
INTRODUCTION
Prostate cancer (PC) is the most frequently diagnosed cancer type in men in the developed world [1]. The disease likely originates from high grade prostatic intra-epithelial neoplasia (HGPIN) and progresses to metastatic PC with bone as the most common site [2]. Organ-confined tumors can be managed through multiple means, such as active surveillance, surgery, or radiation. Patients with advanced and metastatic disease can be treated with androgen deprivation therapy (ADT), a strategy pioneered by Charles Huggins in the 1940s [3, 4] and remains the current standard of care. The treatment however is not curative, as resistant tumors known as castration resistant PCs (CRPCs) inevitably develop. While CRPC development is likely mediated by multiple mechanisms, the best understood property of this progression is its dependency of androgen receptor (AR) signalling. Despite low serum testosterone levels following castration (< 50 ng/ml), persistent AR signalling continues, making it a critical contributing factor in CRPC development [5–7]. However, it appears that AR-independent mechanisms are also involved in CRPC acquisition, like in the case of AR-negative neuroendocrine PCs (NEPCs) [8], an aggressive type of CRPC. ADT [9, 10] and inhibition of AR signalling through both abiraterone and enzalutamide [11, 12] has been shown to induce NEPCs. Nevertheless, our understanding of mechanisms underlying PC progression (metastasis and CRPC development) remains incomplete.
The chromosomal locus 8q24 is one of the most frequently amplified and altered regions in a variety of cancer types, including ovarian [13], colorectal [14–17], breast [18–22], prostate [23–28], and others. The 8q24.21 locus is particularly attractive, in part attributable to its containment of the most commonly amplified gene in cancer, MYC. As expected, the 8q24.21 region is amplified in PCs [29, 30]. Variations in the locus associate with an increased risk of PC development [15, 24–26, 31, 32]. The 8q24.21 locus contains a 1.2Mb gene desert bounded by FAM84B and MYC at the centromeric and telomeric end, respectively [15, 21, 24, 25, 27]. A pseudogene of POU5F1, POU5F1P1/POU5F1B, lies within this gene desert [33] and elevations of POU5F1P1 at both the mRNA and protein levels were observed in PCs [34]. While alterations in MYC is well demonstrated during PC tumorigenesis [29, 35], changes in this locus for FAM84B in PCs remain unclear.
We demonstrate for the first time that the FAM84B gene is amplified and expression is upregulated during prostate tumorigenesis, and follows PC progression. FAM84B upregulation was observed in DU145 cell-derived prostate cancer stem-like cells (PCSLCs) in comparison to non-PCSLCs and in prostate carcinoma compared to normal prostate tissues. The upregulation and FAM84B gene amplifications are particularly clear in CRPCs.
RESULTS
Increased levels of FAM84B in PCSLCs
MYC resides in 8q24.21 and is one of four genes that were initially identified to program fibroblast cells into induced pluripotent stem cells (iPSCs) [36]. Considering MYC and FAM84B are located at either end of the 8q24.21 gene desert (Supplementary Figure 1), a potential association of FAM84B with PCSLCs was examined. We have recently established PCSLCs grown as spheres in suspension from DU145 monolayer cells [37]. To examine FAM84B expression in spheres cells versus monolayer cells, we analyzed their gene expression profiles by conducting three separate affymetrix microarrays. A significant increase in FAM84B mRNA in spheres was observed (Figure 1A), while no changes in both MYC and POU5F1P1 were detected, suggesting that the upregulation of FAM84B was specific. This elevation was confirmed by real time PCR and Western blot (Figure 1B, 1C). In comparison to DU145 cells, PC3 and LNCaP cells express higher levels of FAM84B at both the protein and mRNA levels (Figure 1C, 1D). We also noticed an elevation of FAM84B mRNA in another androgen-sensitive PC line (22Rv1) and an immortalized prostate epithelial cell line (BPH-1) compared to DU145 cells (Figure 1D). The underlying reason for increased levels of FAM84B in immortalized BPH-1 cells as well as androgen sensitive LNCaP and 22Rv1 cells compared to more aggressive androgen resistant DU145 cells remains unclear (see Discussion for details). To further examine PCSLC-associated FAM84B upregulation, we analyzed its expression in xenograft tumors produced by either DU145 monolayer or sphere cells. Despite a significantly higher level of FAM84B expression in sphere cells in vitro (Figure 1A. 1B), there were no clear differences in FAM84B protein expression between the two types of xenograft tumors (Figure 1E, Supplementary Figure 2). These observations suggest that the mechanisms underlying FAM84B upregulation in DU145 PCSLCs are attributable to epigenetic changes instead of gene amplification, which may contribute to only FAM84B’s increased expression among the three genes (FAM84B, POU5F1P1, and MYC) in the 8q24.21 gene desert (Supplementary Figure 1).
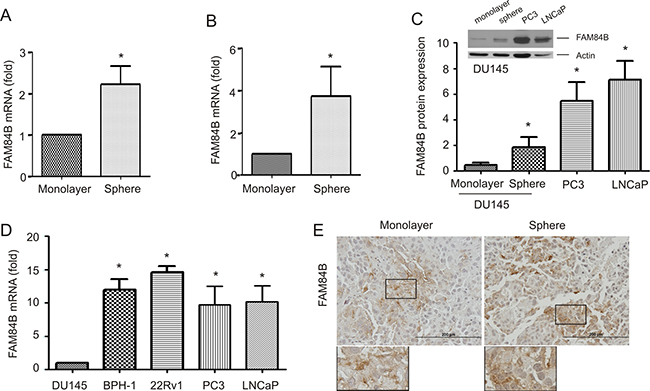
Figure 1: Upregulation of FAM84B in prostate cancer stem-like cells (PCSLCs). (A) Affymetrix profiling gene expression in DU145 monolayer and sphere cells. The profiling was carried out three times with duplicates for each repeat. FAM84B mRNA is expressed as fold change compared to monolayer cells. Mean±SD (standard deviation) are graphed. Statistical analysis was performed using 2-tailed Student’s t-test. (B, D) Real-time PCR analysis of FAM84B mRNA in DU145 monolayer, DU145 sphere cells (PCSLCs), and the indicated PC lines. β-actin was used as an internal control. Experiments were repeated three times. FAM84B mRNA abundance is graphed as a fold change to DU145 monolayer cells; mean ± SD are graphed. *:p < 0.05 by a 2-tailed Student’s t-test. (C) Western blot examination of FAM84B protein expression in DU145 monolayer and sphere, PC3, and LNCaP cells. Experiments were repeated three times; typical images from a single repeat are shown (inset). FAM84B protein levels were normalized to actin; mean±SD are graphed. *p < 0.05 by a 2-tailed Student’s t-test in comparison to DU145 monolayer cells. (E) FAM84B protein expression in xenograft tumors produced by either DU145 monolayer or sphere cells. The overall patterns of FAM84B expression at low magnification are presented in Supplementary Figure 2.
FAM84B upregulation associates with PC tumorigenesis
We have previously shown that DU145 sphere cells (PCSLCs) display a 100-fold higher tumorigenic ability [37]. While we did not detect elevated FAM84B expression in sphere cell-derived xenograft tumors (see Discussion for details) (Figure 1E, Supplementary Figure 2), it remains possible that FAM84B upregulation associates with PC tumorigenesis and/or progression. To investigate this scenario, we produced subcutaneous (s.c.) xenograft tumors and lung metastasis (via tail vein injection) using DU145 monolayer cells [38]. FAM84B expression was heterogeneously detected in both tumor types (Supplementary Figure 3A); FAM84B is largely a cytosolic protein (Figure 2A, enlarged images), and is clearly expressed at an elevated level in lung metastasis (Figure 2A, 2B; Supplementary Figure 3A). Interestingly, the staining showed higher levels of expression in the tumor edges adjacent to normal mouse lung tissue (Supplementary Figure 3A), suggesting a role of FAM84B in facilitating PC metastasis. This concept is supported by our demonstration of FAM84B expression in 2 out of 4 human PC bone metastases (Figure 2C, Supplementary Figure 3B). Collectively, these observations support an association of FAM84B upregulation with PC progression.
