
INTRODUCTION
Genetic variation contributes to the phenotype and prognosis of breast cancer, as high-penetrance mutations and common variants correlate with various histopathological features, most notably estrogen receptor status [1]. The prognosis and indicated treatment for breast cancer is influenced by tumor grade, stage, HER2 expression, and hormone receptor status, and it is plausible that genetic variants associated with these features would be of prognostic and predictive interest. Additionally, genetic variation may contribute to breast cancer survival independently of these markers, potentially by affecting the efficacy of the treatment. For example, prognostic and predictive SNPs have been discovered in the TP53 gene and its regulatory network, as well as in genes involved in oxidative stress [2-6].
TP53 is a key tumor suppressor involved in several cellular stress response pathways that regulate the cell cycle, apoptosis, senescence, and DNA repair. Somatic TP53 mutations are common in most types of cancer, including breast cancer where TP53 mutations have been estimated to occur in 20-30% of cases [7-9]. These mutations are most commonly dominant-negative missense mutations; truncating loss of function mutations are seen in less than 5% of breast cancers [10-12]. The dominant-negative missense mutations lead to the accumulation of mutated TP53 protein in cell nuclei, which is generally detectable by immunohistochemistry, although it must be noted that the concordance between immunohistochemical detection and sequencing is less than 75% when accounting for truncating mutations and missense mutations outside the conserved regions of the protein [12, 13].
Mutated TP53, detected either by immunohistochemistry or by sequencing, has been reported to associate strongly with aggressive tumor phenotypes, e.g. estrogen receptor negativity and high grade, and poor breast cancer survival [2, 7, 8, 10, 11, 14]. The prognostic value of TP53 mutation status appears to be particularly strong in ER-positive cases, however [11]. Furthermore, TP53 mutations may also influence disease outcome depending on the type of treatment, at least in the case of endocrine therapy for ER-positive cancer [15]. TP53 mutations have also been suggested to modulate sensitivity to anthracycline-cyclophosphamide combination chemotherapy, possibly in interaction with ER status, although the clinical significance of these findings remains inconclusive [12, 16, 17, 18]. We hypothesize that genetic variants that influence TP53-related biological processes may have a TP53-dependent association with breast cancer survival. Such effects could be masked by TP53 mutations or occur exclusively in TP53-mutated cancers. To test this hypothesis, we have utilized a two-stage study design to search for genetic variants associated with survival in TP53-related breast cancer.
RESULTS
Rs10916264 and TP53 IHC have an interactive association with survival in ER-positive cases
First, we performed an initial genome-wide screen in the HEBCS-GWS data set (N = 572) for candidate SNPs that may be associated with survival in a TP53-dependent manner. In this analysis, 111 SNPs met our selection criteria and were also represented on the BCAC iCOGS array and therefore selected for further validation (Supplementary Table 1). In total, 136 BCAC SNPs matched the 111 HEBCS-GWS candidate SNPs at a LD threshold of r2 > 0.8.
Next, we performed an interaction analysis between SNP genotypes and TP53 immunohistochemistry (IHC) status in the BCAC validation series (N = 4610) (Table 1). At this stage, five closely linked SNPs emerged as statistically significant, the strongest being rs10916264 (p(interaction) = 3.44 x 10-5; p = 0.0011 after Benjamini-Hochberg adjustment) among ER-positive tumors. These SNPs are in high LD with each other (minimum r2 0.93, D’ 0.97) and therefore represent the same association signal. This interaction remained statistically significant after adjustment for standard prognostic factors (tumor size, grade, and node status). The Cox proportional hazards models for the rs10916264:TP53 interaction in ER-positive cases are presented in Table 2. The hazard ratio of the interaction term 2.06 (1.47 – 2.91) is consistent with the corresponding HEBCS-GWS SNP rs6604887 (r2 0.93, D’ 0.97) despite the low power in the ER-positive HEBCS-GWS subset (HR 1.94, 95% C.I. 0.82 – 4.02). In a comparison of the Cox models in ER-positive and ER-negative BCAC cases, the interaction terms suggested opposite effects and this difference was statistically significant (p = 0.0022, z-test for heterogeneity). The same was seen in the HEBCS-GWS data set, even though the interactions themselves did not reach statistical significance in either ER-based subgroup (heterogeneity p = 0.04).
Table 1: Description of the data sets used in this study.
a T-stage for HEBCS-GWS. For BCAC, categorized based on tumor size: 1 = < 20 mm, 2 = 20-50 mm, 3 = >50 mm.
b In BCAC, approximately 5% of methotrexate-treated cases also received anthracycline treatment, and 2% of anthracycline-treated cases also received methotrexate.
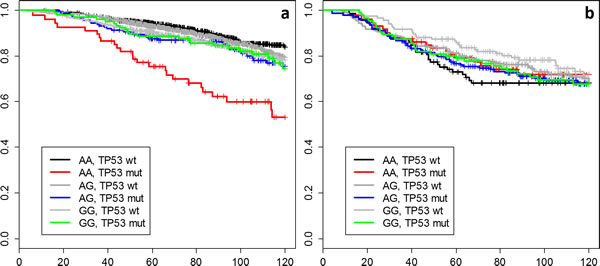
Figure 1: Kaplan-Meier curves for all combinations of rs10916264 genotype and TP53 status among pooled a) ER-positive and b) ER-negative BCAC cases.
To better illustrate the interaction results, we plotted Kaplan-Meier curves of all genotype-IHC combinations (Figure 1). The plots indicate that specifically the rs10916264 AA ancestral genotype (genotype frequency 23.2%, allele frequency 48.0%; used as reference allele in the interaction analysis) associates with worse survival among ER-positive, TP53-positive cases, while all other genotype-IHC combinations are clustered together. We therefore also analyzed this SNP using the recessive genetic model, using G as the reference allele, and calculated Cox proportional hazard models in TP53- and ER-based subgroups. In ER-positive, TP53-positive breast cancer cases, the homozygous rs10916264 AA genotype associated with a HR of 2.36 (95% confidence interval [C.I.] 1.45 – 3.82; Figure 2a), while in ER-positive, TP53-negative cases, the homozygous genotype did not associate with a difference in survival at all (HR 0.80, 95% C.I. 0.62 – 1.02; Figure 2a). When the rs10916264:TP53 interaction model was calculated separately for each study, all studies were in agreement on the direction of the interaction term (I2 = 11.72%, Q test for heterogeneity p = 0.3927; Figure 3). The interaction remained statistically significant (p = 2.6 x 10-4) when adjuvant endocrine therapy was included in the Cox model, and the interaction exists at a nominally significant level even among ER-positive cases not treated with endocrine therapy (n = 843, 140 events, p = 0.04). The interaction was also independent of adjuvant chemotherapy treatment with nominally significant interaction p-values in both chemotherapy-treated and untreated groups (p = 0.033 and 0.028, respectively).
Table 2: Proportional hazards models depicting the interaction between rs10916264 genotype (additive model) and TP53 immunohistochemistry in ER-positive cases, and between rs798755 genotype (recessive model) and adjuvant anthracycline chemotherapy.
