
INTRODUCTION
Although the globalization of tobacco use began centuries ago, the public health response to the consequences that it has caused is less than 50 years old [1]. Exposure to tobacco is primarily associated with cigarette smoke, which contains carcinogens that cause lung cancers [2, 3]. Lung cancer, an aggressive and heterogeneous disease caused mainly by smoking [4], results in more than 1 million deaths annually [5]. Although the association between lung cancer and cigarette smoke has been studied for several decades, the tumorigenic process caused by cigarette smoke remains largely unclear.
c-Myc, activated in various human malignancies, is an oncogenic transcription factor [6]. The c-Myc protein, often overexpressed in tumors, is essential for various cellular functions and is associated with poor cancer outcomes [7, 8]. In normal cells, the levels of c-Myc are strictly controlled; however, its deregulated expression results in uncontrolled cell proliferation and the formation of various cancers, including those of the lung [7]. Therefore, it is essential to understand the molecular networks involved, in particular, whether c-Myc participates in the tumorigenic process caused by cigarette smoke.
Many diseases are associated with an altered transcription pattern, which is not restricted to the production of aberrant levels of protein-coding RNAs, but includes dysregulation of the expression of noncoding members [9]. Although more than 90% of DNA is transcribed, most transcribed RNAs are noncoding [10–12]. Many are processed to generate small RNAs, including microRNAs (miRNAs), the best-known class of small RNAs [13, 14]. Other transcripts (long noncoding RNAs or lncRNAs) have, in their mature form, more than 200 nucleotides [15, 16]. Although the roles of miRNAs in the translation of protein-coding transcripts (mRNAs) have been studied, the influence of lncRNAs upon miRNA functions is only now being understood [17]. The interplay between lncRNAs and miRNAs during the tumorigenic process provides new insight into the regulatory mechanisms underlying noncoding RNA classes relevant to cancer [9]. The lncRNA, CCAT1 (colon cancer-associated transcript-1), also named CARLo-5 (cancer-associated region lncRNA-5), was first found to be upregulated in colon cancer [18]. CCAT1 is now known to be deregulated in various cancer types, including lung cancer [19–22]. Therefore, we propose that CCAT1 modulates the functions of miRNAs in cigarette smoke-induced carcinogenesis.
Although expression profiles for lncRNAs and transcription factors correlate with tumor growth [23–25], limited information is available regarding mechanisms by which alterations in lncRNAs and transcription factors contribute to cigarette smoke-induced carcinogenesis. In the present study, we investigated the relationship between CCAT1 and c-Myc in the tumorigenic process induced by cigarette smoke extract (CSE). The results reveal a feedback loop between CCAT1 and c-Myc, acting through let-7c, that promotes the CSE-induced tumorigenic process and presents a previously unknown mechanism by which CCAT1 and c-Myc contribute to CSE-induced carcinogenesis.
RESULTS
CSE increases the levels of c-Myc, affecting the malignancy of transformed HBE cells
c-Myc, a transcription factor, is correlated with tumor aggression and poor clinical outcomes [26, 27]. In human malignancies, dysregulated expression or function of c-Myc is a common abnormality [28]. In the present study, we measured c-Myc levels in HBE cells exposed to 20 μg/mL CSE for 0, 6, 12, or 24 h or for 0, 20, 30, or 40 passages. With increased time of exposure to CSE in acute or chronic treatment of HBE cells, there was greater expression of c-Myc (Figure 1A–1D). Such changes were not evident in control cells. In assessing the function of cMyc in CSE-transformed cells, we found that, in the presence of c-Myc siRNA, the numbers of colonies and the invasion/migration capacities of CSE-transformed HBE cells were decreased (Figure 1E–1G). These results indicate that CSE induces increases of c-Myc levels, which enhance neoplastic activity in the CSE-induced malignant transformation of HBE cells.
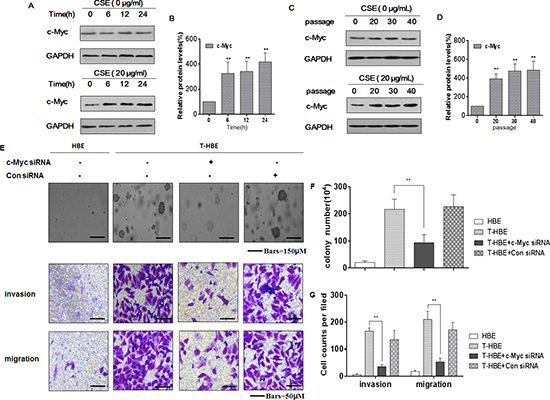
Figure 1: CSE increases the levels of c-Myc, affecting the degree of malignancy and the invasion/migration capacity of transformed HBE cells. Abbreviations: HBE, passage-control HBE cells; T-HBE, CSE-transformed HBE cells. Densities of bands were quantified by Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. HBE cells were exposed to CSE (0 or 20 μg/mL) for 0, 6, 12, or 24 h. (A) Western blots were performed, and (B) relative protein levels (means ± SD, n = 3) of c-Myc were determined. ** P < 0.05, different from control HBE cells. HBE cells were exposed to 0 or 20 μg/mL CSE for 0, 20, 30, or 40 passages. (C) Western blots were performed, and (D) relative protein levels (means ± SD, n = 3) of c-Myc were determined. *P < 0.05, different from passage-control HBE cells. T-HBE cells were transfected for 24 h with c-Myc siRNA or control siRNA at a final concentration of 100 ppm. (E) Representative images of colony formation in soft agar (upper, bars = 150 μm), cell invasion (middle, bars = 50 μm), and cell migration (lower, bars = 50 μm) were prepared. The numbers (means ± SD, n = 3) of colonies formed (F) and of invading or migrating cells (G) were quantified. **p < 0.05, different from T-HBE cells in the absence of c-Myc siRNA.