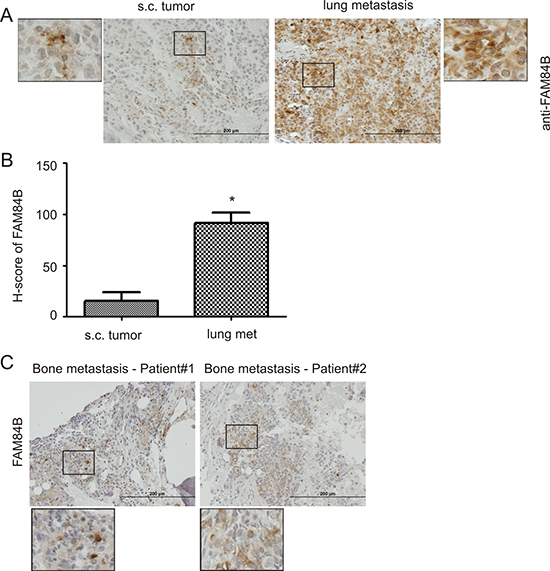
Figure 2: Upregulation of FAM84B in metastatic PC. DU145 subcutaneous (s.c.) xenograft tumors and lung metastasis were produced in NOD/SCID mice (5 mice per group). (A) Typical IHC staining for FAM84B in s.c. tumors and lung metastasis (see Supplementary Figure 3A for overall staining). The indicated regions are enlarged three fold and placed on the side of the original panel. (B) IHC staining was quantified through ImageScope software. Average HScores±SDs are graphed. *p < 0.05 by a 2-tailed Student’s t-test. (C) IHC staining for FAM84B was performed on four human bone metastases; two tumors with positive staining are shown here (see Supplementary Figure 3B for additional images). Indicated regions are enlarged 2.5 fold and placed beneath the original panel.
To further investigate this association, we determined FAM84B mRNA levels in 7 pairs of PC and benign prostate tissues using Nanostring technology. The PC tissues contained 60–80% cancer tissue. This collection of samples revealed PTEN reductions and ERG increases (indicative of TMPRSS2-ERG fusion) in 5/7 of the PC cases (Table 1), which validated these tissues for analysis of PC-associated gene expression. Impressively, an elevation of FAM84B mRNA was detected in all carcinomas compared to the matched non-tumor prostate tissues (Table 1; p = 0.002 by a 2-tailed Student’s t-test), supporting FAM84B upregulation in PCs.
Table 1: Nanostring analysis of gene expression in primary prostate cancer tissues
Genes |
P1a |
P2a |
P3a |
P4a |
P5a |
P6a |
P7a |
|---|---|---|---|---|---|---|---|
FAM84B |
+1.3 |
+3 |
+1.2 |
+2.6 |
+1.6 |
+2.7 |
+2.1 |
TMPRSS2-ERGb |
+16.2 |
+30.2 |
Nc |
N |
+17 |
+25.3 |
+27.3 |
PTENb |
–1.4 |
–1.4 |
N |
N |
–1.4 |
–1.3 |
–2.6 |
a:patients 1-3 (GS6), patients 4-6 (GS7, 4+3 for P4,5, and 3+4 for P6), and P7 (GS4+4).
b:TRPRSS2-ERG and PTEN were used as positive controls for upregulated and downregulated genes in PC. downregulated or upregulated genes.
c:no downregulation or upregulation.
To further examine this concept, we analyzed gene expression data available from three major databases; Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) [39], OncomineTM (Compendia Bioscience, Ann Arbor, MI), and cBioPortal [40, 41] (http://www.cbioportal.org/index.do). We extracted FAM84B mRNA expression data from GSD2546 (dataset2546) (http://www.ncbi.nlm.nih.gov/sites/GDSbrowser) in GEO [42, 43] (Supplementary Figure 4). The dataset contained 17 normal prostate, 59 tumor adjacent normal prostate tissues (n = 76), 66 primary PCs, and 25 metastases (lymph node, liver, kidney, and others) (Supplementary Figure 4). A significant increase in FAM84B mRNA can be demonstrated following PC progression from normal prostate tissues to carcinoma and to metastasis (Figure 3A). Elevated FAM84B mRNA in primary PCs over normal prostate tissues was also observed using the FAM84B mRNA data extracted from the Taylor, Grasso, Lapointe, and Vanaja datasets [44] (Figure 3B–3E) within the OncomineTM database [45–47]. While metastatic PCs expressed higher levels of FAM84B than the normal prostate tissues (Figure 3B–3D), a significant increase in metastases over primary tumors can only be demonstrated in the Taylor dataset (Figure 3B). Together the five datasets contain 181 normal prostates and 343 primary PCs (Figure 3A–3E), and validate the association of FAM84B upregulation with prostate tumorigenesis. This concept is further supported by their respective receiver-operating characteristic (ROC) curves that separate normal prostates from primary PCs via an area under curve (AUC) value ranging from 0.69 to 0.9 (Figure 3F–3H). Nonetheless, the concept of whether FAM84B upregulation correlates with PC progression to metastatic disease warrants further investigations.
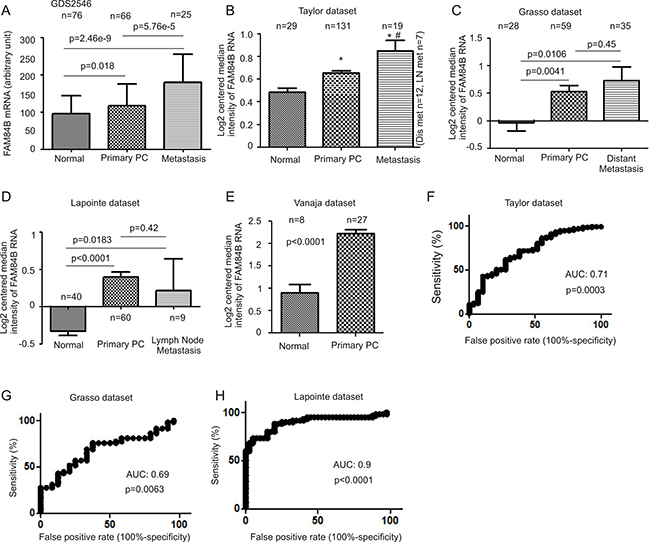
Figure 3: FAM84B upregulation associates with PC tumorigenesis. Data were extracted from GDS2546 (Gene Expression Omnibus) (A), and OncomineTM (Compendia Bioscience, Ann Arbor, MI) datasets Taylor (B), Grasso (C), Lapointe (D), and Vanaja (E) for analysis of changes in FAM84B mRNA. Mean ± SD are graphed. *p < 0.05 by a 2-tailed Student’s t-test in comparison to Normal; #p < 0.05 in comparison to primary PC (2-tailed Student’s t-test). (F–H) A receiver-operating characteristic (ROC) curve of primary prostate tumors versus metastatic PCs was derived from the indicated datasets. AUC: area under the curve.
Higher levels of FAM84B in primary PCs may be in part attributable to increases in gene copy number (GCN). In an analysis of FAM84B GCN data extracted from the TCGA dataset within OncomineTM, FAM84B GCN is significantly increased in 171 primary PCs compared to 61 normal prostate tissues (Figure 4A). The differences separate the two groups with an AUC value of 0.86 (p < 0.0001) (Figure 4B).
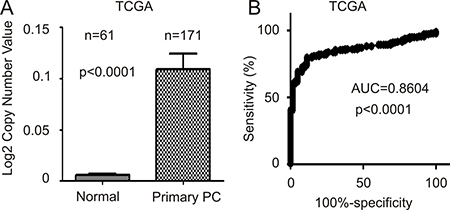
Figure 4: FAM84B gene amplification in prostate tumors. FAM84B gene copy variation data were extracted from the TCGA dataset within the OncomineTM database. (A) Mean ± SD are graphed. Statistical analysis was performed using 2-tailed Student t-test. (B) A ROC curve of primary PC versus normal prostate tissues was calculated.
FAM84B expression does not correlate with primary PC progression
Primary PCs can be divided into and analyzed as low grade (Gleason score 6–7/GS6-7) and high grade (GS8-10) tumors [48]. Data extracted from the Taylor dataset [45] indicated FAM84B mRNA levels in both low and high grade PCs are significantly higher than in normal prostates (Figure 5A). Although the number of cases with high grade PCs was limited, high grade PCs do not seem to display higher levels of FAM84B than low grade PCs (Figure 5A). We further examined FAM84B protein expression via immunohistochemistry in our limited patient cohort consisting of 4 low and 18 high grade prostate carcinomas (Supplementary Table 1). The FAM84B protein was clearly detected in low and high grade PCs (Figure 5B). However, we could not demonstrate higher levels of FAM84B expression in high grade PCs (Figure 5C). While FAM84B is largely a cytosolic protein in PC cells (Figure 5B), nuclear staining was also observed in high grade cases (Supplementary Figure 5).