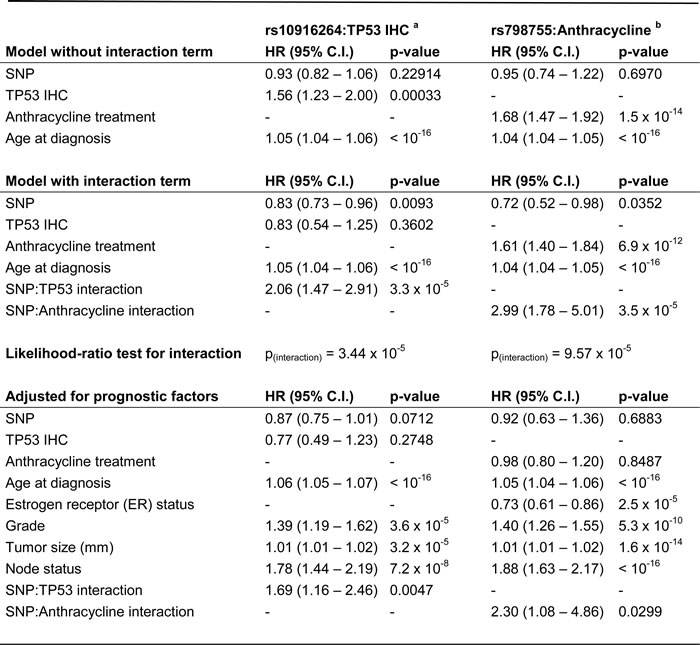
a Additive model, among ER-positive cases only
b Recessive model, all cases
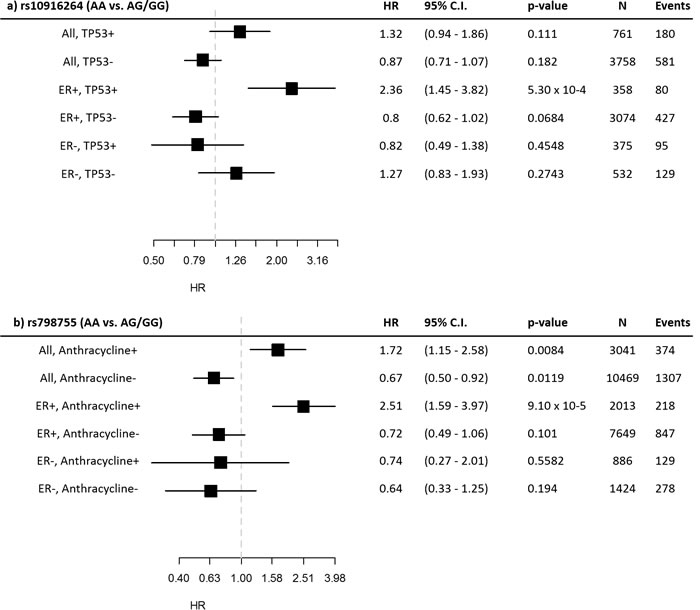
Figure 2: Subgroup statistics of the SNPs detected in the interaction analyses. Hazard ratios and confidence intervals are displayed for the recessive model in the indicated subgroups for a) rs10916264 and b) rs798755.
No statistically significant SNP:P53 interactions were detected in the full BCAC data set (when ER-status was ignored), nor in ER-negative cases. A complete listing of all interaction test p-values can be found in Supplementary Table 2. See also Supplementary Table 3 for a list of all candidate SNPs from the HEBCS-GWS pilot, the corresponding BCAC SNPs, and linkage disequilibrium statistics between the two.
Rs798755 associates with survival after anthracycline therapy
As positive TP53 IHC correlates with aggressive tumor characteristics that often indicate adjuvant chemotherapy treatment in the clinic, some SNPs observed to interact with TP53 IHC in our pilot study may reflect response to treatment rather than a true biological interaction with TP53. We therefore also conducted a treatment-based interaction analysis irrespective of TP53 status, restricting the interaction test to anthracycline-based regimens as this was the commonly used treatment type in both HEBCS-GWS and BCAC data sets. One statistically significant SNP emerged under the recessive genetic model: rs798755 (p(interaction) = 9.57 x 10-5; p = 0.0130 after Benjamini-Hochberg adjustment) (Supplementary Table 2). The Cox proportional hazards models describing this interaction are presented in Table 2. Upon further analysis of genotype-specific hazards, the rs798755 homozygous AA-genotype (genotype frequency 4.0%, allele frequency 19.7%) was associated with an increased hazard in anthracycline-treated cases (HR 1.72, 95% C.I. 1.15 – 2.58), and a decreased hazard in cases not treated with anthracyclines (HR 0.67, 95% C.I. 0.50 – 0.92; includes also cases that received no adjuvant chemotherapy). In the HEBCS-GWS data set, the corresponding SNP rs798766 (r2 1.0, D’ 1.0) also associated with an increased hazard in anthracycline-treated cases (HR 2.62, 95% C.I. 1.30 – 5.29), although we saw no evidence of a protective effect in untreated cases (HR 1.54, 95% C.I. 0.88 – 2.72).
An analysis restricted to BCAC cases receiving non-anthracycline chemotherapy (instead of all cases not treated with anthracyclines) suffers from low statistical power, but the HR estimate is similar to the above (HR 0.65, 95% C.I. 0.29 – 1.47). The interaction between rs798755 and anthracycline therapy may be dependent on ER status: an increased hazard was seen in ER-positive, anthracycline-treated cases (HR 2.51, 95% C.I. 1.59 – 3.97), but not in ER-negative, anthracycline-treated cases (HR 0.74, 95% C.I. 0.27 – 2.01). (Figure 2b)
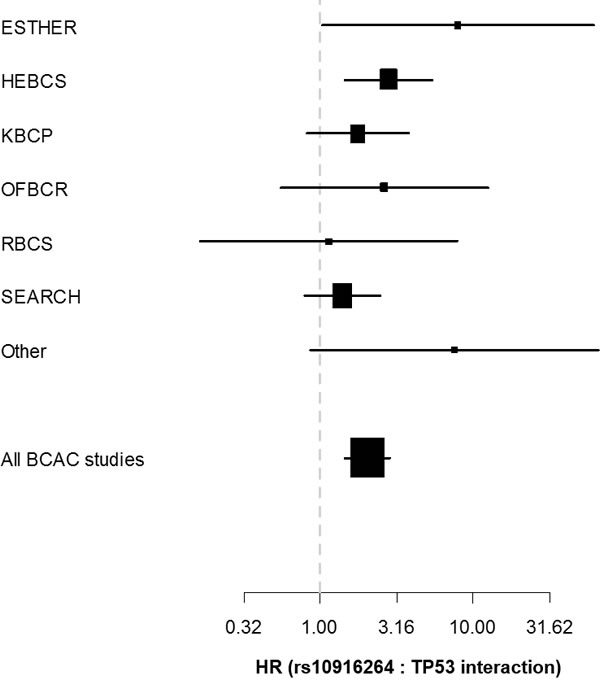
Figure 3: Forest plot depicting the hazard ratio (x-axis) and corresponding confidence intervals for the rs10916264:TP53 interaction term (additive genetic model among ER-positive cases) separately in each eligible BCAC study. Studies with fewer th were pooled into the “Other” category.