CSE induces increases of CCAT1 levels and decreases of let-7c levels in HBE cells
Various lncRNAs may function in tumor progression and metastasis [29, 30]. As shown in our previous research, exposure of cells to CSE affects levels of lncRNAs, and the lncRNA CCAT1 is related to the malignant characteristics of CSE transformed-HBE cells [31–33]. miRNAs can be used as biomarkers for exposure to environmental factors, including cigarette smoke, air pollution, nanoparticles, and diverse chemicals [34]. In the present study, we verified the expression of CCAT1 and measured various miRNAs associated with cigarette smoking in HBE cells exposed to 20 μg/mL CSE for 0, 6, 12, or 24 h. With longer times of exposure to CSE, there were greater expressions of CCAT1, miR-21, and miR-155 and lower expressions of let-7c and miR-218 (Figure 2A and 2B). Since the expression of let-7c was changed, and, in hepatocellular carcinomas and lung adenocarcinoma, CCAT1 promotes the proliferation and migration of cancer cells through functioning as a let-7 sponge [19, 35], we focused on CCAT1 and let-7c for further study. HBE cells were exposed to 0 or 20 μg/mL CSE for 0 to 40 passages. With longer times of exposure, there were increases of CCAT1 levels and decreases of let-7c levels (Figure 2C and 2D). Such changes were not found in control cells, indicating that their expressions were affected by CSE. These results show that, in HBE cells, CSE induces up-regulation of CCAT1 and down-regulation of let-7c.
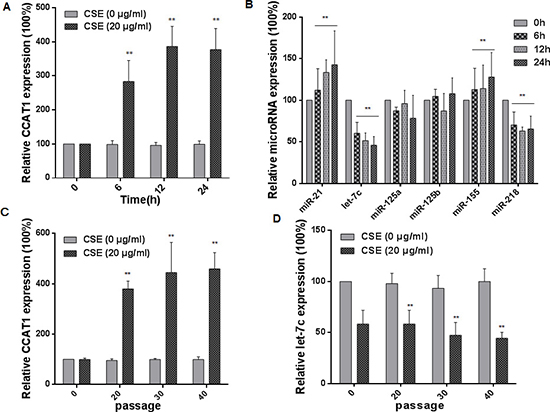
Figure 2: CSE induces increases of CCAT1 levels and decreases of let-7c levels in HBE cells. HBE cells were exposed to CSE (0 or 20 μg/mL) for 0, 6, 12, or 24 h. The levels (means ± SD, n = 3) of CCAT1 (A) miR-21, let-7c, miR-125a, miR-125b, miR-155, and miR-218 (B) were determined by quantitative RT-PCR. ** P < 0.05, different from control HBE cells. HBE cells were exposed to 0 or 20 μg/mL CSE for 0, 20, 30, or 40 passages. The levels (means ± SD, n = 3) of CCAT1 (C) and let-7c (D) were determined by quantitative RT-PCR. **P < 0.05, different from passage-control HBE cells.
c-Myc increases CCAT1 expression via binding to the promoter of CCAT1 in HBE cells
Various transcription factors are involved in regulation of lncRNA transcription [15, 16]. To determine how transcription of CCAT1 is controlled, we searched for potential transcription factor binding sites in the promoter of CCAT1 (http://jaspar.genereg.net) and found one E-box element that could be recognized by c-Myc (Figure 3A). After they were transfected with c-Myc-specific siRNA or control siRNA for 24 h, HBE cells were exposed to CSE for 48 h. The transfection efficiency was assessed by Western blots (Figure 3B and 3C). After depletion of c-Myc, there were lower levels of CCAT1 compared with levels in cells exposed to CSE (Figure 3D). To explore the mechanism for c-Myc regulation of CCAT1, ChIP assays were performed for HBE cells exposed to CSE. For control and CSE-treated cells, the c-Myc antibody was used to immunoprecipitate chromatin-containing DNA fragments that included the promoter region of CCAT1. The results of ChIP and RT-PCR assays of HBE cells exposed to CSE showed that the amount of c-Myc binding to the promoter of CCAT1 was more than that in control HBE cells, which confirmed the interaction between c-Myc and the CCAT1 promoter (Figure 3E). These results indicate that, in CSE-treated HBE cells, c-Myc activates the expression of CCAT1 via binding to its promoter region.
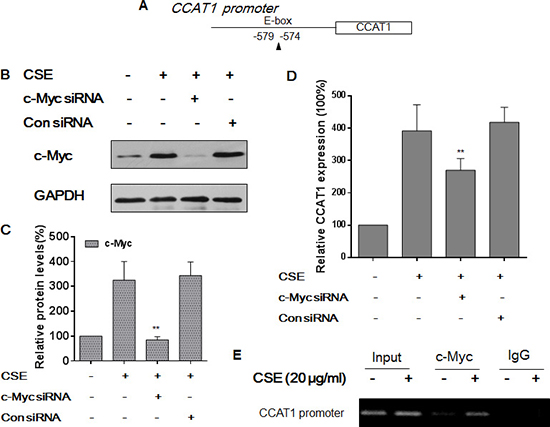
Figure 3: c-Myc regulates CCAT1 expression by binding to the promoter of CCAT1 in HBE cells. Densities of bands were quantified by Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. (A) Schematic graph illustrating binding sites of c-Myc in the promoter of CCAT1. HBE cells were exposed to CSE (0 or 20 μg/mL) in the absence or presence of 100 ppm c-Myc siRNA or control siRNA for 24 h. (B) Western blots and (C) relative protein levels (means ± SD, n = 3) of c-Myc were determined. (D) The levels (means ± SD, n = 3) of CCAT1 were determined by quantitative RT-PCR. **P < 0.05, different from CSE-treated HBE cells in the absence of the c-Myc siRNA. HBE cells were exposed to CSE (0 or 20 μg/mL) for 24 h. (E) the binding of c-Myc to promoters of CCAT1 was measured by a ChIP assay after chromatin was immunoprecipitated with an antibody against c-Myc.
let-7c suppresses CSE-induced increases of c-Myc expression in HBE cells
Various studies have shown that miRNAs, such as miR-155, let-7a, let-7c, and miR-145, regulate c-Myc expression [36–40]. Here, we used StarBase and microRNA.org (http://www.microrna.org/) to predict that let-7c forms complementary base pairing with c-Myc through binding to its 3′-UTR (Figure 4A). When the expression of let-7c was decreased in CSE-exposed HBE cells, the expression of c-Myc was increased (see Figure 1 and 2). To determine if let-7c inhibits c-Myc expression through binding to its 3′-UTR, luciferase reporter assays were conducted. The let-7c mimic reduced the luciferase activities of the cells co-transfected with pmirGLO-c-Myc-3′UTR-WT (Figure 4B). A let-7c mimic was transfected into HBE cells for 24 h, then the cells were exposed to CSE for 24 h. The transfection efficiency was assessed by quantitative RT-PCR (Figure 4C). Ectopic expression of let-7c attenuated the CSE-induced increase of c-Myc levels (Figure 4D). These results indicate that, in HBE cells, let-7c suppresses CSE-induced up-regulation of c-Myc.