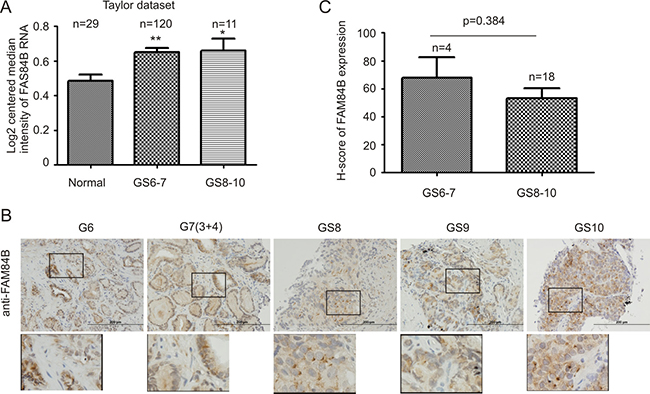
Figure 5: FAM84B upregulation does not associate with Gleason score (GS) advancement. (A) FAM84B mRNA expression data were extracted from the Taylor dataset in the OncomineTM database, and were analyzed as low grade (GS6-7) or high grade (GS8-10) PCs. *p < 0.05 and **p < 0.01 in comparison to normal prostate tissues (2-tailed Student’s t-test). (B) IHC staining for FAM84B in 22 primary PC tissues (Supplementary Table 1). Typical images for tumors with the indicated GS are shown. Scale bars represent 200 μm (See Supplementary Figure 5 for additional images). (C) IHC staining was quantified through ImageScope software. Average HScores ± SDs are graphed (for detailed scores, See Supplementary Table 1). Statistical analyses were performed using Student’s t-test.
FAM84B upregulation correlates with CRPC development
A critical form of PC progression is the development of CRPC. To examine whether FAM84B upregulation associates with this, we determined FAM84B expression in vitro and in vivo under androgen deprivation conditions. Androgen free media for 24 hours robustly reduced prostate specific antigen (PSA) expression in LNCaP cells as expected (Supplementary Figure 6A), however, FAM84B mRNA levels were only slightly reduced (Supplementary Figure 6B). This suggests that FAM84B transcription is not directly controlled by AR signaling.
To further investigate FAM84B expression in the process of CRPC development, we generated xenograft tumors using LNCaP cells in intact and castrated NOD/SCID mice (Figure 6A). Castration delayed tumor growth, which was followed by the generation of castration resistant tumors as indicated by changes in PSA levels following castration (Figure 6A). In comparison to xenograft tumors in intact mice, castration resistant tumors demonstrated increases in FAM84B mRNA expression (Figure 6B).
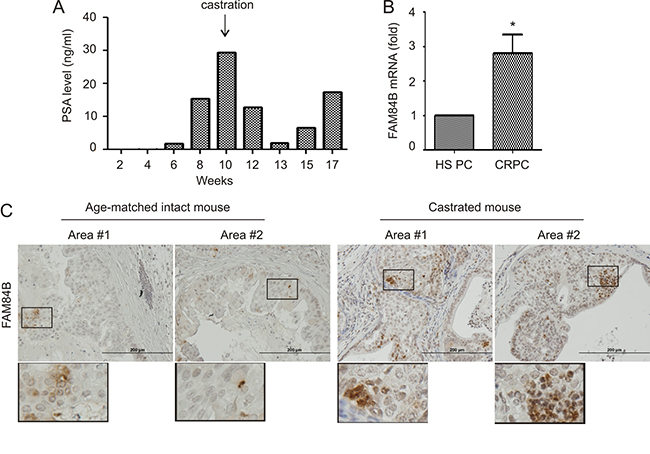
Figure 6: Alterations of FAM84B expression in animal models of castration resistant PC. (A) PSA levels in NOD/SCID mice (n = 3) bearing LNCaP cell-derived xenograft tumors prior to and after castration; a typical pattern from a single mouse is shown. (B) Real time PCR analysis of FAM84B mRNA in hormone sensitive (HS) and castration resistant PC (LNCaP xenograft tumors). *p < 0.05 by a 2-tailed Student’s t-test in comparison to hormone naive xenograft tumors. HS PC: hormone sensitive prostate cancer. (C) IHC staining of FAM84B in prostate tumors produced in intact and castrated PTEN–/– mice (n = 3; see Supplementary Figure 7 for the production of prostate specific PTEN–/– mice). Indicated regions are enlarged 2.5 fold and placed beneath the original panel.
We also examined FAM84B expression in PCs generated in prostate-specific PTEN–/– mice with and without castration. Prostate specific PTEN–/– mice were castrated at 23 weeks and euthanized after 13 weeks, and presented with CRPC (Supplementary Figure 7A, 7B). In comparison to hormone naive tumors, prostate tumors produced in castrated PTEN–/– mice exhibited an elevated level of FAM84B protein expression (Figure 6C). Interestingly, FAM84B is heterogeneously expressed and the positive cells can be clearly detected as cell clusters particularly in castrated mice (Figure 6C; Supplementary Figure 7C), suggesting that FAM84B-positive cells may contribute to the clonal expansion of CRPC cells. Additionally, they were located at the edges of tumor adjacent to normal prostate tissue (Supplementary Figure 7C, image #3). Taken together, the above observations support a correlation between FAM84B upregulation and CRPC development.
To continue examining this correlation, we extracted FAM84B mRNA information from cBioPortal datasets and performed analyses using tools provided on the website [40, 41]. There are currently 5 datasets containing mRNA data in cBioPortal, which cover primary prostate tumors and metastatic castration resistant prostate cancers (mCRPCs) (cBioPortal/ http://www.cbioportal.org/index.do) [45, 49–52]. In two datasets with 549 primary PCs, increases in FAM84B mRNA were detected in 12% (11–13%) of PCs (Table 2); in comparison, FAM84B upregulation occurred in 18% (9–27%) of mCRPCs in three datasets consisting of 412 mCRPCs (Table 2). The differences in the distribution of tumors with FAM84B upregulation in primary PCs and mCRPCs are statistically significant (p < 0.025, Table 2). Collectively, our comprehensive analyses (xenograft tumors, PCs generated in prostate specific PTEN–/– mice, and primary PCs) strongly support the notion that FAM84B upregulation correlates with CRPC development.
Table 2:a Upregulation of FAM84B mRNA following PC progression
Dataset |
PC type |
Casesb |
Upregulationc (%) |
Ref |
|---|---|---|---|---|
CGARN |
Primary PC |
333 |
11% (38/333) |
Cell 163, 1011–25, 2015 |
Taylor et al |
Primary PC |
216 |
13% (29/216) |
Cancer Cell 18, 11–22, 10 |
Total |
549 |
12% (67/549) |
||
Kumar et al |
mCRPCd |
176 |
9% (16/176) |
Nat Med 22, 369–28, 16 |
Robinson et al |
mCRPC |
118 |
22% (26/118) |
Cell 161, 1215–28, 2015 |
Beltran et al |
mCRPC |
114 |
27% (31/114) |
Nat Med 22, 298–305, 16 |
Total |
412 |
18% (73/412)* |
a: data were extracted from cBioPortal; b: number of cases; c: upregulation was defined by z-score ≥ 2; d: metastatic castration resistant prostate cancer; *p < 0.025 in comparison to the average rate of FAM84B upreglation by Fisher’s Exact Test.
FAM84B gene amplification correlates with CRPC development
We subsequently analyzed FAM84B gene amplification and its association with CRPC. The Taylor dataset within OncomineTM contains GCN data for 181 primary PCs and 37 metastatic prostate tumors [45]. FAM84B GCN is significantly increased in metastatic PCs compared to primary tumors (Figure 7A, left panel). The metastatic PCs consist of mCRPC and lymph node (LN) metastases [45]; the significant increases in FAM84B GCN observed in the metastatic PC population were attributed to mCRPCs (Figure 7B, left panel). Additionally, the respective ROC curves differentiate primary PCs from mCRPCs (distant metastases) with increased accuracy (AUC = 0.82; Figure 7B, right panel) compared to all metastases including LNs (AUC = 0.75; Figure 7A, right panel).
