designed as well (Supplementary Table 1 and 2). The mutation-specific padlock probes were designed with identical target sequences except for the last nucleotide in the 3´-end that differ depending on genotype (Fig. 1A). Mismatches at this position are not accepted by the DNA ligase used and single nucleotide differences, like point mutations, are therefore efficiently discriminated [13]. To distinguish the RCPs from each other using detection probes labeled with different fluorescence dyes, e.g. green and red, two different sites for detection probes for wild-type and mutant padlocks were incorporated. We also included detection of the ACTB transcript in our assays, detected by an additional fluorophore, as an internal reference having a relative constant expression between cell types. A comparison of the ACTB signals across samples provided an estimation of the detection efficiency in different samples. The ACTB data was useful during the development phase of this assay, but turned out to be dispensable for mutation scoring and tissue classification. Before applying the padlock probes onto cell lines or tissues they were evaluated in vitro with synthetic templates to assure similar hybridization and ligation efficiency.
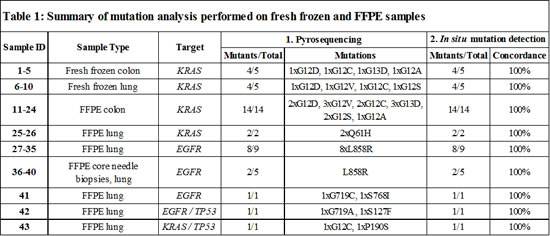
Validation of KRAS in situ mutation detection in colon and lung cancer tissues with known KRAS status
The selectivity of the padlock probes was first tested in situ on KRAS wild-type- and mutant cell lines (Supplementary Fig. 1). After confirmation of the quality of the probes, the in situ genotyping method was applied to ten fresh frozen human colon and lung cancer tissues with known KRAS status (Fig. 2A-D and Supplementary Fig. 2 and 3). In this validation phase, each mutation specific probe-pair was tested individually. The samples represented all codon 12 and 13 mutations except for the rarest one, G12R (Table 1), but the performance of the G12R mutation assay was verified on one of the tested cell lines (Supplementary Fig. 1F). Scoring for KRAS status was done by microscopic inspection in a fashion similar to regular fluorescent in situ hybridization (FISH). Scoring criteria are further discussed in Supplementary Note 2.
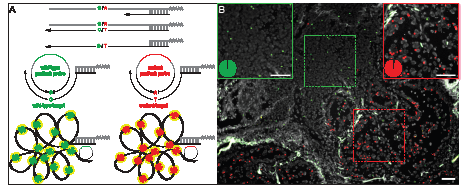
Figure 1: In situ genotyping with padlock probes and target-primed RCA. (A) Schematic overview of the method. Target cDNA (black) is created by reverse transcription with an LNA-primer. Target mRNA (grey) is degraded by RNase H, except for the region that is hybridized to the LNA-part of the primer that is protected from degradation, anchoring the created cDNA to the target. Target specific padlock probes (wild-type:green, mutant:red), with similar target sites except for the single point mutated base (e.g. G/A), are hybridized to the cDNA and circularized by target-dependent ligation. The targeted transcripts act as primer for RCA and the resulting RCPs are labeled with fluorescence-labeled detection probes and visualized as bright spots in the cells or tissue. (B) In situ detection of a KRAS codon 12 (G12S) mutation in fresh frozen lung tumor tissue. Green RCPs represent wild-type KRAS transcripts (GGT), red RCPs represent mutant KRAS (AGT). The green dashed square indicates normal lymphocytes (magnified in solid green square) and the red dashed square indicates tumor cells with an activating G12S KRAS mutation (magnified in solid red square). The pie charts indicate the ratio between wild-type (green) and mutant (red) signals in respective square. Cell nuclei are shown in grey. Scale bar, 50 μm.
The colon and lung sections with KRAS mutations displayed both wild-type and mutant signals (Fig. 2A, C and Supplementary Fig. 2A-D and 3A-D), whereas wild-type tissues almost exclusively showed wild-type signals (Fig. 2B, D and Supplementary Fig. 2E and 3E). By visually examining the ten samples we could clearly see variations in KRAS expression levels both within and between the tissues. Most cases displayed both wild-type and mutant KRAS signals in the tumor cell areas, indicating the presence of a heterozygous mutation and expression of both alleles. In contrast, one lung sample predominantly displayed mutant signals in the tumor regions, potentially reflecting KRAS homozygous mutation or loss-of-heterozygosity (LOH), while the few existing wild-type signals originated from the stroma. (Fig. 1B and Supplementary Fig. 3D).