Characterization of the rs10916264 locus
The rs10916264 locus is located at 1q42.11 in a promoter-flanking regulatory region (Ensembl regulatory region ID ENSR00001772409) between the genes TP53BP2 and FBXO28. The maximum LD region (r2 > 0.1) around the SNP contains the genes CAPN8, TP53BP2, FBXO28, DEGS1, CNIH4, and WDR26. Computational annotation of correlated SNPs using multiple sources of genomic data revealed several potentially functional variants. Intersection of variants with genomic features relevant to target gene prediction methods suggested several potential target genes. For example, SNPs correlated (at r2>0.8) with rs10916264 overlap regulatory marks associated with CHIA-PET signals that interact with the promoters of CNIH4 and FBXO28. Enhancers associated with expression of FBXO28 and DEGS1 also harbor highly correlated SNPs. Many of these variants overlap annotated regulatory features (such as Roadmap and ENCODE enhancers, promoters and transcription factor binding sites) and consequently exhibit RegulomeDB scores suggestive of functional impact. These results are presented in detail in Supplementary Table 4.
Our eQTL analyses of METABRIC gene expression data indicate that rs10916264 and its tagging SNPs associate with the expression of FBXO28 also in breast tumor tissue (p = 0.0016). The rs10916264 A-allele was associated with higher expression of FBXO28. We also noted two additional cis-eQTLs between the rs10916264 locus and the genes TP53BP2 (p = 0.0087) and CNIH3 (p = 0.0023). Of these, FBXO28 and CNIH3 were predicted to be regulatory targets of the variants in this region. No statistically significant trans-eQTLs were detected.
Analysis of Kaplan-Meier Plotter data indicates that high FBXO28 expression is associated with poor survival among ER-positive breast cancer cases (HR 1.57, 95% C.I. 1.35 – 1.81; p = 9.5 x 10-10). Similar to the rs10916264 SNP, this effect is not seen in ER-negative cases: the calculated HR for high FBXO28 expression would in fact suggest a protective effect (HR 0.81, 95% C.I: 0.63 – 1.03) although the difference is not statistically significant (p = 0.081). Restricting the analysis to cases with known sequence-based TP53 mutation status greatly reduces statistical power, but the association between FBXO28 and survival is consistent with the SNPs in the rs10916264 locus. High FBXO28 expression was associated with poor survival in ER-positive, TP53-mutated cases (HR 2.35, 95% C.I. 1.17 – 4.72, p = 0.0133), but not in ER-positive TP53 wild-type cases (HR 1.27, 95% C.I. 0.82 – 1.96, p = 0.2886). A similar result was seen for TP53BP2, where high expression had a protective effect in ER-positive, TP53-mutated cases (HR 0.27, 95% C.I. 0.12 – 0.63, p = 0.001), but not in ER-positive TP53-wild type cases (HR 0.82, 95% C.I. 0.53 – 1.27, p = 0.383) or in ER-negative cases (HR 1.19, 95% C.I. 0.91 – 1.56, p = 0.191). The direction of the hazard ratios for TP53BP2 and FBXO28 were consistent with the directions of the eQTLs observed for the survival-associated SNPs in the region. Unlike FBXO28 and TP53BP2, the expression level of CNIH3 did not associate with survival.
Characterization of the rs798755 locus
The rs798755 LD region (r2 > 0.1) on chromosome 4 contains the genes FAM53A, SLBP, TMEM129, and TACC3, all of which are in eQTL association with the SNPs in the region in numerous cell types, although no such correlation has been reported specifically in breast tissue. The SNP is located in a regulatory region (ENSR00002001253) containing multiple active promoter and enhancer histone marks in a variety of tissues including breast. At this locus, SNPs correlated with rs798755 (r2 > 0.8) overlap with multiple MCF7 ChIA-PET signals that interact with the promoters of the above four genes as well as a number of other genes further along the chromosome: FGFR3, MXD4, WHSC1, RP11-572O17.1 and NELFA. Correlated SNPs also intersect with computationally predicted enhancers linked to expression of TACC3 and SLBP. Many highly correlated variants coincide with regulatory elements in normal and tumor breast cell types, including enhancers, promoters and transcription factor binding sites. In our METABRIC eQTL analysis of breast tumor data, SNPs correlated with rs798755 were associated with the expression of TACC3 (p = 0.00099). See Supplementary Table 4 for details on the predicted regulatory sites in this region. Gene expression based survival analyses were not feasible for these genes, since data on specific adjuvant chemotherapy regimens was not available in the Kaplan-Meier Plotter database.
siRNA knockdown of FAM53A expression influences doxorubicin sensitivity in breast cancer cells
To test whether the expression levels of the genes surrounding rs798755 influence anthracycline response in breast cancer cell lines (CAL-120, MCF7, MDA-MB-231, and MDA-MB-361), we tested whether siRNA knockdown of these genes influences the doxorubicin dose response of the cells. Of the genes in the rs798755 region, FAM53A knockdown resulted in increased doxorubicin resistance (lower DSS) compared to the negative control siRNA in the triple-negative, TP53-mutated MDA-MB-231 cells (p < 10-16). In CAL-120 cells, a similar but statistically non-significant effect was seen between FAM53A siRNAs and negative controls (p = 0.162). In the Luminal-B MDA-MB-361 and Luminal-A MCF7 cell lines, FAM53A knockdown instead resulted in increased sensitivity to doxorubicin (p = 0.005 and p = 0.017, respectively). See Figure 4 for a visual comparison of DSS scores between FAM53A siRNAs and negative controls in the four cell lines. Knockdown of SLBP, TMEM129, or TACC3 did not influence doxorubicin sensitivity in any of the cell lines.
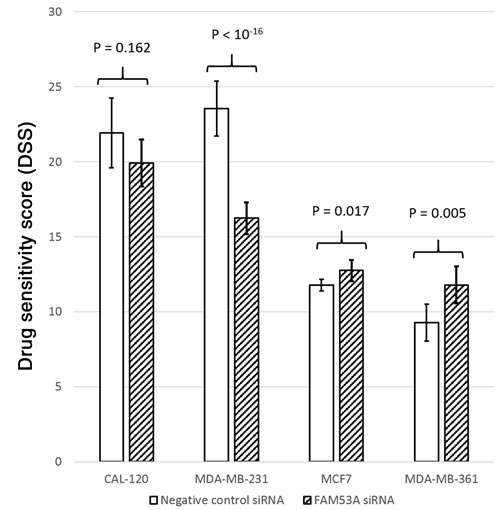
Figure 4: Drug Sensitivity Scores (DSS) for cells transfected with FAM53A siRNAs and negative control siRNAs in each of the four cell lines. Higher DSS scores indicate better sensitivity to doxorubicin.
DISCUSSION
We have performed a two stage SNP association study for the purpose of discovering genetic variants that may influence breast cancer survival in a TP53-dependent manner. The general workflow of the study is illustrated in Figure 5. In an interaction test between SNP genotypes and TP53 overexpression, one locus (represented by rs10916264 and linked SNPs) emerged as statistically significant among ER-positive cases, independently of conventional prognostic factors; this effect differed significantly from ER-negative cases. Although initially detected using the additive genetic model, the effect appears to be recessive: specifically the rs10916264 AA-genotype is associated with poor survival in TP53- and ER-positive cases. The survival difference in this group of patients is remarkable: rs10916264 appears to distinguish a subgroup of TP53-positive cases with poor prognosis among ER-positive cases (Figure 1a). No association of rs10916264 with survival was seen in the ER-positive, TP53-negative cases, nor in ER-negative cases. The interaction was not seen in the main analysis of all BCAC cases, but this is consistent with the effect direction differing between ER-positive and ER-negative cases. This effect appeared to be independent of both endocrine and adjuvant chemotherapy treatment, although more detailed analyses of specific chemotherapy regimens were not carried out due to lack of statistical power.