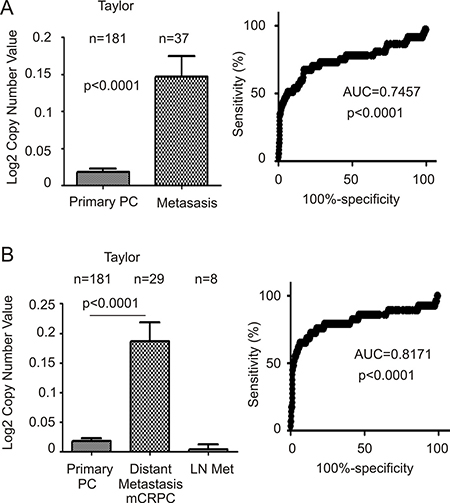
Figure 7: FAM84B is increased in metastatic castration resistant prostate cancer (mCRPC). Data related to FAM84B gene copy number was extracted from the Taylor dataset within OncomineTM (Compendia Bioscience, Ann Arbor, MI). (A) mean ± SD (left panel) and a ROC curve (right panel) of primary versus metastatic PC were calculated and graphed. Statistical analyses were performed using Student’s t-test. (B) The same data were analyzed by separating out distant metastasis (Dis met)/mCRPC from lymph node metastases (LN met). An ROC curve for this arrangement is shown.
The association of FAM84B gene amplification was also demonstrated using seven large datasets within the cBioPortal database [45, 46, 50–54] (Table 3). While the FAM84B gene was amplified in 4.8% (0.9–7%) of 546 primary PCs, an average rate of 26% (13–44%) was found in 467 mCRPCs (Table 3, p < 0.0001).
Table 3:a FAM84B gene amplification in CRPCs
Dataset |
PC type |
Casesb |
Upregulationc (%) |
Ref |
|---|---|---|---|---|
Barbieri et al |
Primary PC |
109 |
0.9% (1/109) |
Nat Genet 44, 685–9, 2012 |
Hieronymus et al |
Primary PC |
104 |
2% (2/104) |
PNAS 111, 11139–44, 2014 |
Taylor et al |
Primary PC |
333 |
7% (23/333) |
Cancer Cell 18, 11–22, 2010 |
Total |
546 |
4.8% (26/546) |
||
Kumar et al |
mCRPCc |
149 |
30% (44/149) |
Nat Med 22, 369–28, 16 |
Robinson et al |
mCRPC |
150 |
13% (20/150) |
Cell 161, 1215–28, 2015 |
Grasso et al |
mCRPC |
61 |
16% (10/61) |
Nature 487, 239–43, 2012 |
Beltran et al |
mCRPC |
107 |
44% (47/107) |
Nat Med 22, 298–305, 16 |
Total |
467 |
26% (121/467)* |
a: data were extracted from cBioPortal.
b: number of cases.
c: metastatic castration resistant prostate cancer.
*p < 0.0001 in comparison to the average rate of FAM84B gene amplification by Fisher’s Exact Test.
Persistent AR signalling though alterations in AR is known to contribute to CRPC progression [5], suggesting a relationship between genomic changes of the FAM84B and AR genes. In support of this possibility, we obtained evidence for a co-occurrence of AR genomic changes and increased FAM84B GCNs; co-occurrence of the two covers a large proportion of the mCRPCs with FAM84B gene amplification in individual datasets (Figure 8A, Supplementary Figure 8A) [46, 50–52]. The co-occurrence also includes AR and FAM84B mRNA upregulation (Figure 8B, Supplementary Figure 8B), suggesting a role of AR signalling in FAM84B’s genomic alterations and upregulation.
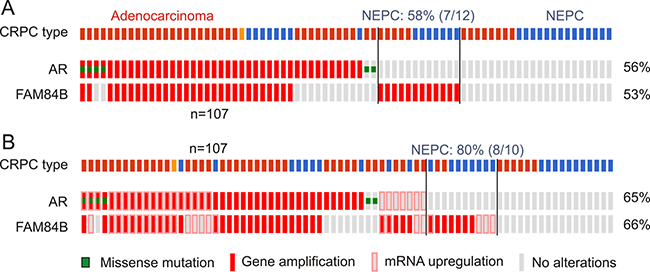
Figure 8: Genomic alterations of the AR and FAM84B genes. Data were extracted from the Beltran dataset within the cBioPortal database [40, 41]. The dataset covers 107 CRPCs derived from 77 patients [52]. The indicated genomic alterations without (A) and with the respective mRNA upregulations (B) in 77 CRPC patients are shown. The percentages of alterations were based on the patient population (n = 77). CRPC tumor types are also indicated; red bars: adenocarcinoma, blue bars: neuroendocrine prostate cancer (NEPC), orange bar: adenocarcinoma mixed with NEPC. Symbols for genomic mutations and mRNA upregulation for the AR and FAM84B genes are indicated.
These changes in FAM84B also take place independently of AR signalling (Figure 8, Supplementary Figure 8). Intriguingly, in those mCRPCs, 58% (7/12) are the neuroendocrine type (NEPC) (Figure 8A); with FAM84B mRNA upregulation included, the NEPC enrichment reaches 80% (8/10) (Figure 8B), suggesting that CRPCs with changes occurring only in FAM84B are likely NEPCs. In the other three datasets, it would be interesting to examine how many NEPC cases are among the 25 mCRPCs with changes detected only in FAM84B (Supplementary Figure 8).
FAM84B genomic changes and mRNA upregulation associate with a reduction in disease free survival (DFS)
CRPC is a major stage of PC evolution. Our observed correlation of FAM84B changes (genomic alterations and mRNA upregulation) with CRPC development indicates a relationship with PC recurrence (DFS). Of note, by extracting and analyzing genomic and mRNA data from 194 prostate tumors with copy number alterations (CNA) from the “MSKCC, Cancer Cell 2010” dataset [45] within cBioPortal, we were able to show that FAM84B gene amplification alone and with its mRNA upregulation (Supplementary Figure 9A) associates with decreases in DFS (Figure 9A). As expected, similar changes in the AR gene (Supplementary Figure 9B) resulted in a rapid PC recurrence (Figure 9B). Although the combination of changes in AR and FAM84B decreases the power of AR-associated changes in predicting PC recurrence (comparing the respective curves and p values in panel B and C, Figure 9), the combination covers more recurrent tumors; 7 for AR versus 9 for AR+FAM84B (left panels of Figure 9B, 9C) and 15 for AR vs 21 for AR+FAM84B (right panels of Figure 9B, 9C). These analyses thus support the notion that genomic changes and mRNA upregulation in FAM84B contribute to AR-associated reduction in DFS, a concept that is in line with the aforementioned changes in FAM84B occurring concurrently and independently of the respective changes in AR in this patient cohort (Supplementary Figure 9C).
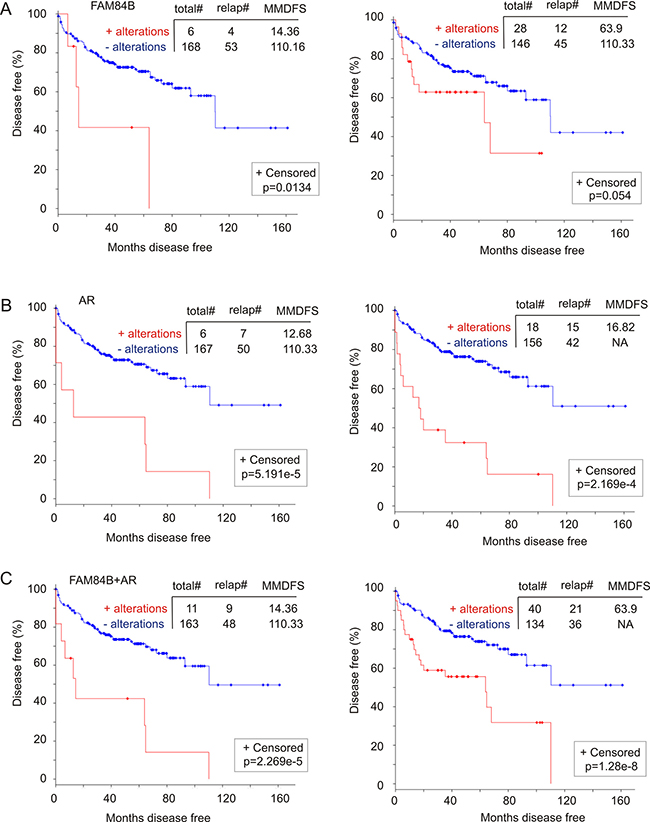
Figure 9: Genomic alterations in the FAM84B gene associate with a reduction in disease free survival (DFS). A dataset of primary PCs [45] (within the cBioPortal database [40, 41]) were used to assess the impact of FAM84B (A), AR (B), and FAM84B+AR (C) genomic changes on DFS without (left panels) and with the respective mRNA upregulation (right panels). The detailed alterations are documented in Supplementary Figure 9. Statistical analysis was performed using Logrank Test. Total#: total number of cases; relap#: number of relapsed cases; MMDFS: median months disease free survival; NA: not available. Censored individuals are indicated; the number of censored individuals is the total individuals minus relapsed patients.