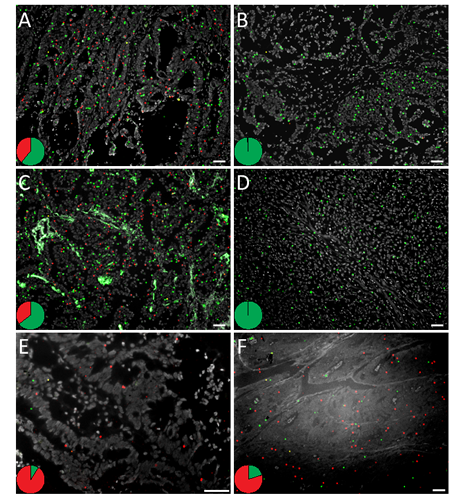
Figure 2: In situ mutation detection of codon 12 and 13 KRAS mutations on (A-D) fresh frozen colon and lung tissues and (E, F) FFPE colon tissues using padlock probes and RCA. The tissues display KRAS mutant (red) and wild-type (green) RCPs. Cell nuclei are shown in grey. KRAS G12D mutation analysis in fresh frozen (A) mutant and (B) wild-type colon tumor tissue, in (C) mutant and (D) wild-type lung tumor tissue, and on FFPE colon tissues with reported (E) G12C or (F) G13D KRAS mutations. The pie charts indicate the ratio between wild-type (green) and mutant (red) signals in respective tissue. The images were acquired with 10x or 20x objective. Scale bar, 50 μm. See Supplementary Fig. 2-4 for the complete set of analyzed samples.
We next assessed whether the in situ padlock probe technique could be applied on FFPE tissues. KRAS mutation analysis was performed on a collection of 14 colorectal FFPE cancer tissues (Fig. 2E, F, Table 1 and Supplementary Fig. 4) with known KRAS mutations in codon 12 and 13. We also designed probes for the most common mutation in codon 61 (Q61H) and tested them in two FFPE colon tumor samples (Table 1 and Supplementary Fig. 5). All tissues displayed a combination of wild-type and mutant signals, however variation in the number of signals (for both KRAS and ACTB) was significant between tissues, which probably reflects the expected difference in tissue quality among FFPE samples.
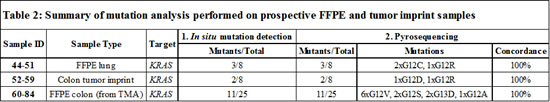
Multiplex in situ detection of KRAS mutations on prospective clinical samples with unknown mutation status
To investigate whether multiple mutations could be tested in the same reaction, we combined all KRAS probes and compared the in situ mutation detection result to that of individual mutation-specific probes (Supplementary Fig. 6). Detection efficiency or selectivity was not affected noticeably when multiple probes competed for the two-codon target site. The combined analysis can thus provide a rapid answer to whether the tumor harbors an activating KRAS mutation or not. Multiplex mutation detection was thereafter demonstrated on eight prospective lung FFPE tissues (Table 2 and Supplementary Fig. 7) with unknown KRAS mutation status. In situ mutation analysis suggested that three of the eight cases were mutated (Fig. 3A and Supplementary Fig. 7A-C). The results were compared to pyrosequencing of DNA extracts from the same tissues, and the suggested genotypes were confirmed to be correct in every case (Supplementary Fig. 12).
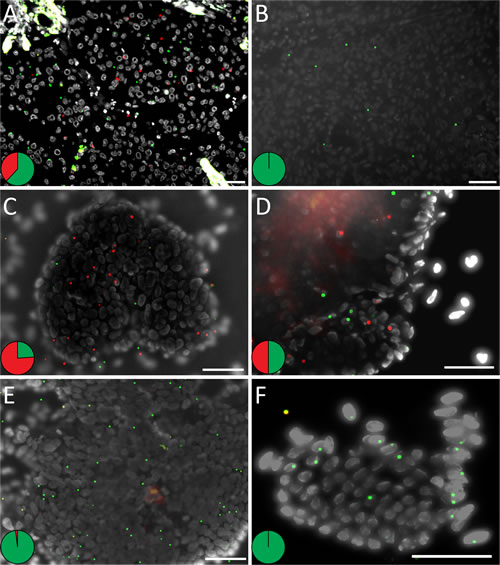
Figure 3: In situ detection of KRAS mutations on prospective clinical samples with unknown mutation status. The tissues display KRAS mutant (red) and wild-type (green) RCPs and cell nuclei are shown in grey. KRAS detection in lung FFPE tissues with (A) a G12C mutation and in (B) a KRAS wild-type sample. Mutation detection in prospective colon touch tumor imprints in (C) G12D and (D) G12R mutated samples and in (E, F) imprint samples with wild-type KRAS. The pie charts indicate the ratio between wild-type (green) and mutant (red) signals in respective tissue. Scale bar, 50 μm. See Supplementaryn Fig. 7 and 8 for the complete set of analyzed samples.
To test the method in a diagnostic setting involving cytology preparations, we prepared tumor imprint slides from eight prospective fresh colon cancer specimens with unknown KRAS mutation status, and subjected them to multiplex KRAS in situ mutation detection (Fig. 3C-F, Table 2 and Supplementary Fig. 8). Two cases were scored positive in the in situ mutation assay (Fig. 3C, D and Supplementary Fig. 8A, B), while the other six only showed wild-type signals (Fig. 3E, F and Supplementary Fig. 8C-H). DNA from corresponding FFPE tumor sections were thereafter tested for KRAS mutations by pyrosequencing, and the results were concordant with the in situ assay (Supplementary Fig. 12).
High-throughput mutation screening on tissue microarrays
Tissue microarrays (TMA) can be used to analyze hundreds of FFPE tumor samples on one slide, and have been used to characterize protein expression by immunohistochemistry (IHC) and gene copy number variations (by FISH) in large patient cohorts [14]. We assayed a TMA containing 25 duplicate FFPE colon samples (normal mucosa, tubular adenomas, serrated adenomas, primary tumors, and matched metastases) of unknown KRAS mutation status for KRAS codon 12 and 13 mutations. Eleven samples, two adenomas, one serrated adenoma, four primary tumors and their matched metastases, were scored KRAS positive (Supplementary Fig. 9). Subsequent mutational analysis of KRAS by pyrosequencing on the corresponding FFPE blocks was concordant with the in situ data (Supplementary Fig. 12).