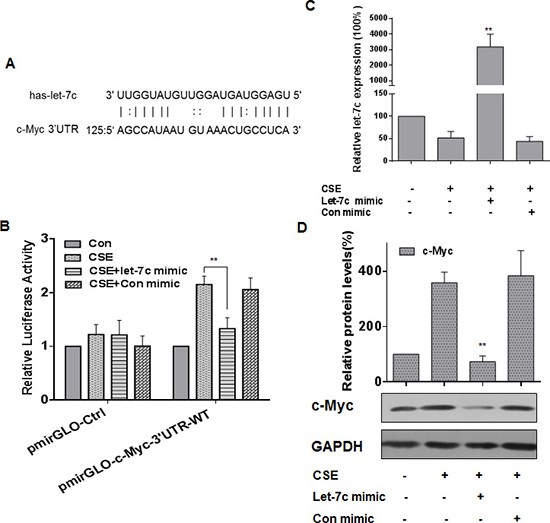
Figure 4: let-7c is involved in CSE-induced increases of c-Myc expression in HBE cells. Densities of bands were quantified by Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. (A) Schematic graph of the binding sites of let-7c in the 3′UTR of c-Myc. HBE cells were exposed to CSE (0 or 20 μg/mL) for 24 h after they were transfected with 50 nM let-7c mimic or control mimic for 24 h. HBE cells were co-transfected with pmirGLO-c-Myc-3′UTR -WT or pmirGLO-Ctrl and with 50 nM let-7c mimic or control mimic for 24 h, then exposed to CSE (0 or 20 μg/mL) for 24 h. (B) Luciferase activity was measured at 24 h after transfection. Means of triplicate assays with standard deviations were presented. (C) The levels (means ± SD, n = 3) of let-7c were determined by quantitative RT-PCR. **P < 0.05, different from CSE-treated HBE cells in the absence of the let-7c mimic. (D) Western blots and relative protein levels (means ± SD, n = 3) of c-Myc were determined. **P < 0.05, different from CSE-treated HBE cells in the absence of the let-7c mimic.
Through let-7c, CCAT1 increases the CSE-induced up-regulation of c-Myc expression in HBE cells.
To investigate the relationship among CCAT1, let-7c, and c-Myc in CSE-treated HBE cells, these cells were treated with CCAT1 siRNA or control siRNA for 24 h, then exposed them to 0 or 20 μg/mL CSE for 24 h. The transfection efficiency was assessed by quantitative RT-PCR (Figure 5A). After CCAT1 silencing, CSE-induced increased levels of c-Myc were attenuated (Figure 5B and 5C). Since lncRNAs participate in regulation of the function of miRNAs [17, 41], we used the online software MiRCode (www.miRCode.org) and MicroInspector (http://bioinfo.uni-plovdiv.bg/ ) to predict that let-7c formed complementary base pairing with CCAT1 (Figure 5D). To confirm the binding between CCAT1 and let-7c, luciferase reporter assays were conducted. The wild-type CCAT1 reporter vector, together with a let-7c mimic or a control mimic, was transfected into HBE cells exposed to CSE (0 or 20 μg/mL). The let-7c mimic reduced the luciferase activities of the cells co-transfected with pGL3-CCAT1-WT (Figure 5E). As let-7c targets c-Myc through binding to its 3′-UTR (see Figure 4), we determined if the c-Myc levels were regulated by CCAT1 through let-7c. After depletion of CCAT1 and let-7c in CSE-exposed HBE cells, the expression of c-Myc was higher than that in cells treated with CCAT1 siRNA (Figure 5F). Relative to CSE-exposed HBE cells, there was no appreciable decrease of let-7c levels after down-regulation of CCAT1 (Figure 5G). The results indicate that CCAT1, at least in part, positively influences the CSE-induced increases of c-Myc levels through binding to let-7c.
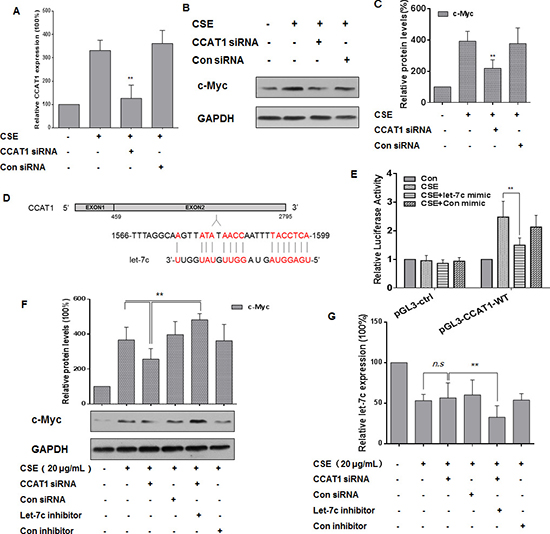
Figure 5: CCAT1 is involved in the CSE-induced elevation of c-Myc expression though let-7c in HBE cells. Densities of bands were quantified by Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. HBE cells were cultured in the presence of control siRNA or CCAT1 siRNA (100 ppm) for 24 h and then exposed to CSE (20 μg/mL) for 24 h. (A) The levels (means ± SD, n = 3) of CCAT1 were determined by quantitative RT-PCR. (B) Western blots and (C) relative protein levels (means ± SD, n = 3) of c-Myc were determined. **P < 0.05, different from CSE-treated HBE cells in the absence of CCAT1 siRNA. (D) Predicted binding sites for let-7c in CCAT1. HBE cells were co-transfected with pGL3-CCAT1-WT or pGL3-ctrl and with 50 nM let-7c mimic or control mimic for 24 h, then exposed to CSE (0 or 20 μg/mL) for 24 h. (E) Luciferase activity was measured at 24 h after transfection. Means of triplicate assays with standard deviations were presented. HBE cells were exposed to CSE (0 or 20 μg/mL) for 24 h after cells were co-transfected with CCAT1 siRNA and let-7c inhibitor for 24 h. (F) Western blots and relative protein levels (means ± SD, n = 3) of c-Myc were determined. **P < 0.05, different from CSE-treated HBE cells in the presence of CCAT1 siRNA. (G) The levels (means ± SD, n = 3) of let-7c were determined by quantitative RT-PCR. ** P < 0.05, different from CSE-treated HBE cells in the presence of CCAT1 siRNA. ‘n.s.’ indicates no significant difference from CSE-treated HBE cells in the presence of CCAT1 siRNA.