To further investigate FAM84B gene amplification-associated reductions in DFS, we analyzed a cohort of 492 patients in the TCGA dataset within the cBioPortal database (http://www.cbioportal.org/) in which the PCs have been examined for copy number variations (CNVs) [49]. FAM84B CNVs (largely amplification) were detected in 8% (37/492) of PCs, while AR CNVs occurred in 1% of prostate tumors (Supplementary Figure 10A). In this patient cohort, FAM84B CNVs correlate with a trend of DFS decreases (Figure 10A) and the combination of FAM84B and AR CNVs increased the prediction power (comparing the p values in Figure 10A to that in Supplementary Figure 10C). However, the differences do not reach the p < 0.05 level of significance (Supplementary Figure 10C).
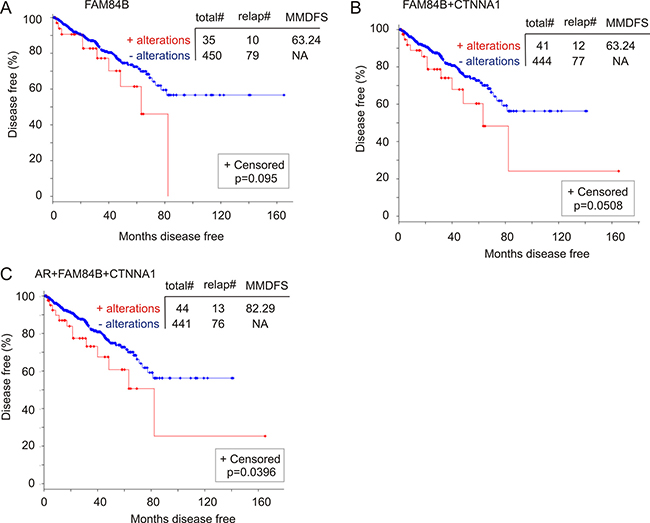
Figure 10: Genomic alterations in the FAM84B network genes associate with a reduction in DFS. The TCGA dataset within the cBioPortal database [40, 41] contains 492 primary prostate tumors with copy number variation determined. The detailed genomic alterations in the FAM84B, CTNNA1, and AR genes are presented in Supplementary Figure 10. The effects of these changes with respect to the FAM84B (A), FAM84B+CTNNA1 (B) or FAM84B+CTNNA1+AR (C) are calculated. Statistical analysis was performed using Logrank Test. Total#: total number of cases; relap#: number of relapsed cases; MMDFS: median months disease free survival; NA: not available. Censored individuals are indicated; the number of censored individuals is the total individuals minus relapsed patients.
FAM84B has not been thoroughly studied in the current literature, which results in our lack of knowledge about the proteins or factors that affect FAM84B function. Nonetheless, the cBioPortal database indicates an interactive network between FAM84B and CTNNA1 (α-catenin 1) (Supplementary Figure 11A). In a mCRPC cohort of 107 tumors obtained from 77 patients [52], the CTNNA1 gene was amplified in 21% (16/77) of patients and 15% (16/107) of mCRPCs (Supplementary Figure 11B). Importantly, CTNNA1 gene amplification displays co-occurrence and independence with FAM84B gene amplification (Supplementary Figure 11B). The co-occurrence is also observed with AR and becomes more apparent when mRNA upregulations are included in the comparisons (Supplementary Figure 11C). These observations together with the different chromosomal locations for FAM84B (8q24.21) and CTNNA1 (5q31.2) collectively support a network relationship between the two proteins. Of note, we noticed that although CTNNA1 genomic alterations were detected in 1% of the TCGA cohort of 492 patients (Supplementary Figure 10A), genomic alterations in the network are likely associated with DFS reductions (Figure 10B). Furthermore, the combination of AR genomic changes with those of the network associates with a decline in DFS (Figure 10C). Taken together, evidence supports an association of FAM84B genomic alterations with decreases in DFS in PC patients.
FAM84B genomic alterations correlate with a reduction in overall survival (OS)
We examined a potential association of FAM84B genomic alterations with OS. Among the 11 datasets related to genomic alteration in PC from cBioPortal [40, 41], two have follow-up data for OS, one for mCRPC patients [46] and another for primary PCs (n = 492) with CNVs determined (TCGA, http://www.cbioportal.org/) [49]. Genomic changes in AR, FAM84B, and CTNNA1 individually or in any combination have no relationship with OS in mCRPC patients (data not shown). In the primary PC patient cohort, the 8% of genomic alterations in the FAM84B gene significantly associate with a reduction in OS (Figure 11A). Although the number of cases associated with PC-related fatality is small (n = 9), a third (3/9) of these deaths occurred in patients with prostate tumors in which the FAM84B gene has been altered (Figure 11A). Combination with either CTNNA1 or AR does not enhance the association (Figure 11B, 11C). In fact, all fatalities in patients with PC harboring either AR or CTNNA1 genomic alterations were observed in patients with tumors containing FAM84B genomic alterations. Taken together, evidence suggests that genomic alterations in the FAM84B gene correlate with poor prognosis in patients with primary prostate tumors.
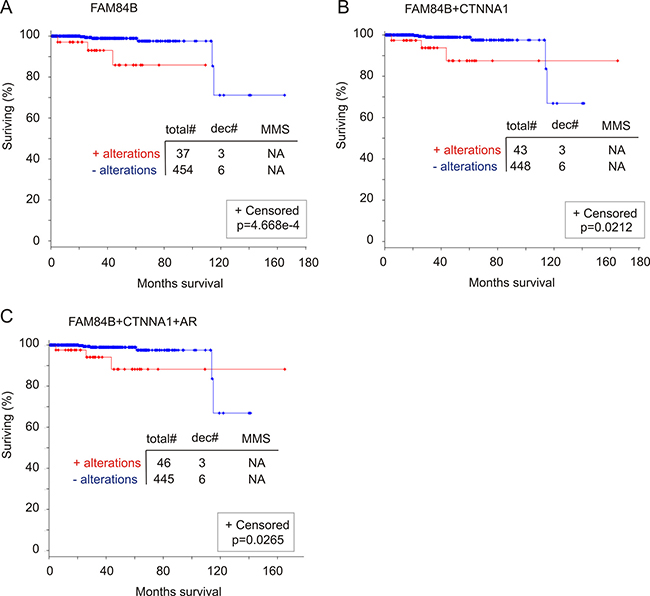
Figure 11: Genomic alterations in the FAM84B gene associate with a reduction in overall survival (OS). The TCGA dataset within the cBioPortal database [40, 41] contains 492 primary prostate tumors with copy number variation determined. The detailed genomic alterations in the FAM84B, CTNNA1, and AR genes are presented in Supplementary Figure 10. The effects of genomic alterations involving the FAM84B (A), FAM84B+CTNNA1 (B) or FAM84B+CTNNA1+AR (C) are calculated. Statistical analysis was performed using Logrank Test. Total#: total number of cases; dec#: number of deceased cases; MMS: median months survival; NA: not available. Censored individuals are indicated; the number of censored individuals is the total individuals minus relapsed patients.