The rs10916264 locus is in a promoter-flanking region upstream of the FBXO28 gene, a predicted target of the regulatory variants in LD with rs10916264. The rs10916264 A-allele correlates with increased expression of FBXO28, which in turn was associated with poor breast cancer survival in the Kaplan-Meier Plotter database. Similar to the SNP, FBXO28 expression was only associated with survival in ER-positive breast cancers with somatic TP53 mutations. A similar but opposite effect was seen for TP53BP2, also consistent with the eQTL direction: low TP53BP2 expression associated with adverse prognosis in ER-positive cases with TP53 mutations. This lends credence to the idea that genetic variation at this locus influences breast cancer survival through regulation of one or both of the genes TP53BP2 and FBXO28.
Our evidence points to FBXO28 as the strongest candidate gene in the region: it is predicted to be a target of the regulatory variants, its expression in breast tumor tissue correlates with these variants, and its expression level also associates with breast cancer survival specifically in ER-positive, TP53-mutated breast cancer. Functional FBXO28 activity has also been shown to be associated with adverse breast cancer prognosis, and correlates with TP53 mutation status specifically in ER-positive breast tumors [19, 20]. FBXO28 belongs to the F-box family of proteins that determine the substrate specificity of the SCF ubiquitin ligase complex, a regulatory system that plays a critical role in tumorigenesis [21, 22]. SCF-FBXO28 specifically ubiquitylates Myc, promoting Myc-p300 transcriptional activity and subsequent oncogenic signaling [19]. Increased FBXO28 gene expression alone is not sufficient for this process: FBXO28 activation requires phosphorylation by the cyclin-dependent kinases CDK1 and CDK2. Since CDK activity is regulated by TP53 through p21 [23], this provides another rationale for why increased FBXO28 expression would influence survival predominantly in TP53-mutated cancer.
While TP53BP2 was not a predicted target in our computational analysis, variants in strong LD with rs10916264 did associate with its expression in breast tumors. This, along with the gene’s well-characterized interaction with TP53, makes it also a strong candidate to functionally connect the regulatory variants in this region to breast cancer survival. The association between low TP53BP2 expression and poor survival in TP53-mutated breast cancer seems counterintuitive at first, because TP53BP2 binds TP53 to induce apoptosis, and this pro-apoptotic cooperation can be defective or absent when TP53 is mutated [24, 25]. However, TP53BP2 also has TP53-independent binding partners and tumor suppressor activities that can partially compensate for defective TP53. TP53BP2 can bind IκB and induce repression of p63 through NF-κB, suppressing tumorigenesis and metastasis in squamous cell carcinoma [26]. It can also inhibit autophagy by binding Ras [27], a mechanism through which TP53BP2 has been shown to enhance oxaliplatin-induced apoptosis in colorectal cancer cells independently of TP53 [28]. Furthermore, TP53BP2 can regulate proliferation and apoptosis in the TP53-mutated MDA-MB-231 breast cancer cell line [29].
Certain caution is advisable in the interpretation of these results, since TP53 immunohistochemistry as used in this study is not a comprehensive method for TP53 mutation detection. The detectable over-abundance of TP53 results from typically dominant-negative TP53 missense mutations that lead to the accumulation of a stable but dysfunctional form of the protein. The study material therefore includes an unknown proportion of cases with other types of somatic mutations; the concordance between TP53 immunohistochemistry and DNA sequencing has previously been estimated to be below 75%, mainly due to truncating mutations [12, 13]. The immunohistochemically detectable dominant-negative mutations may be of particular interest, however, as they have been reported to confer oncogenic activity to TP53 [30]. It is also of note that the initial SNP selection in this study relies on a small sample set with relatively little statistical power, which may have led to missed SNPs as well as an inflated number of false positives in the initial candidate SNP set. Our goal was to offset this by the increase in statistical power achieved by focusing the validation analysis on a fairly small set of SNPs, resulting in a lesser degree of multiple testing and therefore more power to detect weak to moderate effect sizes (HRs). Given that positive TP53 status was strongly associated with aggressive tumor characteristics and therefore correlated strongly with the administration of adjuvant chemotherapy, we speculated that the initial TP53-based signal in HEBCS-GWS might in fact reflect an interaction with treatment. When we tested for interaction between candidate SNP genotypes and anthracycline treatment, the SNP rs798755 emerged as statistically significant. This locus (rs798766, r2 = 1) has previously been shown to associate with urinary bladder cancer risk and recurrence [31]. Our breast tumor eQTL and target prediction analyses pointed to TACC3 as the gene most likely affected by variants in this region. Overexpression of TACC3 has previously been associated with oncogenic activity, defective DNA repair, and poor survival in breast and lung cancer [32]. This would be consistent with the association of the rs798755 minor allele with high TACC3 expression in our eQTL analysis of breast tumor data. However, TACC3 siRNA knockdown did not influence doxorubicin sensitivity in the breast cancer cell lines we tested, although we cannot rule out the possibility that this gene may contribute to the observed association with survival in vivo.
Previously published studies have identified rs798755 as a cis-eQTL locus for the FAM53A gene in a wide variety of tissue types. FAM53A belongs to a vertebrate-specific family of three homologous genes of largely unknown function: FAM53A, FAM53B, and FAM53C [33]. In the siRNA experiment, FAM53A knockdown affected doxorubicin response significantly in three out of four tested cell lines. The TP53-mutated, basal MDA-MB-231 cells were rendered more doxorubicin-resistant by FAM53A depletion, and a similar but statistically non-significant effect was seen in the similarly basal and TP53-mutated CAL-120 cell line, while the luminal MDA-MB-361 (truncating TP53 mutation) and MCF7 (TP53 wild-type) cells became more sensitive to doxorubicin. It is tempting to speculate that this effect is caused by the basal-luminal difference, and/or the presence of gain-of-function TP53 mutations in the two basal cell lines, but more cell lines would have to be examined to support such conclusions, and further study is required to elucidate the underlying biological mechanisms and their in vivo relevance. These findings are consistent with our SNP-based clinical findings and eQTL data, however: the rs798755 A-allele associates with poor prognosis in ER-positive, anthracycline-treated cases, as well as with higher FAM53A expression, which based on our siRNA experiments correlates with anthracycline resistance in ER-positive cell lines. It can therefore be speculated that FAM53A may play a role in the commonly seen resistance to anthracyclines and other chemotherapy drugs in ER-positive breast cancer [34-36].