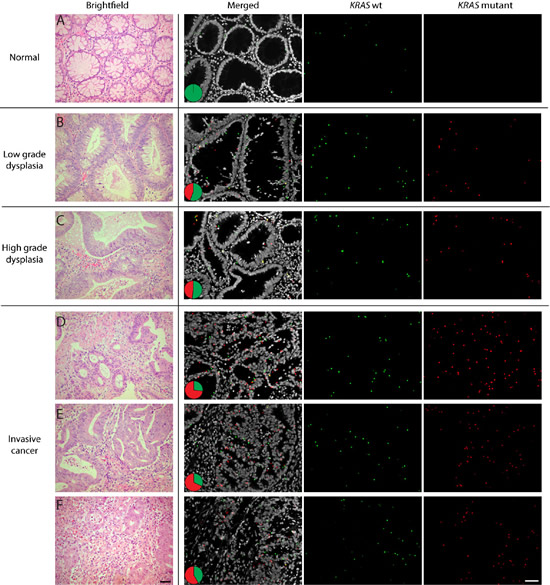
Figure 4: Detection of the Q61H KRAS point mutation in a FFPE colon sample. Images show representative regions of (A) normal colon, (B) low grade dysplasia, (C) high grade dysplasia and (D-F) invasive cancer. The figure presents brightfield microscopy images from corresponding tumor areas in a consecutive H&E-stained slide to the left and fluorescent images to the right in a merged format as well as in respective color to show the distribution of the target transcripts. Red RCPs show mutant KRAS and wild-type RCPs are shown as green spots. The pie charts indicate the ratio between wild-type (green) and mutant (red) signals in respective tissue area. Nuclei are shown in grey. Scale bars, 50 µm.
Differential expression of mutated oncogene alleles related to tumor progression and histological heterogeneity
Variable expression of a mutated oncogene across a tumor could potentially result in a variable response to targeted therapy in different areas of a tumor. We therefore screened cases analyzed by the in situ assay for distinct patterns of expressed mutations. In one colon cancer case with a codon 61 mutation, the histological progression from normal colon mucosa to low-grade and high-grade dysplasia and invasive carcinoma could be visualized on a single slide (Fig. 4 and Supplementary Fig. 5A). There was a clear increase in the expression of mutated mRNA transcripts, relative to the wild-type transcript and in absolute numbers in a given tumor cell area, along with transition from adenomatous to invasive tumor growth. Thus, one can speculate if the level of resistance to EGFR inhibitors would follow the expression levels of mutant KRAS transcripts in the different tumor compartments.
We also targeted the EGFR L858R mutation in a set of nine surgical FFPE lung cancer specimens in which eight were known to be positive (Table 1). The results from the in situ mutation assay were concordant with the DNA sequencing data. Even though some of the lung samples were aged more than a decade, we observed high detection efficiency with high numbers of signals, especially mutant signals, which might reflect high mRNA expression from amplified EGFR in the tumor (Supplementary Fig. 10). In one lung sample we observed a great histological heterogeneity with regard to tumor growth patterns (Fig. 5 and Supplementary Fig. 10A). In normal bronchial epithelium only wild-type EGFR signals were detected (Fig. 5A). In areas with bronchioalveolar/lepidic growth pattern (Fig. 5B) the expression of mutated EGFR was low, and equaled the expression of the wild-type allele. The expression of the mutant allele was increased in more poorly differentiated glandular areas, both in absolute numbers and relative to the wild-type allele (Fig. 5C) and peaked in areas with solid growth pattern (Fig. 5D). Thus, if the expression of L858R transcripts affects the sensitivity of a tumor clone for EGFR-TKI (tyrosine kinase inhibitor) therapy, as would be expected based on IHC data [15], the poorly differentiated areas would be expected to respond better than the well differentiated areas in this individual tumor. To evaluate if the method is applicable to small diagnostic samples, we analyzed five core needle biopsies from lung cancers, of which two were known to be positive for the L858R mutation (Supplementary Fig. 10J-N). Despite low tumor cell content, as low as 10% in one of the cases, all five biopsies were correctly scored using the in situ assay (Table 1). The specificity and sensitivity of the assay was further evaluated in experiments in which cells from a KRAS mutant (G12S) cell line (A-549) were mixed in different ratios with a KRAS wild-type cell line (ONCO-DG-1), and then analyzed with probes for the mutation. The experiment was performed both on preparations of fresh cells grown directly on microscopy slides, and on sections of formalin fixed and paraffin embedded cells. Samples with a ratio of as low as 1% KRAS mutant cells in an otherwise wild-type cell background were successfully detected (Supplementary Note 3). The effect of FFPE preparation and the importance of spatial context are also discussed in Supplementary Note3.