CCAT1, via let-7c, enhances the degree of malignancy and the invasion/migration capacity of CSE transformed-HBE cells
CCAT1, which is activated by the oncogene c-Myc, is involved in tumor development [22, 42, 43]. In the present study, we found that, in CSE-transformed HBE cells, the let-7c inhibitor reversed the CCAT1 siRNA-induced down-regulation of c-Myc (Figure 6A). And depletion of CCAT1 decreased colony formation and the invasion and migration capacities of CSE-transformed HBE cells; after co-transfection with CCAT1 siRNA and a let-7c inhibitor, these effects were reversed (Figure 6B–6D). These results establish that CCAT1 controls the degree of malignancy and the invasion/migration capacity of CSE transformed-HBE cells through regulating the function of let-7c.
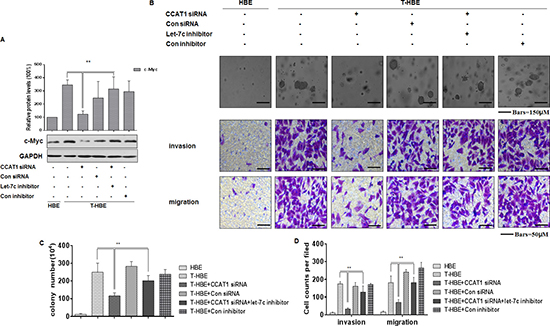
Figure 6: CCAT1, via let-7c, influences the degree of malignancy and the invasion/migration capacity of CSE transformed-HBE cells. Abbreviations: HBE, passage-control HBE cells; T-HBE, CSE-transformed HBE cells. T-HBE cells were exposed to CCAT1 siRNA or control siRNA and to let-7c inhibitor or control inhibitor for 24 h. (A) Western blots and relative protein levels (means ± SD, n = 3) of c-Myc were determined. **P < 0.05, different from CSE-treated HBE cells in the presence of CCAT1 siRNA. (B) Representative images of colony formation in soft agar (upper, bars = 150 μm), cell invasion (middle, bars = 50 μm), and cell migration (lower, bars = 50 μm) were prepared. The numbers (means ± SD, n = 3) of colonies formed (C) and invading/migrating cells (D) were quantified. **P < 0.05, different from T-HBE cells in the presence of CCAT1 siRNA.
Further, in CSE-induced neoplastic transformation of HBE cells, over-expression of c-Myc increases CCAT1 expression through binding to its promoter region; in turn, CCAT1 increases c-Myc expression by binding free let-7c, which negatively regulates the expression of c-Myc through binding to its 3′-UTR (Figure 7). These results provide evidence that a positive feedback loop is formed to ensure CSE-induced CCAT1 and c-Myc expression, which are involved in transformation of cells and carcinogenesis.
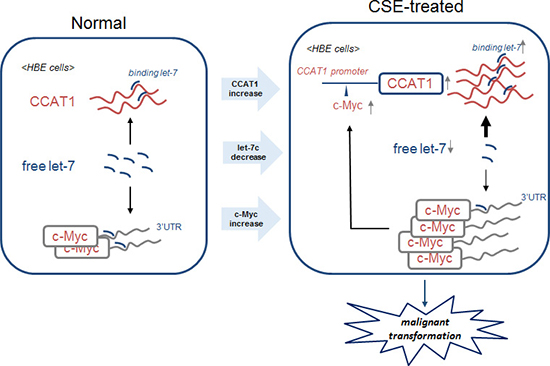
Figure 7: Schematic model of the feedback circuitry between c-Myc and CCAT1 acting via let-7c in CSE-induced transformation of HBE cells. In HBE cells, CSE induces increases of CCAT1 levels and binding of let-7c and decreases of free let-7c levels, which causes increases of c-Myc. The enhancement of c-Myc promotes the expression of CCAT1 via binding to the promoter of CCAT1. The feedback circuitry, via let-7c between c-Myc and CCAT1, is involved in CSE-induced malignant transformation of HBE cells.
DISCUSSION
Lung cancer is a leading cause of death, and, in countries with high tobacco consumption, the proportion of cases attributable to smoking has reached up to 90% [4,44]. Cigarette smoke contains more than 4000 compounds, of which forty are carcinogenic [45]. Among these, nicotine, polycyclic aromatic hydrocarbons, and nicotine-derived nitrosamine ketone induce lung cancer through various signaling pathways [46–50]. In line with evaluations of individual compounds in cigarette smoke, studies with CSE have elucidated various signaling pathways and factors involved in the enhancing effect of cigarette smoke on lung cancer [31, 51–55]. Although cigarette smoke has effects on tumor growth and metastasis [45], the molecular events in carcinogenesis caused by cigarette smoke remain largely unclear.
c-Myc is a helix–loop–helix leucine zipper transcription factor that regulates an estimated 10–15% of genes in the human and Drosophila genomes [56]. As an oncoprotein, it regulates various cellular functions, such as cell division, growth, apoptosis, and differentiation. In human cancers, its expression is frequently elevated, and it is associated with tumor aggression and poor clinical outcomes [57]. In the present study, we found that the expression of c-Myc was increased in HBE cells exposed to CSE, and c-Myc silencing reduced the neoplastic capacity of CSE-transformed HBE cells. These results indicate that, in these cells, c-Myc acts as an oncogene. Further, we demonstrated the underlying mechanism by which c-Myc is involved in the CSE-induced transformation of HBE cells.