The functions of FAM53A and its paralogues are not well understood. The best-known member of the family is FAM53B, a controller of tissue development and cell proliferation [33, 37]. FAM53B is required for Wnt signaling, a pathway involved in epithelial-to-mesenchymal transition and subsequent metastasis in breast cancer [38-40]. FAM53B has also been identified as a critical gene in the prognosis of multiple myeloma in a transcriptional network analysis [41]. FAM53B has also been shown to bind 14-3-3 chaperones, a family of proteins known to play a key role in cellular resistance to anticancer drugs, including doxorubicin [33, 42-44]. FAM53A is therefore a plausible candidate to influence anthracycline response in breast cancer, even though the mechanism of action cannot be speculated on in any detail, and the results concerning rs798755 and its associated genes must be considered hypothesis-generating without immediate clinical relevance. If confirmed, however, these results may aid researchers in understanding the mechanisms of anthracycline resistance in ER-positive breast cancer, which in turn can facilitate clinical research towards improved individualized therapy.
In conclusion, we have identified a regulatory genetic locus in 1q42.11, represented by rs10916264 and other SNPs in its LD region, which distinguishes a group with poor survival after breast cancer specifically in cases with TP53 overabundance in ER-positive tumors. This genetic variation may influence the expression and/or regulation of several genes in the region, with the evidence pointing most strongly to FBXO28 and TP53BP2. This result may provide useful clues to the relationship between TP53 and ER signaling in breast cancer as well as to the way this interplay influences tumor progression and the outcome of the disease. We have also identified a cis-eQTL variant for FAM53A that may associate with response to anthracycline treatment in ER-positive breast cancer. Both of these genetic loci may provide useful prognostic and/or predictive genetic markers that, if validated, may be of clinical use in identifying cases likely to benefit from specific treatments or more aggressive treatment regimens. Detailed investigation of the biological and clinical significance of these variants requires further study.
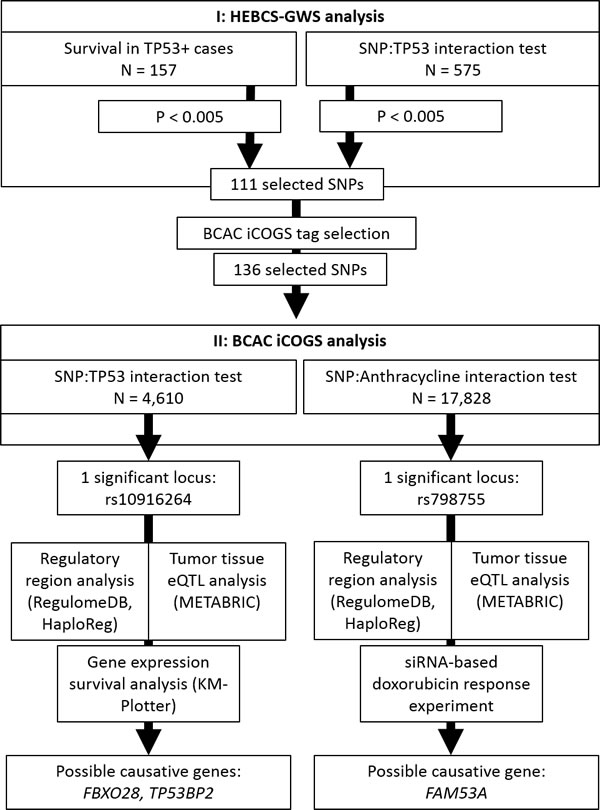
Figure 5: Diagram of the workflow of this study.
MATERIALS AND METHODS
Discovery GWS (HEBCS-GWS)
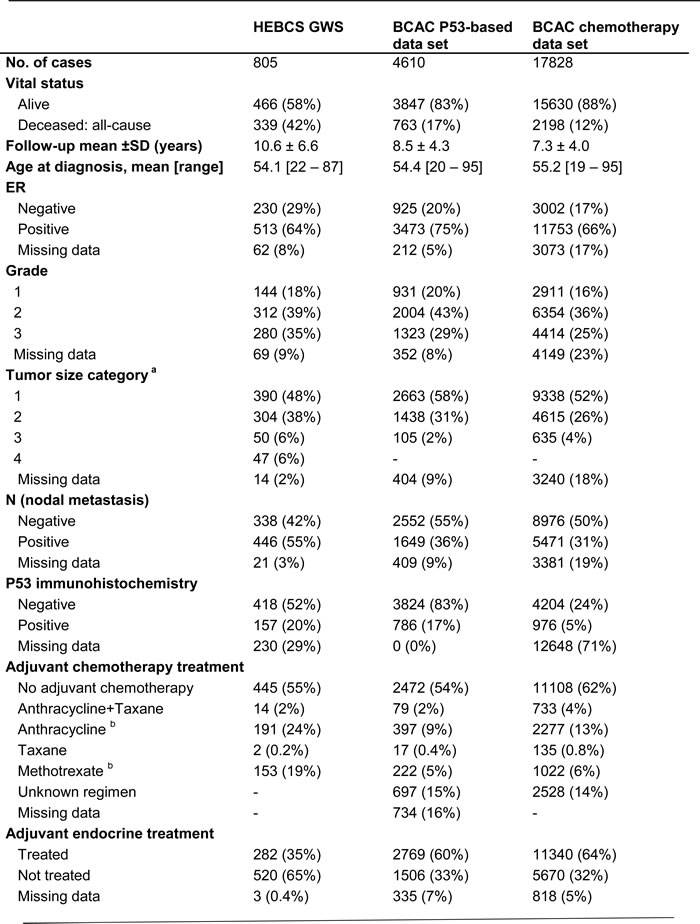
The collection and genotyping of the HEBCS-GWS series has been previously described [45]. In total, genotype information was obtained from a study series consisting of 805 Finnish breast cancer cases (HEBCS-GWS), enriched for cases with distant metastasis or death at the time of the initiation of the study in 2008: the series includes 312 breast cancer specific events, and 339 any-cause mortality events. TP53 immunohistochemistry data was available for 575 cases [2]; these cases comprise the discovery series. All cases were female, ascertained for their first primary invasive breast cancer. See Table 1 for a detailed description of this data set.
Validation (BCAC)
Candidate SNPs identified in the HEBCS GWAS analysis were included on a custom Illumina Infinium array (iCOGS) for large-scale genotyping of a data set of 50,927 individuals from 52 Breast Cancer Association Consortium (BCAC) member studies [46]. Each of the host institutions of the respective study recruited under ethically approved protocols by the local institutional review boards. BCAC studies represented on the iCOGS chip were included in survival analysis if sufficient follow-up data was available, with a minimum requirement of at least ten survival events (deaths from any cause) per study. Additionally, we included only studies with cases from predominantly European ancestry (in total 99.7% of individuals known to be of European origin), and that provided data on either adjuvant treatment or TP53 immunohistochemistry. See Supplementary Table 5 for a description of these studies. Clinicopathological data was collected as previously described [47], including TP53 immunohistochemistry data from 3,476 cases. Tumor tissue microarray (TMA) samples from an additional set of 1,134 individual cases (from five BCAC studies) were stained for TP53 and scored centrally at the Helsinki University Hospital using monoclonal mouse anti-human DO-7 antibody (Dako Inc., Carpinteria, CA 93013, USA) at a 1:200 dilution. Slides were pre-treated for 60 minutes in CC1 buffer (Ventana Inc., Tucson, AZ 85755, USA) and stained using the Ventana Benchmark XT system (Ventana, USA). The Ultraview Universal DAB Detection kit 760-500 (Ventana, USA) was used for detection. Scoring was categorized into a positive/negative score based on a cutoff of >20 % clearly positive tumor nuclei. See Supplementary Table 6 for a listing of the TP53 staining and scoring methods for each study.