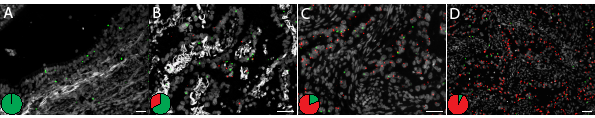
Figure 5: Detection of the EGFR L858R mutation in a FFPE lung tumor tissue sample using mutant and wild-type specific L858R padlock probes and RCA. Red RCPs represent mutants and green RCPs wild-type EGFR. Cell nuclei are shown in grey. The figure illustrates (A) a small bronchus with respiratory epithelium with expression of wild-type EGFR transcripts and (B-D) great heterogeneity within a tumor with regard to expression of the mutant EGFR L858R transcript. The pie charts indicate the ratio between wild-type (green) and mutant (red) signals in respective tissue area. Scale bar, 50 μm.
Expression patterns in tumors with multiple mutations
To further study intratumor heterogeneity, we designed probes for tumors that were known to harbor multiple point mutations. As a proof-of-concept that intratumor heterogeneity can be studied, we established individualized in situ mutation assays for screening of FFPE cases carrying unique combinations of mutations in EGFR, KRAS, and TP53 (Table 1). One lung cancer case was positive for the activating EGFR mutation G719C, as well as the EGFR S768I mutation that is associated with resistance to anti-EGFR therapy (Fig. 6 and Supplementary Fig. 11A). The expression of the G719C transcript was high compared to the S768I transcript throughout the tumor section. This balance between the expressed mutated alleles might be expected as this case represents a patient that had not received anti-EGFR therapy so no selection pressure for increased expression of the resistance mutation was present.
A second lung cancer FFPE sample was assayed for a G719A EGFR mutation in combination with a S127F mutation of the tumor suppressor gene TP53 (Fig. 7A, B and Supplementary Fig. 11B). The in situ analysis showed cells in stromal regions that only expressed the wild-type form of TP53 while no expression of any of the EGFR alleles could be detected. H&E staining of this tissue sample confirmed that the cell populations with wild-type TP53 were lymphocytes. The TP53 S127F mutation-positive tumor regions displayed signals from both the wild-type EGFR and G719A padlock probes but none from the wild-type TP53 padlock probe, indicating TP53 LOH.
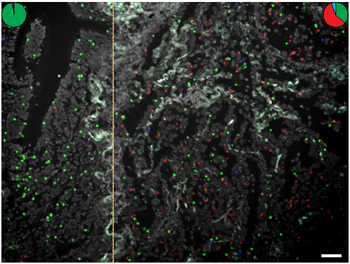
Figure 6: Detection of two EGFR point mutations, G719C and S768I, using mutation-specific padlock probes giving rise to differently colored RCPs; green RCPs represent wild-type EGFR, red RCPs G719C mutants and blue RCPs S768I mutants. Cell nuclei are shown in grey. The left part of the tissue represents benign respiratory epithelium of a small bronchus that expresses wild-type EGFR whereas the right part shows a representative tumor area with cancer cells displaying RCPs from the EGFR wild-type, G719C and S768I padlock probes. The pie charts indicate the ratio between wild-type (green), G719C mutant (red) and S768I (blue) signals in two different areas of the tissue (dotted line denotes border). Scale bar, 50 μm.
Finally, we applied a set of padlock probes on a FFPE lung tissue sample with reported KRAS G12C and TP53 P190S mutations. In contrast to the previous case, in which the wild-type and mutant TP53 signals were located in different compartments (stroma and tumor respectively), here the mutant and wild-type TP53 transcripts were expressed in a heterozygous fashion in the tumor compartment (Fig. 7C). Similarly, the wild-type and mutant KRAS signals were evenly distributed across the tumor areas with a higher expression of mutant compared to wild-type KRAS alleles. This difference in expression pattern of the wild-type and mutant alleles in the two cases would not have been identified unless an in situ technique was included as a complement to DNA sequencing. Moreover, since this in situ assay reveals information on a single cell level, unique information (e.g. expression of more than one mutation in the same cell (Fig. 7D-I)), can be identified and studied in detail. Due to the relatively low density of signals for these transcripts in this tissue, such co-occurrences are relatively rare in our data. However, co-localization of different alleles in the same cell provides strong evidence of their co-existence in cells in the tumor while absence of co-localization does not prove that they are not co-expressed in a certain cell-lineage. Even though all four alleles were not detected in any of these cells, some cells displayed mutations in both KRAS and TP53 and the most likely interpretation of the staining pattern in Fig. 7C-I is that the KRAS mutation is carried by all TP53 mutation-positive cells.