Most diseases, including human cancer, are associated with an altered transcription pattern [9]. lncRNAs, which are noncoding members with little or no protein-coding capability, function in a variety of biological processes and disease states [16, 58]. Moreover, deregulation of lncRNAs results in aberrant gene expression that contributes to the progression of cancers, including lung cancer [29, 59–62]. As confirmed in the present study, exposure of cells to CSE increases the expression of CCAT1, an oncogenic lncRNA [63]. CCAT1, acting via the transcription factor, c-Myc, is involved in pathogenesis of several malignancies, such as gastric carcinoma [22], hepatocellular carcinoma [20], and colorectal cancer [42, 43]. In the present study, we demonstrated that, in HBE cells, CSE-induced upregulation of c-Myc promotes CCAT1 expression through its binding to the promoter of CCAT1. Additionally, ChIP assays established that c-Myc, increased by CSE, binds to the promoter of CCAT1, indicating that c-Myc activates CCAT1 expression.
Acting by various mechanisms, most of which are repressive, miRNAs regulate the patterns of expressed proteins [64]. They regulate rates of gene transcription, inhibit the initiation and elongation of target mRNAs, promote decay of target mRNAs, and reduce the stability of proteins newly synthesized from target mRNAs [17, 34]. We investigated several miRNAs associated with cigarette smoking, including miR-21, let-7c, miR-125a, miR-125b, miR-155, and miR-218 [34]. In HBE cells, CSE induced a decrease of let-7c levels. The let-7 family is involved in the proliferation, apoptosis, and invasion of cancer cells [65]. The RNA-binding protein, HuR, inhibits c-Myc expression by recruiting let-7c-loaded RISC (RNA miRNA-induced silencing complex) to the c-Myc 3′-UTR [37]. In our study with HBE cells, a let-7c mimic inhibited the increase of c-Myc levels induced by CSE, which may have occur partly through binding to the c-Myc 3′-UTR.
The present results show that CCAT1 silencing reduces the increased expression of c-Myc caused by CSE. There is cross-regulation between miRNAs and lncRNAs [17], and lncRNAs affect the function of miRNAs [17]. It has been proposed that the concentration of miRNAs in the cytoplasm (and likely the nucleus) is titrated by abundant lncRNAs that harbor similar miRNA target sequences and hence sequester miRNAs away from mRNA [17, 66]. Since studies on the role of lncRNA-miRNA interactions in carcinogenesis are limited, we investigated the functions of CCAT1, along with those of miRNAs, which, in HBE cells, could contribute to the processes of malignant transformation caused by CSE. It has been reported that CCAT1 promotes the proliferation and migration of cancer cells depending on the sponging of let-7c [19, 35]. Here, we also found binding sites between CCAT1 and let-7c, and knockdown of CCAT1 did not appreciably change let-7c levels in CSE-treated cells, but, together with depletion of let-7c levels, the effect of CCAT1 silencing on the expression of c-Myc in CSE-exposed HBE cells was recovered, supporting the hypothesis that CCAT1 increases c-Myc expression though sponging of let-7c. We also demonstrated that, through binding let-7c, CCAT1 inhibits the function of let-7c, reducing c-Myc expression, which promotes proliferation and invasion/migration of CSE-transformed HBE cells. This supports the hypothesis that CCAT1 affects the expression of c-Myc, at least partially through regulating the function of let-7c in CSE-exposed HBE cells. The precise mechanism underlying the effect of cigarette smoke on lung cancer requires further investigation.
Thus, we have shown that CCAT1, an lncRNA, and c-Myc, a transcription factor, are over-expressed in the HBE cells exposed to CSE. Presenting further insight into the complex network underlying cigarette smoke-induced lung cancer, we have provided evidence for a previously unknown function for an lncRNA and a transcription factor in CSE-induced lung carcinogenesis. let-7c negatively regulates c-Myc, and CCAT1 increases c-Myc expression through sponging of let-7c. Our findings highlight the role of CCAT1 in regulation of the function of let-7c during exposure of HBE cells to CSE, which contributes to the overexpression of c-Myc. In turn, c-Myc transcriptionally activates CCAT1, thus forming a positive feedback loop to ensure CSE-induced CCAT1 and c-Myc expression, which are involved in malignant transformation of HBE cells. In sum, CCAT1 and c-Myc have a positive correlation, which expands understanding of the carcinogenic potential of cigarette smoke.
MATERIALS AND METHODS
Cells, cell culture methods, and reagents
Human bronchial epithelial (HBE) cells were obtained from the Shanghai Cell Bank of Chinese Academy of Sciences on 1th Jan 2014. Simian virus 40 (SV40)-transformed HBE cells are nontumorigenic and retain features of parent HBE cells. They are useful for studies of multistage bronchial epithelial carcinogenesis [67]. These cells were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). HBE cells were maintained in Eagle’s minimum essential medium (MEM) with 10% fetal bovine serum (FBS, Life Technologies/Gibco, Grand Island, NY), 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies/Gibco, Gaithersburg, MD) and incubated in 5% CO2 at 37°C. At a concentration of 20 μg/mL, CSE does not change cell viability [54]. For chronic exposure, 1 × 106 cells were seeded into 10-cm (diameter) dishes for 24 h and exposed to 0 or 20 μg/mL of CSE for 24–48 h per passage. This process was continued for 40 passages (about 20 weeks). CSE-transformed HBE (T-HBE) cells were those exposed to 20 μg/mL of CSE for 40 passages, during which they undergo malignant transformation [31, 54]. The cells in passage 0 (normal HBE cells) were not exposed to CSE. This process and the preparation of CSE were performed as described previously [54].