Two partially overlapping subsets of the BCAC data set were used. For the primary TP53 interaction analysis, we selected all cases with available genotype, TP53 immunohistochemistry, and survival data (N = 4,610). For an analysis of chemotherapy-related effects, we included all cases with available survival and adjuvant chemotherapy information, irrespective of TP53 data availability (N = 17,828). These data sets are described in detail in Table 1.
Statistical analysis
Survival statistics were calculated using Cox proportional hazards models. The preliminary HEBCS-GWS survival analysis was carried out using two different endpoints in parallel: five-year BDDM (breast cancer death or distant metastasis), and 10-year overall survival (death from any cause). Two different approaches were used: (i) survival analyses restricted to TP53-positive cases only (N = 157), and (ii) interaction analyses between SNP genotypes and TP53 immunohistochemistry in all cases (N = 575). Only the log-additive genetic model was analyzed at this stage. SNPs associating with survival at p < 0.005 in either of the two tests were selected as candidates for validation in the next stage. If the SNP itself was not present on the iCOGS genotyping chip, tagging SNPs were selected using 1000genomes r2 > 0.8 as the minimum LD threshold for proxy SNP selection.
At the validation stage (BCAC), 10-year overall survival (death from any cause) was used as the end point in all survival analyses. Follow-up times were left-truncated to account for case recruitment latency. All Cox models were adjusted for age at diagnosis and stratified by study. The primary analysis consisted of an interaction test between SNP genotypes and TP53 immunohistochemistry using the log-additive genetic model; statistical significance was determined by a likelihood ratio test. The following groups were analyzed: all cases, ER-positive cases, and ER-negative cases. For statistically significant SNPs, we also calculated multivariate Cox interaction models that included the following standard prognostic factors in addition to age: estrogen receptor status (ER), histological grade, nodal metastasis (N), and tumor size.
In the anthracycline treatment based analyses, interaction terms of the form SNP*Anthracycline were introduced to the Cox models. To take advantage of the greater statistical power in this data set, both the additive and recessive genetic models were analyzed. Statistically significant hits from the BCAC interaction analysis were tested for consistency of effect direction in HEBCS-GWS within identically defined subgroups. Taxane- and methotrexate-based regimens were not investigated, as these regimens are less representative of the original HEBCS-GWS material, and the numbers were deemed to be too small for an adequately powered interaction analysis. Benjamini-Hochberg correction was used to confirm statistical significance in the presence of multiple testing [48].
eQTL analysis
To determine if the survival-associated SNPs or other SNPs in the LD region (r2 > 0.1) associate with gene expression in breast cancer, we utilized the publicly available METABRIC data set [49]. The METABRIC gene expression data was generated by the Illumina Human WG6 v3 platform. Tumor tissue genotyping had been carried out using the Affymetrix Genome Wide Human SNP array 6.0. METABRIC consists of 1,328 breast tumor samples with both genotype and gene expression data. eQTL analysis was carried out by calculating linear models between genotype and gene expression using the R package ‘MatrixEQTL’ [50]. We searched for cis-eQTLs within regions defined as ±1 Mb from any SNP in the LD region. Any genes outside these regions were analyzed for trans-eQTL and subjected to transcriptome-wide multiple testing correction (Benjamini-Hochberg).
Further in silico evaluation of SNPs and candidate genes
It is likely that the prognostic SNPs identified in this study are merely linkage disequilibrium proxies for other, functionally significant variants in their genomic vicinity. In an effort to identify the SNPs and genes with a direct functional impact on breast cancer survival, we utilized a number of public databases. For these analyses, regions of interest were defined as regions containing SNPs in any linkage disequilibrium (LD) with the survival-associated SNPs at r2> 0.1. Linked SNPs in these regions were analyzed for their impact on regulatory features using a target gene prediction pipeline that utilizes publicly available chromosome conformation capture, promoter prediction, super-enhancer, and enhancer promoter cap data to connect regulatory variants to likely target genes in breast-derived cell types as previously described [51]. These data were intersected with the breast cancer specific eQTL results from METABRIC to identify the most likely target genes. For a detailed listing of the target prediction resources, see Supplementary Table 4. Candidate genes in the regions surrounding the prognostic SNPs were analyzed in Kaplan-Meier plotter, a gene expression and survival database [52]. These analyses were performed using 10-year relapse-free survival, as this was the most widely available end-point, and optimized break points for the binarization of gene expression levels.
siRNA transfection / drug response measurement of breast cancer cell lines
In the case of SNPs that associated with survival in anthracycline-treated cases, we performed a siRNA knockdown based drug response experiment to test whether the genes in the LD regions surrounding the SNPs influence doxorubicin response in breast cancer cell lines. The target genes and selected siRNAs were FAM53A, SLBP, TACC3, and TMEM129. Three different siRNAs were used per gene (see Supplementary Table 7 for details), and assayed separately, each with five replicates. The cell lines used were MCF7 (luminal A, TP53 wild-type), MDA-MB-361 (luminal B, TP53 truncating mutation), MDA-MB-231 (triple-negative, TP53 missense mutation), and CAL-120 (triple-negative, TP53 missense mutation). Cells were transfected with siRNAs and 24h later supplemented with five different doxorubicin concentrations: 0.83, 10, 100, 500, and 1000 nM. The number of viable cells was measured after 71h doxorubicin treatment. Based on cell viability at increasing doxorubicin concentrations, drug response curves were calculated and converted into Drug Sensitivity Score (DSS) statistics [53]. DSS scores for each target gene were then pooled across replicates and siRNAs and compared to cells transfected with a target-less negative control siRNA in twelve replicates using Student’s t-test.
Data availability
The relevant BCAC SNP genotype data underpinning these analyses can be accessed by applying to the BCAC consortium (http://bcac.ccge.medschl.cam.ac.uk/). The dataset can be made available by the BCAC coordinating centre upon request to the corresponding authors and with the permission of BCAC Data Access Coordination Committee.