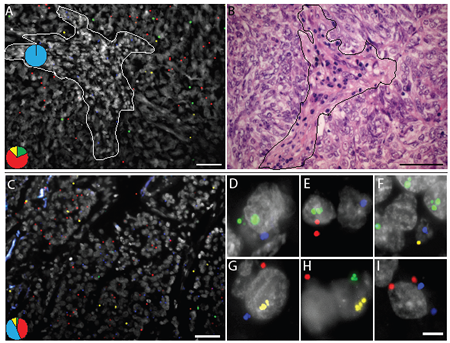
Figure 7: In situ mutation analysis of FFPE lung tumor samples known to harbor (A, B) an EGFR G719A and a TP53 S127F mutation or (C-I) a reported KRAS G12C and a TP53 P190S mutation. (A) Green RCPs represent wild-type EGFR, yellow RCPs mutant EGFR G719A, blue RCPs wild-type TP53 and red RCPs represent mutant TP53 S127F. The image represent cells in stromal regions (dotted line) in which only the wild-type form of TP53 is expressed while the tumor regions displayed RCPs originating from the S127F padlock probe as well as the wild-type EGFR and G719A padlock probes. The pie charts indicate the ratio between wild-type EGFR (green), EGFR G719A mutant (yellow), wild-type TP53 (blue) and mutant TP53 S127F (red) signals in two different areas of the tissue (dotted line denotes border). Cell nuclei are shown in grey. (B) Brightfield image from the same tumor region in a consecutive H&E-stained section. Scale bars, 50 μm. (C) Merged image showing a homogenous expression pattern in a tumor area of mutated and wild-type TP53 and KRAS alleles. Green RCPs represent wild-type KRAS, yellow RCPs mutant KRAS G12C, blue RCPs wild-type TP53 and red RCPs represent mutant TP53 P190S. The pie chart indicates the ratio between wild-type KRAS (green), KRAS G12C mutant (yellow), wild-type TP53 (blue) and mutant TP53 P190S (red) signals in the tissue. Cell nuclei are shown in grey. Scale bar, 50 μm. (D-I) Panel of single cells displaying different signal patterns zoomed in from the same FFPE lung sample case. (D) Wild-type KRAS, wild-type TP53, (E) wild-type KRAS, mutant TP53, (F) mutant KRAS, wild-type TP53, (G) mutant KRAS, wild-type and mutant TP53, (H) wild-type and mutant KRAS and (I) wild-type and mutant TP53. Scale bar, 5 μm.
DISCUSSION
We here report the establishment of a multiplex in situ assay that specifically targets point mutations on tumor tissue sections and on cytological preparations. cDNA copies of transcripts, synthesized by reverse transcription of mRNA in situ, are targeted with mutant- or wild-type specific padlock probes and amplified to a detectable level with RCA. The resulting wild-type and mutated products are thereafter labeled with fluorophores of different colors.
To our knowledge, this padlock probe-based assay demonstrate for the first time that mutation analysis for molecular cancer diagnostics can be performed directly on tumor tissue sections. As a proof-of-concept we developed and validated a multiplexed in situ assay for the activating point mutations in KRAS codon 12 and 13 that are associated with resistance to anti-EGFR therapy in colorectal cancer. First, the selectivity of the probes was tested individually, and there was a clear-cut distinction between the KRAS mutant and wild-type samples and the genotypes were easily determined by simple microscopic visualization of the corresponding fluorescent signals. Second, we used unfixed tumor cells on touch imprints from the fresh cut tumor surface, and demonstrated that a validated KRAS mutation status could be obtained on the day of sample arrival. This could potentially be extended to analyses of other cytological preparations, such as smears after fine needle aspiration or immobilized circulating tumor cells, using the same protocol. Third, to enable routine clinical applications of the technology, we developed a procedure for mutational analyses in FFPE tissue sections.
Paraffin-embedded tissue blocks are used globally in routine surgical pathology and can be preserved for years in tissue archives. However, crosslinking of biomolecules induced by formalin results in fragmentation of DNA and RNA. The short length of the padlock probe, in combination with the requirement of dual recognition sites and ligation makes this assay ideal for fixed histopathology specimens. Using a protocol optimized for formalin-fixed tissues we achieved in situ mutation detection in routine FFPE sections, and prospective surgical cancer specimens with unknown KRAS status were successfully characterized. A promising prospect for this assay is that hundreds of FFPE cancer samples can be screened simultaneously in TMAs for presence of mutations. Thus, for biomarker discovery in retrospective patient cohorts with available TMAs, high-throughput screening for point mutations could be performed along with IHC for protein expression and FISH-analysis for chromosomal aberrations [16].
Despite the fact that the 84 patient samples (10 fresh frozen, 8 touch imprints and 66 FFPE tissues) assayed in this study had been collected at different time points during the last two decades and represented many different types of clinically relevant tissue material with different degree of RNA preservation, they all proved to qualify as suitable tissue material. The samples were not selected on a priori knowledge about RNA preservation, and they were not quality tested before analysis. All samples that entered the study are presented in this study, none were excluded. Furthermore, padlock probes were successfully applied for in situ detection of totally 14 different point mutations which have made us confident that our mutation assay offers robustness and can easily be adapted for detection of other mutations on tissue material from various sources.
Our in situ RNA detection approach yields data that can be quantified by digital spot counting, and has previously been shown to correlate well with the RNA content of fixed cells from cell cultures as determined by quantitative PCR (qPCR) [2]. In cell lines, the detection efficiency (i.e. the fraction of mRNA molecules present in the cell that is detected) has been estimated to 30%. In tissue sections, this number is probably lower due to loss and degradation of RNA upon preservation, storage, sectioning, and slide preparation. The quality of sections will vary from tissue to tissue, and perhaps most prominently in FFPE tissues. The loss of RNA due to issues with preservation is likely to affect different mRNAs at similar rate, so the quality of the tissue section can be assessed by ACTB or similar housekeeping gene staining. Indeed, we note that the density of signals varies from tissue to tissue, and in some FFPE sections the density of signals is quite low. However, the relative abundance of our signals for the different transcripts seems to be similar, and they agree well with published RNA level estimates in normal colon, normal lung, and lung cancer tissue (Supplementary Note 4) [17].