Quantitative real-time PCR
Total RNA was isolated by use of TRIzol (Invitrogen) according to the manufacturer’s instructions. Briefly, total RNA was prepared, and its concentration and integrity were assessed as previously described [54]. For detection of mature let-7c, 2 μg of total RNA, miRNA-specific stem-loop RT primers, and MMLV reverse transcriptase (Promega Corp., Madison, WI) were used in reverse transcription following the manufacturer’s protocol. For measurement of CCAT1, the isolated RNA was reverse-transcribed into cDNA using a reverse transcription kit (Takara, Dalian, China). Quantitative real-time PCR was performed with an Applied Biosystems 7300HT machine and MaximaTM SYBR Green/ROX qPCR Master Mix (Fermentas). U6 snRNA and 18 S RNA were used as internal controls to determine relative miRNA and lncRNA expressions. Relative gene expression was calculated by the formula 2−(ΔΔCt) [68].
The following primers were used: miR-21-F, 5′- ACACTCCAGCTGGGTAGCTTATCAGACTGA -3′, miR-21-R, 5′-TGGTGTCGTGGAGTCG-3′; let-7c-F, 5′- ACA CTCCAGCTGGGTGAGGTAGTAGGTTGT -3′, let-7c-R, 5′-TGGTGTCGTGGAGTCG-3′; miR-125a-F, 5′-ACACT CCAGCTGGGTCCCTGAGACCCTTTAAC-3′, miR-125a -R, 5′-TGGTGTCGTGGAGTCG-3′; miR-125b-F, 5′- ACACTCCAGCTGGGTCCCTGAGACCCTAAC-3′; miR-125b-R, 5′-TGGTGTCGTGGAGTCG-3′; miR-155-F, 5′- AC ACTCCAGCTGGGTTAATGCTAATCGTGAT-3′; miR-155- R, 5′-TGGTGTCGTGGAGTCG-3′; miR-218-F, 5′-ACAC TCCAGCTGGGTTGTGCTTGATCTAA-3′; miR-218-R, 5′-TGGTGTCGTGGAGTCG-3′; U6-F, 5′-CGCTTCGG CAGCACATATACTAAAATTGGAAC-3′; U6-R, 5′-GCT TCACGAATTTGCGTGTCATCCTTGC-3′; CCAT1-F, 5′-T TTATGCTTGAGCCTTGA-3′; CCAT1-R, 5′- CTTGCC TGAAATACTTGC-3′; 18S-F, 5′-GTAACCCGTTGAACC CCATT-3′; and 18S-R, 5′- CCATCCAATCGGTAGTAG CG-3′.
Cell transfection
CCAT1 siRNA and control siRNA were purchased from Santa Cruz Biotechnology. The let-7c mimic/mimic control and the let-7c inhibitor/inhibitor control were purchased from Genechem, Shanghai, China. Cells were grown on six-well plates and transfected by use of Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. At 24 h after transfection, cells were treated and subsequently harvested for qRT-PCR or Western blot analyses. All assays were performed in triplicate. The final concentrations employed were as follows: CCAT1 siRNA/negative control siRNA, 100 ppm; CCAT1-wt/CCAT1-ctrl, 50 nM; let-7c mimic/mimic control, 50 nM; and let-7c inhibitor/inhibitor control, 50 nM.
Western blots
Western blot analyses to assess c-Myc and GADPH expression were accomplished as described previously [33]. Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was purchased from Sigma, and an antibody for c-Myc was purchased from Abcam (Cambridge, MA). For densitometric analyses, the bands on the blots were measured by Eagle Eye II.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed by use of a Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (Millipore) following the manufacturer’s protocol. c-Myc antibody (Abcam, Cambridge, MA) and isotype IgG were used for immunoprecipitation. This process was performed as described previously [69].The specific sequences of immunoprecipitated and input DNA were determined by PCR primers for the CCAT1 promoter containing E-box: CCAT1 promoter forward, 5′-AGTCACTGGTGTTCTTGC-3′, and reverse, 5′-GGTATGCGTAGGTGATAGT-3′.
Luciferase reporter gene assays
The 3′-UTR of c-Myc was sub-cloned into the luciferase reporter plasmid (pmirGLO-c-Myc-3′UTR-WT), and a 206-bp portion of CCAT1 (from 1491 nt to 1697 nt) was sub-cloned into a luciferase reporter plasmid (pGL3-CCAT1-WT), which together with a pmirGLO-Ctrl and pGL3-ctrl luciferase construct, were purchased from GeneChem (Shanghai, China). The pmirGLO-c-Myc-3′UTR-WT/pGL3-CCAT1-WT or pmirGLO-Ctrl/pGL3-ctrl was co-transfected with the let-7c mimic or mimic control into cells by Lipofectamine® 2000 (Invitrogen)-mediated gene transfer according to the manufacturer’s protocol. Approximately 24 h after transfection, the cells were washed three times with PBS (pH 7.4). Then the cells were lysed with 1× passive lysis buffer (Promega). The lysates from each well were analyzed in a 96-well plate illuminometer (Berthold Detection System, Pforzheim, Germany). The amounts of luciferase and Renilla luciferase were measured with the Dual-Luciferase Reporter Assay System kit (Promega) following the manufacturer’s instructions. Transfection experiments were performed in quadruplicate and were repeated at least three times. The relative luciferase activity was normalized to Renilla luciferase activity, as reported previously [70].
Colony formation assays
The cells were disassociated, suspended in MEM medium containing 0.35% agar, and plated onto 0.7% agar in triplicate at a density of 1 × 104 cells/6-cm dish. The numbers of colonies that were > 0.5 mm in diameter were counted 14 days later, as reported previously [33].