Abbreviations
BCAC Breast Cancer Association Consortium
SNP Single Nucleotide Polymorphism
ER Estrogen Receptor
eQTL Expression Quantitative trait Loci
HR Hazard Ratio
C.I. Confidence Interval
FDR False Discovery Rate
GWS Genome-Wide association Study
IHC Immunohistochemistry
siRNA Small Interfering RNA
LD Linkage Disequilibrium
CDK Cyclin-Dependent Kinase
TMA Tumor Tissue Microarray
N Lymph Node metastasis
ACKNOWLEDGMENTS
We thank Johanna Kiiski, Kirsimari Aaltonen, Karl von Smitten, Irja Erkkilä (HEBCS-GWS and HEBCS); Swapnil Potdar, John-Patrick Mpindi, Bhagwan Yadav, and the High Thoughput Biomedicine unit (FIMM); Maggie Angelakos, Judi Maskiell, and Gillian Dite (ABCFS);; Sten Cornelissen, Frans Hogervorst, Emiel Rutgers, Annegien Broeks, (ABCS); Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, Katja Butterbach (ESTHER); Eija Myöhänen, Helena Kemiläinen (KBCP); Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab (kConFab/AOCS); Gilian Peuteman, Dominiek Smeets, Thomas Van Brussel and Kathleen Corthouts (LMBC); Anja Rudolph, Dieter Flesch-Janys, Petra Seibold, Judith Heinz, Nadia Obi, Alina Vrieling, Sabine Behrens, Ursula Eilber, Muhabbet Celik, and Til Olchers (MARIE); Teresa Selander, Nayana Weerasooriya (OFBCR); Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner (PBCS); Petra Bos, Jannet Blom, Ellen Crepin, Elisabeth Huijskens, Annette Heemskerk, the Erasmus MC Family Cancer Clinic (RBCS); The Swedish Medical Research Counsel (SASBAC); The SEARCH and EPIC teams (SEARCH); and all study participants, clinicians, family doctors, researchers and technicians for their contributions and commitment to this study (SKKDKFZS).The BCAC iCOGS study would not have been possible without the contributions of the following: Andrew Lee, Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Javier Benitez, Anna Gonzalez-Neira and the staff of the CNIO genotyping unit, Jacques Simard and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Stig E. Bojesen, Sune F. Nielsen, Borge G. Nordestgaard, and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility. We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
The Breast Cancer Association Consortium (BCAC) is funded by Cancer Research UK [C1287/A10118, C1287/A12014] and by the European Community´s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). Funding for the iCOGS genotyping infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112 - the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund.
The Australian Breast Cancer Family Study (ABCFS; John L. Hopper, Melissa C. Southey) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. M.C.S. is a NHMRC Senior Research Fellow.
The ABCS study (Marjanka K. Schmidt, Renske Keeman) was supported by the Dutch Cancer Society [grants NKI 2007-3839; 2009 4363].
The work of the BBCC (Matthias W. Beckmann, Peter A. Fasching) was partly funded by ELAN-Fond of the University Hospital of Erlangen.
The ESTHER study (Hermann Brenner, Bernd Holleczek) was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe).
The HEBCS (Rainer Fagerholm, Sofia Khan, Päivi Heikkilä, Maral Jamshidi, Kristiina Aittomäki, Carl Blomqvist, Heli Nevanlinna) was financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society, The Nordic Cancer Union, and the Sigrid Juselius Foundation.
Financial support for KARBAC (Annika Lindblom, Sara Margolin) was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation.
The KBCP (Veli-Matti Kosma, Arto Mannermaa) was financially supported by the special Government Funding (EVO) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland.
kConFab (Jonathan Beesley, Georgia Chenevix-Trench) is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command [DAMD17-01-1-0729], Cancer Council Victoria, Queensland Cancer Fund, Cancer Council New South Wales, Cancer Council South Australia, The Cancer Foundation of Western Australia, Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC; 400413, 400281, 199600). G.C.T. and P.W. are supported by the NHMRC. RB was a Cancer Institute NSW Clinical Research Fellow.
LMBC (Giuseppe Floris, Diether Lambrechts) is supported by the ‘Stichting tegen Kanker’ (232-2008 and 196-2010). Diether Lambrechts is supported by the FWO and the KULPFV/10/016-SymBioSysII.
The MARIE study (Sabine Behrens, Jenny Chang-Claude) was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402].
The MCBCS (Fergus J. Couch, Emily Hallberg) was supported by the NIH grants CA192393, CA116167, CA176785 an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [CA116201], and the Breast Cancer Research Foundation and a generous gift from the David F. and Margaret T. Grohne Family Foundation.
The Ontario Familial Breast Cancer Registry (OFBCR; Irene L. Andrulis, Gord Glendon) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR.
The PBCS (Montserrat García-Closas, Jonine Figueroa) was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA.
The RBCS (Maartje J. Hooning, Agnes Jager) was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318).
The SASBAC (Kamila Czene, Per Hall) study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation.
SEARCH (H. Raza Ali, Fiona M. Blows, Qi Guo, Elena Provenzano, Mitul Shah) is funded by a programme grant from Cancer Research UK [C490/A10124] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge.
SKKDKFZS (Ute Hamann, Maria Kabisch) is supported by the DKFZ.
Author contributions
Study design: Rainer Fagerholm, Carl Blomqvist, Heli Nevanlinna
Statistical analyses and data management at the analysis stage: Rainer Fagerholm, Sofia Khan.
Manuscript preparation: Rainer Fagerholm, Sofia Khan, Marjanka K. Schmidt, Georgia Chenevix-Trench, Carl Blomqvist, Heli Nevanlinna.
Histopathology data acquisition and coordination: Montserrat García-Closas, Päivi Heikkilä
In vitro siRNA transfection and drug response experiments: Jani Saarela
Genotype and phenotype data acquisition and management: all authors
Manuscript review: all authors.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. 2010; 4: 174-91.
2. Tommiska J, Eerola H, Heinonen M, Salonen L, Kaare M, Tallila J, Ristimäki A, von Smitten K, Aittomäki K, Heikkilä P, Blomqvist C, Nevanlinna H. Breast cancer patients with p53 Pro72 homozygous genotype have a poorer survival. Clin Cancer Res. 2005; 11: 5098-103.
3. Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrjäkoski K, Kallioniemi A, Kilpivaara O, Mannermaa A, Kosma VM, Uusitupa M, Eskelinen M, Kataja V, et al. NAD(P)H: quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat Genet. 2008; 40: 844-53.
4. Schmidt MK, Tommiska J, Broeks A, van Leeuwen FE, Van’t Veer LJ, Pharoah PD, Easton DF, Shah M, Humphreys M, Dörk T, Reincke SA, Fagerholm R, Blomqvist C, et al. Combined effects of single nucleotide polymorphisms TP53 R72P and MDM2 SNP309, and p53 expression on survival of breast cancer patients. Breast Cancer Res. 2009; 11: R89.
5. Seibold P, Hall P, Schoof N, Nevanlinna H, Heikkinen T, Benner A, Liu J, Schmezer P, Popanda O, Flesch-Janys D, Chang-Claude J. Polymorphisms in oxidative stress-related genes and mortality in breast cancer patients--potential differential effects by radiotherapy? Breast. 2013; 22: 817-23.
6. Jamshidi M, Schmidt MK, Dörk T, Garcia-Closas M, Heikkinen T, Cornelissen S, van den Broek AJ, Schürmann P, Meyer A, Park-Simon TW, Figueroa J, Sherman M, Lissowska J, et al. Germline variation in TP53 regulatory network genes associates with breast cancer survival and treatment outcome. Int J Cancer. 2013; 132: 2044-55.
7. Borresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003; 21: 292-300.
8. Olivier M, Langerød A, Carrieri P, Bergh J, Klaar S, Eyfjord J, Theillet C, Rodriguez C, Lidereau R, Bièche I, Varley J, Bignon Y, Uhrhammer N, et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006; 12: 1157-67.
9. Hamroun D, Kato S, Ishioka C, Claustres M, Beroud C, Soussi T. The UMD TP53 database and website: update and revisions. Hum Mutat. 2006; 27: 14-20.
10. Fernández-Cuesta L, Oakman C, Falagan-Lotsch P, Smoth KS, Quinaux E, Buyse M, Dolci MS, Azambuja ED, Hainaut P, Dell’orto P, Larsimont D, Francis PA, Crown J, et al. Prognostic and predictive value of TP53 mutations in node-positive breast cancer patients treated with anthracycline- or anthracycline/taxane-based adjuvant therapy: results from the BIG 02-98 phase III trial. Breast Cancer Res. 2012; 14: R70.