The sensitivity of mutation assays based on DNA-extraction in combination with sequencing or allele-specific PCR is directly linked to the tumor cell content of the sample. In contrast, the padlock probe-based RCA method in principle, as other in situ assays such as IHC or FISH, is independent of the relative tumor cell fraction, as a correct mutation score will rely on the microscopical identification of a representative group of tumor cells with a sufficient number of specific signals. In order to define the practical sensitivity in a clinical setting, a side-by-side comparison with a standard diagnostic mutation test needs to be performed in a large consecutive patient cohort. However, as mutations in a wide range of sample types were correctly scored in the present study, including core needle biopsies with a tumor cell fraction as low as 10% and even lower in many of the TMA cores, we are confident that this assay will perform in tissue materials relevant to routine cancer diagnostics.
Padlock probes display very little cross-reactivity [18] and could thus be used as a general mutation analysis tool with multiplex detection of a set of relevant cancer targets. The only known limitation is the number of available fluorophores to separate the targets from each other. An alternative is therefore to design padlock probes with many detection sites and identify the targets sequentially [19], or by in situ sequencing [20]. The in situ protocol can be adapted for automation as any conventional FISH-assay, facilitating implementation of the assay for routine use. The fluorescence readout can also be changed to a histochemical staining for brightfield imaging if desired, similar to what was done in a recent in situ RCA-approach [21].
The concept of tumor heterogeneity is complex with one aspect being the variable content of cancer cells with acquired somatic mutations and genetically normal stromal and inflammatory cells. A second aspect is the morphological, and possibly genetic, variation within the tumor compartment with regard to pre-neoplastic versus invasive components, high-grade versus low-grade areas, invasion front versus central tumor area, and variable differentiation patterns. A third aspect is that the expression of a mutated allele can be influenced by promoter and splicing mutations, epigenetic alteration, or gene copy number aberrations, e.g. amplifications, deletions and LOH, in different parts of the tumor. These may be challenging to analyze on a genomic level, but they will in many instances be evident on an mRNA level. Loss of expression of tumor suppressor genes due to bi-allelic deletions, nonsense mediated decay or promoter methylation can be detected as loss of signals compared to a housekeeping gene, but such analysis will probably be less sensitive than the detection of gain of function alleles, where just a few signals will be enough to score positivity. The described in situ technique allows studies of all these features of intratumor heterogeneity.
Heterozygous and homozygous expression of mutated and wild-type alleles can be appreciated in tumor cells and demonstrate one form of fundamental information about a particular tissue specimen that probably would have gone undetected with PCR-based techniques resulting in an average value of the extracted mixture of mutant tumor and wild-type cells. We show increased expression of a mutated KRAS codon 61 allele along with tumor progression in a colon cancer sample (Fig. 4). In a case of lung adenocarcinoma, the expression of an activating EGFR mutation was demonstrated to differ between areas with distinctive histological patterns (Fig. 5). The technique allows dissection of how multiple different mutations are distributed across a tumor lesion, as illustrated by two lung cancer cases where mutated TP53 alleles could be visualized together with activating mutations in EGFR and KRAS respectively (Fig. 7). Thus, mutation analysis in situ can help to dissect processes such as cancer initiation, tumor progression and metastasis. An intriguing application will be studies of resistance mutations after targeted therapy [22]. Here we present a case with a double mutation in EGFR where low expression of the resistance mutation was seen in parallel with expression of the mutation associated with treatment response, as might be expected in a patient with a de novo resistance mutation (Fig. 6). Analysis of a follow-up sample after anti-EGFR treatment could reveal a patient-specific response on a histological level regarding the expression of the two mutations.
As opposed to the heterogeneity with regard to expression levels in cancer cells we could not detect any isolated clones in the sections with distinct mutation patterns reading KRAS, EGFR and TP53 status. This is perhaps not surprising as these mutations can be expected to arise early during tumor development and thus be universally expressed. However, in future studies the in situ technique could be used, instead of laser microdissection, to explore late branching events during tumor evolution, such as the recently described emergence of KRAS mutated clones during anti-EGFR treatment of KRAS wild-type colorectal carcinoma [23], expansion of clones with resistance mutations in EGFR-treated lung carcinoma [24], and development of secondary mutations during mTOR (mammalian target of rapamycin) inhibition in renal cell carcinoma [25].
In conclusion, we believe that the presented padlock probe and RCA technology will be an important assay for at least three distinct types of applications in cancer research and diagnostics. First, it can serve as a very useful complement to next-generation sequencing for studies of histologic-genotypic correlations in complex tumor tissues and to elucidate intra-tumoral genetic heterogeneity identified by sequencing. Second, it can be used for high-throughput targeted mutation screening in TMAs and biopsy sections, and finally for diagnostic molecular pathology, directly combining histological examination and molecular diagnostics. All these applications are important aspects of translational biomarker research and development of companion diagnostics.