DICUSSION
The chromosome locus 8q24 is one of the most frequently modified loci in cancer and these alterations are associated with the development of PC and other cancer types [27, 33]. The tumorigenic functions conferred by the 8q24 regions can be attributable to the changes in a gene desert at 8q24.21 that is flanked by FAM84B and MYC [27, 28, 33, 55, 56]. While it has been puzzling as to why the non-coding region is frequently targeted during tumorigenesis, a recent development revealed the gene desert as being a regulatory hub in causing genomic alterations in other chromosome loci via physical interaction, including CD96 at 3q13, during prostate tumorigenesis [57]. While this research sheds light on the functional consequences of those alterations in the gene desert, genomic changes of MYC have been well demonstrated in promoting tumorigenesis. However, the association of FAM84B, another major component of the gene desert at the 8q24.21 locus (Supplementary Figure 1), with tumorigenesis remains unclear.
This research is the first attempt to thoroughly investigate the correlation of FAM84B with PC tumorigenesis and progression. By using comprehensive experimental systems involving tissue culture, xenograft tumors, and transgenic mice, by examining alterations in the FAM84B gene and mRNA levels, and by analyzing multiple large datasets, this investigation reports several novel observations: FAM84B upregulation occurs in PCSLCs, FAM84B expression is elevated in primary PCs compared to normal prostate tissues, and there is a significant increase in FAM84B mRNA and gene amplification in CRPC development.
The upregulation of FAM84B could only be detected in DU145-derived PCSLCs in vitro. In xenograft tumors produced by PCSLCs and non-PCSLC DU145 cells, elevation of FAM84B expression could not be demonstrated in the prior (Figure 1E, Supplementary Figure 2). This observation in xenograft tumors is likely attributable to PCSLC-mediated tumor regeneration, in which non-PSCLCs are produced. In supporting this possibility, we observed a reduction in PCSLCs self-renewal ability following culture in medium containing 10% fetal bovine serum [37] and also a reduction in PCSLC-associated proteins in xenograft tumors derived from them [38]. However, these tumors do contain PCSLCs which can be re-isolated from the tumor tissue [37]. As a result, we suggest that FAM84B upregulation could not be detected in PCSLC-produced xenograft tumors due to the tumor regeneration process in which the number of PCSLCs are reduced.
The significant upregulation of FAM84B in primary prostate tumors over normal prostate tissues suggests a contribution of FAM84B in PC initiation. However, the mechanisms underlying these changes in FAM84B are likely complex. Nonetheless, our observed FAM84B upregulation in DU145 cell-derived PCSLCs suggests PCSLC-associated plasticity as being a contributor to FAM84B upregulation during PC initiation. This is a possibility that is well in line with the concept of how PCSLCs are also referred to as PC initiating cells. The contribution of PCSLCs to FAM84B upregulation is also supported by the heterogeneous expression of FAM84B protein in xenograft tumors (Figures 1E, 2A) and CRPCs produced in PTEN–/– mice (Figure 6C). PCSC-mediated FAM84B upregulation during PC tumorigenesis is also in accordance with an elevation of FAM84B mRNA during CRPC development. It is now well-regarded that the plasticity of cancer stem cells drives therapeutic resistance. This plasticity likely contributes to the acquisition of new properties underlying CRPC development. Thus, PCSC-derived plasticity is likely a contributing factor to FAM84B upregulation.
The functional impact of FAM84B on tumorigenesis in general and prostate oncogenesis in particular remains to be explored. Nonetheless, evidence suggests an important role of FAM84B in PC progression. We observed an association of FAM84B genomic alteration and mRNA upregulation with PC recurrence (Figure 9). These results support the theme of this work when they are considered in the context of the research. However, this data should be cautiously interpreted, as the number of patients with alterations is limited (Figure 9A). We also noticed a high level of FAM84B protein expression in the edge regions of DU145 cell-derived lung metastasis and CRPC produced in PTEN deficient mice (Supplementary Figure 3A; Supplementary Figure 7C, image #3), indicating a possible role of FAM84B in mediating PC cell invasion. This concept is supported by recent observations in which knockdown of FAM84B reduced esophageal squamous cell carcinoma cell invasion in vitro and from forming xenograft tumors in vivo [58, 59]. Whether FAM84B performs a similar function in PC tumorigenesis, particularly in PC metastasis, will be examined in the future.
A possible role of FAM84B in PC progression lies in its potential contributions to CRPC. This possibility is supported by the specific FAM84B upregulation mentioned here (Table 2); this increase occurred uniquely in CRPC and was observed in several independent and large cohorts (Table 2). Furthermore, in our xenograft model for CRPC and transgenic CRPC mouse model, CRPCs exhibited elevated levels of FAM84B compared to hormone naïve tumors (Figure 6). It is thus tempting to propose a role of FAM84B in promoting CRPC development. This hypothesis is supported by a robust elevation in the rate of FAM84B gene amplification from an average 4.8% among 546 primary prostate tumors to 26% in 467 mCRPCs (Table 3). However, it should be stressed that an association of FAM84B upregulation with CRPC is certainly more complex than the aforementioned statements above. For example, FAM84B mRNA is not expressed at higher levels in androgen-resistant DU145 and PC3 cells in comparison to androgen-sensitive LNCaP and 22Rv1 cells or even immortalized human prostate epithelial BPH-1 cells (Figure 1D). The inconsistency between these cell line-based observations and the in vivo results (animal models) may be attributable to different systems. Clearly, additional research is required to understand these differences; particularly functional studies need to be performed to evaluate the impact of FAM84B on CRPC development.
While detailed mechanisms leading to FAM84B gene amplification specifically in CRPC remain to be investigated, it is likely that the gene desert, which is bounded by FAM84B at the centromeric side, plays a role. The genomic regulatory elements within the 8q24.21 gene desert were reported to physically associate with FAM84B in a region from 266–440 kb away from the 3′ end of the FAM84B gene in multiple PC cell lines [57]. It is also likely that AR signaling during androgen deprivation also contributes to FAM84B gene amplification. This is in accordance with the importance of persistent AR signalling in CRPC development [5] and in causing genomic instability [60–62]. Furthermore, this possibility is supported by the observed concordance between AR gene amplification and that of FAM84B (Figure 8).
MATERIALS AND MATHODS
Cell culture and generation of DU145 spheres (PCSLCs)
LNCaP, PC3, and DU145 cells were purchased from American Type Culture Collection (ATCC), and cultured in RPMI-1640 (LNCaP), F12 (PC3) and MEM (DU145) media supplemented with 10% FBS (Sigma Aldrich) and 1% Penicillin-Streptomycin (Thermo Fisher Scientific). DU145 spheres were generated and cultured according to our published conditions [37]. Briefly, DU145 monolayer cells (non-PCSLCs) were individualized and seeded at a density of 5,000 cells/mL in serum-free (SF) media (3:1 DMEM/F12 mixture) (Thermo Fisher Scientific) containing 0.4% bovine serum albumin (BSA) (Bioshop Canada Inc.) supplemented with 0.2× concentration of B27 minus Vitamin A (Thermo Fisher Scientific) and 10 ng/ml EGF (Sigma Aldrich), in T75 flasks. Typical spheres were formed in 10 to 12 days.
Collecting primary prostate cancer
Prostate biopsies and radical prostatectomy tissues were obtained at St. Joseph’s Hospital in Hamilton, Ontario, Canada under approval from the local Research Ethics Board (REB# 11-3472) and with patient consent.
Xenograft tumor formation
DU145 (106) or LNCaP cells (5 × 106) were resuspended in 0.1 ml culture media/Matrigel mixture (BD) (1:1 volume), followed by subcutaneous implantation into the flanks of 8 week-old male NOD/SCID mice (The Jackson Laboratory). Tumors were assessed through observation and palpation, and tumor growth was measured weekly using calipers. Tumor volume was determined using the formula V = L × W2 × 0.52. Animals were sacrificed once tumors reached a volume ≥ 1000 mm3. For the generation of lung metastasis, 106 DU145 cells were resuspended into 0.3 mL of PBS and injected through the tail vein of NOD/SCID mice. Lungs were harvested at 16 weeks post injection. All animal work was carried out according to experimental protocols approved by the McMaster University Animal Research Ethics Board.
Generation of castration resistant prostate cancer in vivo
LNCaP cells (5 × 106) were s.c. implanted into NOD/SCID mice. Tumor growth was monitored by measuring serum PSA levels (PSA kit, Abcam) and physically with calipers as described above. When tumor volume approached 100–200 mm3, mice were surgically castrated. Serum PSA was measured prior to and following castration. CRPC was defined when serum PSA rose while tumors continued to grow. Animals were sacrificed once tumors reached a volume ≥ 1000 mm3.