Transwell assays
The CSE-transformed HBE cells were suspended in serum-free medium at a density of 1 × 105 cells/mL. Subsequently, 200 μL of cell suspensions were added to the upper chambers of Transwell plates with 8-μm pore membranes (Corning, Inc., Corning, NY, USA) with 35 μL of Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). MEM media (500 μL) supplemented with 10% FBS was added to the lower chambers. After incubation for 24 h at 37°C, the cells on the upper surfaces of the microporous membranes were removed with cotton swabs, whereas cells on the lower surface of the membranes were fixed with 4% paraformaldehyde, stained with crystal violet solution for 30 min, and washed twice with PBS. Images of the stained cells from five selected views were captured under a microscope (high-power field), and the numbers of cells that migrated through the membranes were averaged. To assess the capacity for migration of transformed HBE cells, transfected cells (5 × 104/100 μL) were added to upper chambers without Matrigel. MEM medium containing 10% FBS was added to the lower chambers. Cells were incubated for 24 h at 37°C. Migrating cells were fixed, stained, and calculated.
Statistical analyses
All experiments were performed at least three times in triplicate for each group. The results are presented as means ± S.D. Comparisons of means among multiple groups were performed by one-way analysis of variance (ANOVA), and a multiple-range least significant difference (LSD) was used for inter-group comparisons. Statistical significance was reached if P < 0.05. All statistical analyses were performed with SPSS 16.0.
ACKNOWLEDGMENTS
The authors wish to thank Donald L. Hill (University of Alabama at Birmingham, USA), an experienced, English-speaking scientific editor, for editing.
CONFLICTS OF INTEREST
The authors declare they have no competing financial interests.
FUNDING
This work was supported by the Natural Science Foundations of China (81473011, 81573199, 81302466), the Natural Science Foundation of Jiangsu Province (BK20161585, BK20151585, BK20151593), the Postgraduate Innovation Project of Jiangsu province (CXZZ14_0421, CXZZ14_0951, and KYLX15_0974), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (2014).
REFERENCES
1. Glynn T, Seffrin JR, Brawley OW, Grey N, Ross H. The globalization of tobacco use: 21 challenges for the 21st century. CA Cancer J Clin. 2010; 60:50–61.
2. Hecht SS, Kassie F, Hatsukami DK. Chemoprevention of lung carcinogenesis in addicted smokers and ex-smokers. Nat Rev Cancer. 2009; 9:476–488.
3. Lee YJ, Kim JH, Kim SK, Ha SJ, Mok TS, Mitsudomi T, Cho BC. Lung cancer in never smokers: change of a mindset in the molecular era. Lung Cancer. 2011; 72:9–15.
4. Pesch B, Kendzia B, Gustavsson P, Jockel KH, Johnen G, Pohlabeln H, Olsson A, Ahrens W, Gross IM, Bruske I, Wichmann HE, Merletti F, Richiardi L, et al. Cigarette smoking and lung cancer—relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int J Cancer. 2012; 131:1210–1219.
5. Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol. 2012; 13:790–801.
6. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008; 40:43–50.
7. Pan J, Deng Q, Jiang C, Wang X, Niu T, Li H, Chen T, Jin J, Pan W, Cai X, Yang X, Lu M, Xiao J, et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene. 2015; 34:3957–3967.
8. Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 2002; 84:81–154.
9. Liz J, Esteller M. lncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. 2016; 1859:169–176.
10. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, et al. Landscape of transcription in human cells. Nature. 2012; 489:101–108.
11. Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, et al. The transcriptional landscape of the mammalian genome. Science. 2005; 309:1559–1563.
12. Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005; 308:1149–1154.
13. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009; 10:126–139.
14. Tuck AC, Tollervey D. RNA in pieces. Trends Genet. 2011; 27:422–432.
15. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013; 193:651–669.
16. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009; 10:155–159.
17. Yoon JH, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol. 2014; 34:9–14.
18. Nissan A, Stojadinovic A, Mitrani-Rosenbaum S, Halle D, Grinbaum R, Roistacher M, Bochem A, Dayanc BE, Ritter G, Gomceli I, Bostanci EB, Akoglu M, Chen YT, et al. Colon cancer associated transcript-1: a novel RNA expressed in malignant and pre-malignant human tissues. Int J Cancer. 2012; 130:1598–1606.
19. Chen J, Zhang K, Song H, Wang R, Chu X, Chen L. Long noncoding RNA CCAT1 acts as an oncogene and promotes chemoresistance in docetaxel-resistant lung adenocarcinoma cells. Oncotarget. 2016; 7:62474–62489. doi: 10.18632/oncotarget.11518.
20. Zhu HQ, Zhou X, Chang H, Li HG, Liu FF, Ma CQ, Lu J. Aberrant Expression of CCAT1 Regulated by c-Myc Predicts the Prognosis of Hepatocellular Carcinoma. Asian Pac J Cancer Prev. 2015; 16:5181–5185.
21. Ma MZ, Chu BF, Zhang Y, Weng MZ, Qin YY, Gong W, Quan ZW. Long non-coding RNA CCAT1 promotes gallbladder cancer development via negative modulation of miRNA-218–5p. Cell Death Dis. 2015; 6:e1583.
22. Yang F, Xue X, Bi J, Zheng L, Zhi K, Gu Y, Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol. 2013; 139:437–445.
23. Ma MZ, Li CX, Zhang Y, Weng MZ, Zhang MD, Qin YY, Gong W, Quan ZW. Long non-coding RNA HOTAIR, a c-Myc activated driver of malignancy, negatively regulates miRNA-130a in gallbladder cancer. Mol Cancer. 2014; 13:156.
24. Hung CL, Wang LY, Yu YL, Chen HW, Srivastava S, Petrovics G, Kung HJ. A long noncoding RNA connects c-Myc to tumor metabolism. Proc Natl Acad Sci USA. 2014; 111:18697–18702.
25. Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS, Penn LZ. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006; 66:5330–5337.
26. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc HK, Pinchback RM, Ligon AH, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899–905.
27. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999; 18:3004–3016.
28. Cole MD, McMahon SB. The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 1999; 18:2916–2924.
29. Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013; 108:2419–2425.
30. Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015; 21:1253–1261.
31. Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W, Shi L, Lu X, Liu Q. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015; 282:9–19.
32. Lu L, Luo F, Liu Y, Liu X, Shi L, Lu X, Liu Q. Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial-mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015; 289:276–285.