MATERIALS AND METHODS
In situ mutation detection on cell lines and tissues
Supplementary Figure 13 is providing detailed experimental protocols for the different tissue types and description of the sample pretreatments (Supplementary Note 1). All the molecular in situ reactions were carried out in Secure-seals (Grace Bio-Labs Inc.) and the reaction volumes for tissues or imprints were either 100 µl (size 13 mm diameter, 0.8 mm deep) or 350 µl (size 22 mm diameter, 0.8 mm deep) depending on the size of the sample. The Secure-seals that were used for cells had a total volume of 50 µl (size 9 mm diameter, 0.8 mm deep). The Secure-Seals were mounted over the cells or tissues and the wells were dehydrated by a brief flush with PBS-T (DEPC-PBS with 0.05% Tween-20 (Sigma)). Postfixation of fresh frozen and FFPE tissues was performed for 45 min compared to 30 min for cell lines and tumor imprints. Also, for postfixation of FFPE tissues 3.7% formaldehyde was used instead of 3% (w/v) paraformaldehyde. Finally, the RCA incubation time performed on tissues was longer (5 h) compared to cultured cells and tumor imprints (2 h). For all reactions slides were incubated in humid chambers.
Primers, padlock probes and detection probes
Oligonucleotide sequences (Supplementary Table 1) were designed using GenBank accession numbers NM_033360 (KRAS), NM_005228 (EGFR), NM_001126114.1 (TP53) and NM_001101.3 (ACTB). All padlock probes were 5’ phosphorylated at a concentration of 10 μM with 0.2 U μl-1 T4 PNK (Fermentas) in PNK buffer A and 1 mM ATP for 30 min at 37 °C, followed by 10 min at 65 °C. The primers, padlock probes and detection probes applied on the different tissue samples and cell lines are summarized in Supplementary Material and shows mutant padlock probes and their distribution of mutant and/or wild-type signals in the tested samples (Supplementary Table 2 and Supplementary Table 3).
In situ mutation detection
One µM of cDNA primer was added to the slides with 20 U µl-1 of RevertAid H minus M-MuLV reverse transcriptase (Fermentas), 500 μM dNTP (Fermentas), 0.2 µg µl-1 BSA (NEB), and 1 U µl-1 RiboLock RNase Inhibitor (Fermentas) in the M-MuLV reaction buffer. Slides were incubated for 3 hours at 37 °C. After incubation slides were washed briefly by flushing the wells in PBS-T, followed by a postfixation step for 45 (fresh frozen and FFPE tissues) or 30 (imprints) min at room temperature. After postfixation the samples were washed by flushing the Secure-seals chambers with PBS-T.
To create single-stranded target cDNA available for padlock probe hybridization, the RNA part of the created RNA-DNA hybrids was degraded with RNase H. This was performed in the same step as hybridization and ligation of the padlock probes. The reaction was carried out with 100 nM of each padlock probe in a mix of 1 U µl-1 Ampligase (Epicentre), 0.4 U µl-1 RNase H (Fermentas), 1 U µl-1 RiboLock RNase Inhibitor, 50 mM KCl, 20% formamide in Ampligase buffer. Incubation was performed first at 37 °C for 30 min, followed by 45 min at 45 °C. After ligation, slides were washed by flushing the chambers with PBS-T. For prospective KRAS mutation detection of unknown tissue samples a cocktail of all KRAS codon 12 and 13 padlock probes was mixed with a final concentration of 10 nM.
RCA was performed with 1 U µl-1 Φ29 DNA polymerase (Fermentas) in the supplied reaction buffer with 1 U µl-1 RiboLock RNase Inhibitor, 250 μM dNTP, 0.2 µg µl-1 BSA, and 5% glycerol. Incubation was carried out for 2 h for tumor imprints as well as for cell lines and approximately 5 h for fresh frozen and FFPE tissues at 37 °C. After RCA the samples were washed by flushing the Secure-seals chambers with PBS-T. RCPs were visualized using 100 nM of each corresponding detection probe in 2× SSC and 20% formamide at 37 °C for 15 min. Slides were then washed again by flushing the chambers in PBS-T, the Secure-seals were removed and the slides were dehydrated using a series of 70%, 85%, and 99.5% ethanol for 30 sec each. The dry slides were mounted with Vectashield (Vector), containing 100 ng ml-1 DAPI to counterstain the cell nuclei.
Image analysis and acquisition
The mutation status in the tested samples was based on visual examination using an AxioplanII epifluorescence microscope (Zeiss), equipped with a 100 W mercury lamp, a CCD camera (C4742-95, Hamamatsu), and a computer-controlled filter wheel with excitation and emission filters for visualization of DAPI, FITC, Cy3, Texas Red and Cy5. The tissue sections were carefully analyzed to ensure correct scoring of mutation in representative tumor areas predefined by histopathological examination of corresponding H&E-stained sections. The in situ scoring procedure of wild-type or mutant tumors is further discussed in Supplementary Material (Supplementary Note2).
For capturing the images, a ×10 (Plan-Apocromat, Zeiss) and a ×20 (Plan-Apocromat, Zeiss) objectives were used for fresh frozen and FFPE tissues, a ×20 objective for tumor imprints and finally a ×63 (Plan-neofluar, Zeiss) objective was used for the cells. Images were collected using the Axiovision software (Release 4.8, Zeiss). Images displayed for illustrations were processed using image editing software for clarity in print. The threshold for different color channels was set using ImageJ 1.42q and for clearer visualization of the RCPs, a maximum filter was applied. Autofluorescence in some samples, e.g. lung tissues with extensive connective tissue, was reduced in the ImageJ software by subtracting background signals. For cell lines, core biopsies and imprints (Fig. 3C-F and Supplementary Fig. 1, 8 and 10J-N), a maximum intensity projection was created in Axiovision using the collected z-stack images.