PTENloxp/loxp mice (C;129S4-Ptentm1Hwu/J) were obtained from The Jackson Laboratory, and PB-Cre4 mice (B6.Cg-Tg(Pbsn-cre)4Prb) were obtained from the NCI Mouse Repository. Prostate specific PTEN–/– mice were produced by crossing the two strains. Surgical castration was performed when PTEN–/– mice were 23 weeks old, and observed for 13 weeks before sacrificing.
Western blot analysis
Cells were lysed in a buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 25 mM sodium pyrophosphate, 1 mM NaF, 1 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM PMSF, 2 μg/ml leupeptin and 10 μg/ml aprotinin. 50 μg of whole cell lysate was separated on SDS-PAGE gel, and transferred onto Hybond ECL nitrocellulose membranes (Amersham), followed by blocking with 5% skim milk at room temperature for one hour. Primary antibodies were incubated overnight at 4°C with agitation, and secondary antibodies incubated for one hour at room temperature. Signals were then developed (ECL Western Blotting Kit, Amersham). Primary antibodies: anti-FAM84B 1:1000 (Proteintech) and anti-Actin 1:1000 (Santa Cruz).
Quantitative real-time PCR analysis of FAM84B expression
Total RNA was isolated from prostate cancer cell lines and xenograft tissues with Isol-RNA Lysis Reagent (5 PRIME), and reverse transcription was carried out using Superscript III (Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems) using SYBR-green (Thermo Fisher Scientific). All samples were run in triplicate. FAM84B (Forward): 5′-GACCCAC CTAAGTTACAAGGAAG-3′, FAM84B (Reverse): 5′- GT AGAACACGGAGCATTCCAC-3′. β-Actin (Forward): 5′-TGAAGGTGACAGCAGTCGGT-3′, and β-Actin (Reverse): 5′-TAGAGAGAAGTGGGGTGGCT-3′.
Immunohistochemistry (IHC)
IHC was performed on 22 paraffin embedded and serially cut prostate cancer tissues obtained from St. Joseph’s Hospital, Hamilton, Ontario, Canada, and on the various human xenograft tissues and PTEN–/– prostates. Slides were deparaffinized in xylene and cleared in an ethanol series. Antigen retrieval was performed in a food steamer for 20 minutes using sodium citrate buffer (pH = 6.0). Tissues were blocked for 1 hour in PBS containing 1% BSA and 10% normal goat serum (Vector Laboratories). FAM84B antibody (1:350, Proteintech) was incubated overnight at 4°C. Secondary antibody biotinylated goat anti-rabbit IgG and Vector ABC reagent (Vector Laboratories) were incubated according to the manufacturer’s instructions. Secondary antibody only was used as negative control. Washes were performed with PBS. Chromogenic reaction was carried out with diaminobenzidine (Vector Laboratories), and slides were counterstained with haemotoxylin (Sigma Aldrich). Image analysis was performed using ImageScope software (Leica Microsystems Inc.). Staining intensity values derived from ImageScope were converted to an HScore using the formula [HScore = (% Positive) x (intensity) + 1]. The HScore was normalized through background subtraction and averaged amongst > 5 images per tissue sample.
Statistical analysis
Statistical analysis is depicted as Mean ± SD or HScores ± SD as stated, and performed using Student’s t-test where p < 0.05 is considered statistically significant. ROC curves were generated using GraphPad Prism 5.0 software. Analysis of data from cBioPortal was performed using Logrank Test.
ACKNOWLEDGMENTS
Mouse strain B6.Cg-Tg(Pbsn-cre)4Prb obtained from the NCI Mouse Repository was kindly donated by Dr. Pradip Roy-Burman from the University of South Carolina. The results shown here are in part based upon data generated by Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), the TCGA Research Network (http://cancergenome.nih.gov/), and by OncomineTM (https://www.oncomine.org/).
CONFLICTS OF INTEREST
All authors declare no conflicts of interest
GRANT SUPPORT
This work was supported in part by a GAP funding from McMaster University and St. Joseph’s Hospital in Hamilton, a Teresa Cascioli Charitable Foundation Research Award in Women’s Health to D.T., as well as by grants from the National Natural Science Foundation of China (Grant No. 81201568), the Natural Science Foundation of Guangdong Province (Grant No.2014A030313749), the National Natural Science Foundation of Heilongjiang Province (grant NO.C2015033), and the Shenzhen Program of Innovation and Entrepreneurship for Overseas Elites (grant No. KQCX20120814150420241) to F.W.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–86. doi: 10.1002/ijc.29210.
2. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, Mason M, Matveev V, Wiegel T, Zattoni F, Mottet N. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014; 65:467–79.
3. Rosenberg J, Small EJ. Prostate cancer update. Curr Opin Oncol. 2003; 15:217–21.
4. Ross JS. The androgen receptor in prostate cancer: therapy target in search of an integrated diagnostic test. Adv Anat Pathol. 2007; 14:353–7. doi: 10.1097/PAP.0b013e31814a52c4.
5. Mitsiades N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res. 2013; 73:4599–605. doi: 10.1158/0008–5472.CAN-12–4414.
6. Mateo J, Smith A, Ong M, de Bono JS. Novel drugs targeting the androgen receptor pathway in prostate cancer. Cancer Metastasis Rev. 2014; 33:567–79. doi: 10.1007/s10555-013-9472-2.
7. Ojo D, Lin X, Wong N, Gu Y, Tang D. Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer. Cancers (Basel). 2015; 7:2290–308. doi: 10.3390/cancers7040890.
8. Pienta KJ, Walia G, Simons JW, Soule HR. Beyond the androgen receptor: new approaches to treating metastatic prostate cancer. Report of the 2013 Prouts Neck Prostate Cancer Meeting. Prostate. 2014; 74:314–20. doi: 10.1002/pros.22753.
9. Palapattu GS, Wu C, Silvers CR, Martin HB, Williams K, Salamone L, Bushnell T, Huang LS, Yang Q, Huang J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate. 2009; 69:787–98. doi: 10.1002/pros.20928.
10. Bitting RL, Schaeffer D, Somarelli JA, Garcia-Blanco MA, Armstrong AJ. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 2014; 33:441–68. doi: 10.1007/s10555-013-9483-z.
11. Burgio SL, Conteduca V, Menna C, Carretta E, Rossi L, Bianchi E, Kopf B, Fabbri F, Amadori D, De Giorgi U. Chromogranin A predicts outcome in prostate cancer patients treated with abiraterone. Endocr Relat Cancer. 2014; 21:487–93. doi: 10.1530/ERC-14-0071.
12. Conteduca V, Burgio SL, Menna C, Carretta E, Rossi L, Bianchi E, Masini C, Amadori D, De Giorgi U. Chromogranin A is a potential prognostic marker in prostate cancer patients treated with enzalutamide. Prostate. 2014; 74:1691–6. doi: 10.1002/pros.22890.
13. Braem MG, Schouten LJ, Peeters PH, van den Brandt PA, Onland-Moret NC. Genetic susceptibility to sporadic ovarian cancer: a systematic review. Biochim Biophys Acta. 2011; 1816:132–46. doi: 10.1016/j.bbcan.2011.05.002.
14. Haerian MS, Baum L, Haerian BS. Association of 8q24.21 loci with the risk of colorectal cancer: a systematic review and meta-analysis. J Gastroenterol Hepatol. 2011; 26:1475–84. doi: 10.1111/j.1440-1746.2011.06831.x.
15. Haiman CA, Le Marchand L, Yamamato J, Stram DO, Sheng X, Kolonel LN, Wu AH, Reich D, Henderson BE. A common genetic risk factor for colorectal and prostate cancer. Nat Genet. 2007; 39:954–6. doi: 10.1038/ng2098.
16. Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, Lubbe S, Spain S, Sullivan K, Fielding S, Jaeger E, Vijayakrishnan J, Kemp Z, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007; 39:1315–7. doi: 10.1038/ng.2007.18.
17. Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007; 39:984–8. doi: 10.1038/ng2085.
18. Hindorff LA, Gillanders EM, Manolio TA. Genetic architecture of cancer and other complex diseases: lessons learned and future directions. Carcinogenesis. 2011; 32:945–54. doi: 10.1093/carcin/bgr056.