33. Lu L, Xu H, Luo F, Liu X, Lu X, Yang Q, Xue J, Chen C, Shi L, Liu Q. Epigenetic silencing of miR-218 by the lncRNA CCAT1, acting via BMI1, promotes an altered cell cycle transition in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2016; 304:30–41.
34. Vrijens K, Bollati V, Nawrot TS. MicroRNAs as potential signatures of environmental exposure or effect: a systematic review. Environ Health Perspect. 2015; 123:399–411.
35. Deng L, Yang SB, Xu FF, Zhang JH. Long noncoding RNA CCAT1 promotes hepatocellular carcinoma progression by functioning as let-7 sponge. J Exp Clin Cancer Res. 2015; 34:18.
36. Chi JQ, Teng M, Yu ZH, Xu H, Su JW, Zhao P, Xing GX, Liang HD, Deng RG, Qu LH, Zhang GP, Luo J. Marek’s disease virus-encoded analog of microRNA-155 activates the oncogene c-Myc by targeting LTBP1 and suppressing the TGF-beta signaling pathway. Virology. 2015; 476:72–84.
37. Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009; 23:1743–1748.
38. Nadiminty N, Tummala R, Lou W, Zhu Y, Zhang J, Chen X, EVere WR, Kung HJ, Evans CP, Gao AC. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of Myc expression in prostate cancer cells. J Biol Chem. 2012; 287:1527–1537.
39. Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007; 67:9762–9770.
40. Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA. 2009; 106:3207–3212.
41. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011; 43:904–914.
42. He X, Tan X, Wang X, Jin H, Liu L, Ma L, Yu H, Fan Z. C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biol. 2014; 35:12181–12188.
43. Kim T, Cui R, Jeon YJ, Lee JH, Lee JH, Sim H, Park JK, Fadda P, Tili E, Nakanishi H, Huh MI, Kim SH, Cho JH, et al. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc Natl Acad Sci USA. 2014; 111:4173–4178.
44. Tobacco smoke and involuntary smoking. IARC Monogr Eval Carcinog Risks Hum. 2004; 83:1–1438.
45. Vu T, Jin L, Datta PK. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J Clin Med. 2016; 5.
46. Minna JD. Nicotine exposure and bronchial epithelial cell nicotinic acetylcholine receptor expression in the pathogenesis of lung cancer. J Clin Invest. 2003; 111:31–33.
47. Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999; 91:1194–1210.
48. Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009; 124:36–45.
49. Lin P, Chang H, Ho WL, Wu MH, Su JM. Association of aryl hydrocarbon receptor and cytochrome P4501B1 expressions in human non-small cell lung cancers. Lung Cancer. 2003; 42:255–261.
50. Belinsky SA, Nikula KJ, Baylin SB, Issa JP. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc Natl Acad Sci USA. 1996; 93:4045–4050.
51. Liu H, Zhou Y, Boggs SE, Belinsky SA, Liu J. Cigarette smoke induces demethylation of prometastatic oncogene synuclein-gamma in lung cancer cells by downregulation of DNMT3B. Oncogene. 2007; 26:5900–5910.
52. Martey CA, Pollock SJ, Turner CK, O’Reilly KM, Baglole CJ, Phipps RP, Sime PJ. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004; 287:L981–L991.
53. Li D, Beisswenger C, Herr C, Hellberg J, Han G, Zakharkina T, Voss M, Wiewrodt R, Bohle RM, Menger MD, Schmid RM, Stockel D, Lenhof HP, et al. Myeloid cell RelA/p65 promotes lung cancer proliferation through Wnt/beta-catenin signaling in murine and human tumor cells. Oncogene. 2014; 33:1239–1248.
54. Zhao Y, Xu Y, Li Y, Xu W, Luo F, Wang B, Pang Y, Xiang Q, Zhou J, Wang X, Liu Q. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol Sci. 2013; 135:265–276.
55. Wang B, Liu Y, Luo F, Xu Y, Qin Y, Lu X, Xu W, Shi L, Liu Q, Xiang Q. Epigenetic silencing of microRNA-218 via EZH2-mediated H3K27 trimethylation is involved in malignant transformation of HBE cells induced by cigarette smoke extract. Arch Toxicol. 2016; 90:449–461.
56. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005; 435:839–843.
57. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012; 151:56–67.
58. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013; 152:1298–1307.
59. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014; 25:666–681.
60. Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, Zornig M, MacLeod AR, Spector DL, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013; 73:1180–1189.
61. Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer. 2012; 11:5.
62. Li H, Yu B, Li J, Su L, Yan M, Zhu Z, Liu B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget. 2014; 5:2318–2329. doi: 10.18632/oncotarget.1913.
63. Xin Y, Li Z, Shen J, Chan MT, Wu WK. CCAT1: a pivotal oncogenic long non-coding RNA in human cancers. Cell Prolif. 2016; 49:255–260.
64. Stahlhut EC, Slack FJ. The role of microRNAs in cancer. Yale J Biol Med. 2006; 79:131–140.
65. Childs G, Fazzari M, Kung G, Kawachi N, Brandwein-Gensler M, McLemore M, Chen Q, Burk RD, Smith RV, Prystowsky MB, Belbin TJ, Schlecht NF. Low-level expression of microRNAs let-7d and miR-205 are prognostic markers of head and neck squamous cell carcinoma. Am J Pathol. 2009; 174:736–745.
66. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014; 505:344–352.
67. Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988; 48:1904–1909.
68. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408.
69. Liu Y, Wang B, Liu X, Lu L, Luo F, Lu X, Shi L, Xu W, Liu Q. Epigenetic silencing of p21 by long non-coding RNA HOTAIR is involved in the cell cycle disorder induced by cigarette smoke extract. Toxicol Lett. 2016; 240:60–67.
70. Luo F, Sun B, Li H, Xu Y, Liu Y, Liu X, Lu L, Li J, Wang Q, Wei S, Shi L, Lu X, Liu Q, et al. A MALAT1/HIF-2alpha feedback loop contributes to arsenite carcinogenesis. Oncotarget. 2016; 7:5769–5787. doi: 10.18632/oncotarget.6806.