Quantification of signals/image was performed manually for figure 1-7 and the results are presented as pie charts illustrating the ratio between the differently colored signals in respective image.
Acknowledgements
We thank L. Lindbom for technical aid and helpful assistance and Joakim Lundeberg and Mats Dahlberg for sharing data from RNA seq experiments. Funding was provided by the Knut and Alice Wallenberg Foundation, the Swedish Cancer Foundation, the Swedish Research Council, the European Union FP7 (FP7/2007-2013) project READNA (grant agreement HEALTH-F4-2008-201418), the VINNOVA funded project “Companion Diagnostics Initiative”, the Innovative Medicines Initiative Joint Undertaking under grant agreement n° 115234 (OncoTrack), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution and the European Community’s 7th Framework Program (FP7/2007-2013) under grant agreement n° 259796 (DiaTools).
References
1. Fisher R, Pusztai L and Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013; 108(3):479-485.
2. Larsson C, Grundberg I, Soderberg O and Nilsson M. In situ detection and genotyping of individual mRNA molecules. Nat Methods. 2010; 7(5):395-397.
3. Larsson C, Koch J, Nygren A, Janssen G, Raap AK, Landegren U and Nilsson M. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat Methods. 2004; 1(3):227-232.
4. Luo J, Bergstrom DE and Barany F. Improving the fidelity of Thermus thermophilus DNA ligase. Nucleic Acids Res. 1996; 24(15):3071-3078.
5. Nilsson M, Barbany G, Antson DO, Gertow K and Landegren U. Enhanced detection and distinction of RNA by enzymatic probe ligation. Nat Biotechnol. 2000; 18(7):791-793.
6. Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP and Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994; 265(5181):2085-2088.
7. Daly TM, Dumaual CM, Miao X, Farmen MW, Njau RK, Fu DJ, Bauer NL, Close S, Watanabe N, Bruckner C, Hardenbol P and Hockett RD. Multiplex assay for comprehensive genotyping of genes involved in drug metabolism, excretion, and transport. Clin Chem. 2007; 53(7):1222-1230.
8. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD and Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26(10):1626-1634.
9. Sundstrom M, Edlund K, Lindell M, Glimelius B, Birgisson H, Micke P and Botling J. KRAS analysis in colorectal carcinoma: analytical aspects of Pyrosequencing and allele-specific PCR in clinical practice. BMC Cancer. 2010; 10:660.
10. John T, Liu G and Tsao MS. Overview of molecular testing in non-small-cell lung cancer: mutational analysis, gene copy number, protein expression and other biomarkers of EGFR for the prediction of response to tyrosine kinase inhibitors. Oncogene. 2009; 28 Suppl 1:S14-23.
11. Micke P, Edlund K, Holmberg L, Kultima HG, Mansouri L, Ekman S, Bergqvist M, Scheibenflug L, Lamberg K, Myrdal G, Berglund A, Andersson A, Lambe M, Nyberg F, Thomas A, Isaksson A, et al. Gene Copy Number Aberrations Are Associated with Survival in Histologic Subgroups of Non-small Cell Lung Cancer. J Thorac Oncol. 2011; 6(11):1833-1840.
12. Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, Sundstrom M, Micke P, Botling J and Soussi T. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci U S A. 2012; 109(24):9551-9556.
13. Landegren U, Kaiser R, Sanders J and Hood L. A ligase-mediated gene detection technique. Science. 1988; 241(4869):1077-1080.
14. Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G and Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998; 4(7):844-847.
15. Azuma K, Okamoto I, Kawahara A, Taira T, Nakashima K, Hattori S, Kinoshita T, Takeda M, Nakagawa K, Takamori S, Kuwano M, Ono M and Kage M. Association of the expression of mutant epidermal growth factor receptor protein as determined with mutation-specific antibodies in non-small cell lung cancer with progression-free survival after gefitinib treatment. J Thorac Oncol. 2012; 7(1):122-127.
16. Camp RL, Neumeister V and Rimm DL. A decade of tissue microarrays: progress in the discovery and validation of cancer biomarkers. J Clin Oncol. 2008; 26(34):5630-5637.
17. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP and Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456(7221):470-476.
18. Hardenbol P, Baner J, Jain M, Nilsson M, Namsaraev EA, Karlin-Neumann GA, Fakhrai-Rad H, Ronaghi M, Willis TD, Landegren U and Davis RW. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat Biotechnol. 2003; 21(6):673-678.
19. Goransson J, Wahlby C, Isaksson M, Howell WM, Jarvius J and Nilsson M. A single molecule array for digital targeted molecular analyses. Nucleic Acids Res. 2009; 37(1):e7.
20. Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, Nilsson M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013 Sep;10(9):857-60.
21. Zieba A, Wahlby C, Hjelm F, Jordan L, Berg J, Landegren U and Pardali K. Bright-field microscopy visualization of proteins and protein complexes by in situ proximity ligation with peroxidase detection. Clin Chem. 2010; 56(1):99-110.
22. Pao W and Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010; 10(11):760-774.
23. Diaz LA, Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS and Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486(7404):537-540.
24. Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, Ho BC, Chang GC, Shih JY, Yu SL and Yang PC. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012; 30(4):433-440.
25. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366(10):883-892.