19. Vargas AC, Lakhani SR, Simpson PT. Pleomorphic lobular carcinoma of the breast: molecular pathology and clinical impact. Future Oncol. 2009; 5:233–43. doi: 10.2217/14796694.5.2.233.
20. Garcia-Closas M, Chanock S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin Cancer Res. 2008; 14:8000–9. doi: 10.1158/1078-0432.CCR-08-0975.
21. Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007; 447:1087–93. doi: 10.1038/nature05887.
22. Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock SJ, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, et al. A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res. 2007; 67:2951–6. doi: 10.1158/0008-5472.CAN-06-3591.
23. Nakagawa H, Akamatsu S, Takata R, Takahashi A, Kubo M, Nakamura Y. Prostate cancer genomics, biology, and risk assessment through genome-wide association studies. Cancer Sci. 2012; 103:607–13. doi: 10.1111/j.1349-7006.2011.02193.x.
24. Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Cazier JB, Sainz J, Jakobsdottir M, Kostic J, Magnusdottir DN, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006; 38:652–8. doi: 10.1038/ng1808.
25. Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, Waliszewska A, Penney K, Steen RG, Ardlie K, John EM, Oakley-Girvan I, Whittemore AS, Cooney KA, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci USA. 2006; 103:14068–73. doi: 10.1073/pnas.0605832103.
26. Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007; 39: 631–7. doi: 10.1038/ng1999.
27. Haiman CA, Patterson N, Freedman ML, Myers SR, Pike MC, Waliszewska A, Neubauer J, Tandon A, Schirmer C, McDonald GJ, Greenway SC, Stram DO, Le Marchand L, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007; 39:638–44. doi: 10.1038/ng2015.
28. Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007; 39:645–9. doi: 10.1038/ng2022.
29. Liu W, Xie CC, Zhu Y, Li T, Sun J, Cheng Y, Ewing CM, Dalrymple S, Turner AR, Sun J, Isaacs JT, Chang BL, Zheng SL, et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia. 2008; 10:897–907.
30. Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, Benediktsdottir KR, Magnusdottir DN, Orlygsdottir G, Jakobsdottir M, Stacey SN, Sigurdsson A, Wahlfors T, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009; 41:1122–6. doi: 10.1038/ng.448.
31. Chung CC, Hsing AW, Edward Y, Biritwum R, Tettey Y, Adjei A, Cook MB, De Marzo A, Netto G, Tay E, Boland JF, Yeager M, Chanock SJ. A comprehensive resequence-analysis of 250 kb region of 8q24.21 in men of African ancestry. Prostate. 2014; 74:579–89.
32. Bensen JT, Xu Z, McKeigue PM, Smith GJ, Fontham ET, Mohler JL, Taylor JA. Admixture mapping of prostate cancer in African Americans participating in the North Carolina-Louisiana Prostate Cancer Project (PCaP). Prostate. 2014; 74:1–9. doi: 10.1002/pros.22722.
33. Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai Z, Driver KE, Pooley KA, Ramus SJ, Kjaer SK, Hogdall E, DiCioccio RA, Whittemore AS, Gayther SA, et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J Natl Cancer Inst. 2008; 100:962–6. doi: 10.1093/jnci/djn190.
34. Kastler S, Honold L, Luedeke M, Kuefer R, Moller P, Hoegel J, Vogel W, Maier C, Assum G. POU5F1P1, a putative cancer susceptibility gene, is overexpressed in prostatic carcinoma. Prostate. 2010; 70:666–74. doi: 10.1002/pros.21100.
35. van Duin M, van Marion R, Vissers K, Watson JE, van Weerden WM, Schroder FH, Hop WC, van der Kwast TH, Collins C, van Dekken H. High-resolution array comparative genomic hybridization of chromosome arm 8q: evaluation of genetic progression markers for prostate cancer. Genes Chromosomes Cancer. 2005; 44:438–49. doi: 10.1002/gcc.20259.
36. Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007; 448:318–24. doi: 10.1038/nature05944.
37. Rybak AP, He L, Kapoor A, Cutz JC, Tang D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochim Biophys Acta. 2011; 1813:683–94. doi: 10.1016/j.bbamcr.2011.01.018.
38. Yan J, Ojo D, Kapoor A, Lin X, Pinthus JH, Aziz T, Bismar TA, Wei F, Wong N, De Melo J, Cutz JC, Major P, Wood G, et al. Neural Cell Adhesion Protein CNTN1 Promotes the Metastatic Progression of Prostate Cancer. Cancer Res. 2016; 76:1603–14. doi: 10.1158/0008-5472.CAN-15-1898.
39. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–10.
40. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. doi: 10.1158/2159-8290.CD-12-0095.
41. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. doi: 10.1126/scisignal.2004088.
42. Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007; 7:64. doi: 10.1186/1471-2407-7-64.
43. Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, McDonald C, Thomas R, Dhir R, Finkelstein S, Michalopoulos G, Becich M, Luo JH. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004; 22:2790–9. doi: 10.1200/JCO.2004.05.158.
44. Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003; 63:3877–82.
45. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22. doi: 10.1016/j.ccr.2010.05.026.
46. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–43. doi: 10.1038/nature11125.
47. Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, Ekman P, DeMarzo AM, Tibshirani R, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA. 2004; 101:811–6. doi: 10.1073/pnas.0304146101.
48. Yan J, De Melo J, Cutz JC, Aziz T, Tang D. Aldehyde dehydrogenase 3A1 associates with prostate tumorigenesis. Br J Cancer. 2014; 110:2593–603. doi: 10.1038/bjc.2014.201.
49. Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015; 163:1011–25. doi: 10.1016/j.cell.2015.10.025.
50. Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, Lucas JM, Brown LG, Dumpit RF, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016; 22:369–78. doi: 10.1038/nm.4053.
51. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–28. doi: 10.1016/j.cell.2015.05.001.
52. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016; 22:298–305. doi: 10.1038/nm.4045.
53. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012; 44:685–9. doi: 10.1038/ng.2279.
54. Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, Heguy A, Huberman K, Bernstein M, Assel M, Murali R, Vickers A, Scardino PT, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci USA. 2014; 111:11139–44. doi: 10.1073/pnas.1411446111.
55. Al Olama AA, Kote-Jarai Z, Giles GG, Guy M, Morrison J, Severi G, Leongamornlert DA, Tymrakiewicz M, Jhavar S, Saunders E, Hopper JL, Southey MC, Muir KR, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet. 2009; 41:1058–60. doi: 10.1038/ng.452.
56. Gudmundsson J, Sulem P, Gudbjartsson DF, Masson G, Agnarsson BA, Benediktsdottir KR, Sigurdsson A, Magnusson OT, Gudjonsson SA, Magnusdottir DN, Johannsdottir H, Helgadottir HT, Stacey SN, et al. A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat Genet. 2012; 44:1326–9. doi: 10.1038/ng.2437.
57. Du M, Yuan T, Schilter KF, Dittmar RL, Mackinnon A, Huang X, Tschannen M, Worthey E, Jacob H, Xia S, Gao J, Tillmans L, Lu Y, et al. Prostate cancer risk locus at 8q24 as a regulatory hub by physical interactions with multiple genomic loci across the genome. Hum Mol Genet. 2015; 24:154–66. doi: 10.1093/hmg/ddu426.
58. Cheng C, Cui H, Zhang L, Jia Z, Song B, Wang F, Li Y, Liu J, Kong P, Shi R, Bi Y, Yang B, Wang J, et al. Genomic analyses reveal FAM84B and the NOTCH pathway are associated with the progression of esophageal squamous cell carcinoma. Gigascience. 2016; 5:1. doi: 10.1186/s13742-015-0107-0.
59. Hsu FM, Cheng JC, Chang YL, Lee JM, Koong AC, Chuang EY. Circulating mRNA Profiling in Esophageal Squamous Cell Carcinoma Identifies FAM84B As A Biomarker In Predicting Pathological Response to Neoadjuvant Chemoradiation. Sci Rep. 2015; 5:10291. doi: 10.1038/srep10291.
60. Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010; 42:668–75. doi: 10.1038/ng.613.
61. Nyquist MD, Dehm SM. Interplay between genomic alterations and androgen receptor signaling during prostate cancer development and progression. Horm Cancer. 2013; 4:61–9. doi: 10.1007/s12672-013-0131-4.
62. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, et al. The genomic complexity of primary human prostate cancer. Nature. 2011; 470:214–20. doi: 10.1038/nature09744.