11. Silwal-Pandit L, Vollan HK, Chin SF, Rueda OM, McKinney S, Osako T, Quigley DA, Kristensen VN, Aparicio S, Børresen-Dale AL, Caldas C, Langerød A. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res. 2014; 20: 3569-80.
12. Geisler S, Lønning PE, Aas T, Johnsen H, Fluge O, Haugen DF, Lillehaug JR, Akslen LA, Børresen-Dale AL. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 2001; 61: 2505-12.
13. Norberg T, Lennerstrand J, Inganas M, Bergh J. Comparison between p53 protein measurements using the luminometric immunoassay and immunohistochemistry with detection of p53 gene mutations using cDNA sequencing in human breast tumors. Int J Cancer. 1998; 79: 376-83.
14. Boyle DP, McArt DG, Irwin G, Wilhelm-Benartzi CS, Lioe TF, Sebastian E, McQuaid S, Hamilton PW, James JA, Mullan PB, Catherwood MA, Harkin DP, Salto-Tellez M. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology. 2014; 65: 340-52.
15. Yamamoto M, Hosoda M, Nakano K, Jia S, Hatanaka KC, Takakuwa E, Hatanaka Y, Matsuno Y, Yamashita H. P53 Accumulation is a Strong Predictor of Recurrence in Estrogen Receptor-Positive Breast Cancer Patients Treated with Aromatase Inhibitors. Cancer Sci. 2014; 105: 81-8.
16. Chrisanthar R, Knappskog S, Løkkevik E, Anker G, Ostenstad B, Lundgren S, Risberg T, Mjaaland I, Skjønsberg G, Aas T, Schlichting E, Fjösne HE, Nysted A, et al. Predictive and prognostic impact of TP53 mutations and MDM2 promoter genotype in primary breast cancer patients treated with epirubicin or paclitaxel. PLoS One. 2011; 6: e19249.
17. Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, Turpin E, Plassa LF, de Roquancourt A, Bourstyn E, de Cremoux P, Janin A, Giacchetti S, et al. P53 in Breast Cancer Subtypes and New Insights into Response to Chemotherapy. Breast. 2013; 22: S27-9.
18. Wang Y, Xu Y, Chen J, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Xie Y. TP53 mutations are associated with higher rates of pathologic complete response to anthracycline/cyclophosphamide-based neoadjuvant chemotherapy in operable primary breast cancer. Int J Cancer. 2016; 138: 489-96.
19. Cepeda D, Ng HF, Sharifi HR, Mahmoudi S, Cerrato VS, Fredlund E, Magnusson K, Nilsson H, Malyukova A, Rantala J, Klevebring D, Viñals F, Bhaskaran N, et al. CDK-mediated activation of the SCF(FBXO) (28) ubiquitin ligase promotes MYC-driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Mol Med. 2013; 5: 999-1018.
20. Győrffy B, Bottai G, Lehmann-Che J, Kéri G, Orfi L, Iwamoto T, Desmedt C, Bianchini G, Turner NC, de Thè H, André F, Sotiriou C, Hortobagyi GN, et al. TP53 mutation-correlated genes predict the risk of tumor relapse and identify MPS1 as a potential therapeutic kinase in TP53-mutated breast cancers. Mol Oncol. 2014; 8: 508-19.
21. Randle SJ, Laman H. F-box protein interactions with the hallmark pathways in cancer. Semin Cancer Biol. 2016; 36: 3-17.
22. Heo J, Eki R, Abbas T. Deregulation of F-box proteins and its consequence on cancer development, progression and metastasis. Semin Cancer Biol. 2016; 36: 33-51.
23. Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008; 10: e19.
24. Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001; 8: 781-94.
25. Tidow H, Veprintsev DB, Freund SM, Fersht AR. Effects of oncogenic mutations and DNA response elements on the binding of p53 to p53-binding protein 2 (53BP2). J Biol Chem. 2006; 281: 32526-33.
26. Tordella L, Koch S, Salter V, Pagotto A, Doondeea JB, Feller SM, Ratnayaka I, Zhong S, Goldin RD, Lozano G, McKeon FD, Tavassoli M, Fritzsche F, et al. ASPP2 suppresses squamous cell carcinoma via RelA/p65-mediated repression of p63. Proc Natl Acad Sci U S A. 2013; 110: 17969-74.
27. Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, Ratnayaka I, Zhong S, Goldin RD, Goemans CG, Tolkovsky AM, Lu X. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A. 2012; 109: 13325-30.
28. Shi Y, Han Y, Xie F, Wang A, Feng X, Li N, Guo H, Chen D. ASPP2 enhances oxaliplatin (L-OHP)-induced colorectal cancer cell apoptosis in a p53-independent manner by inhibiting cell autophagy. J Cell Mol Med. 2015; 19: 535-43.
29. Song Q, Song J, Wang Q, Ma Y, Sun N, Ma J, Chen Q, Xia G, Huo Y, Yang L, Li B. miR-548d-3p/TP53BP2 axis regulates the proliferation and apoptosis of breast cancer cells. Cancer Med. 2016; 5: 315-24.
30. Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P. Understanding wild-type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 2011; 18: 2-11.
31. Kiemeney LA, Sulem P, Besenbacher S, Vermeulen SH, Sigurdsson A, Thorleifsson G, Gudbjartsson DF, Stacey SN, Gudmundsson J, Zanon C, Kostic J, Masson G, Bjarnason H, et al. A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer. Nat Genet. 2010; 42: 415-9.
32. Ha GH, Kim JL, Petersson A, Oh S, Denning MF, Patel T, Breuer EK. TACC3 deregulates the DNA damage response and confers sensitivity to radiation and PARP inhibition. Oncogene. 2015; 34: 1667-78.
33. Thermes V, Candal E, Alunni A, Serin G, Bourrat F, Joly JS. Medaka simplet (FAM53B) belongs to a family of novel vertebrate genes controlling cell proliferation. Development. 2006; 133: 1881-90.
34. Berry DA, Cirrincione C, Henderson IC, Citron ML, Budman DR, Goldstein LJ, Martino S, Perez EA, Muss HB, Norton L, Hudis C, Winer EP. Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA. 2006; 295: 1658-67.
35. Shien T, Iwata H, Aogi K, Fukutomi T, Inoue K, Kinoshita T, Takahashi M, Matsui A, Shibata T, Fukuda H. Tamoxifen versus tamoxifen plus doxorubicin and cyclophosphamide as adjuvant therapy for node-positive postmenopausal breast cancer: results of a Japan Clinical Oncology Group Study (JCOG9401). Int J Clin Oncol. 2014; 19: 982-8.
36. International Breast Cancer Study Group (IBCSG). Endocrine responsiveness and tailoring adjuvant therapy for postmenopausal lymph node-negative breast cancer: a randomized trial. J Natl Cancer Inst. 2002; 94: 1054-65.
37. Kizil C, Otto GW, Geisler R, Nüsslein-Volhard C, Antos CL. Simplet controls cell proliferation and gene transcription during zebrafish caudal fin regeneration. Dev Biol. 2009; 325: 329-40.
38. Ki Kizil C, Küchler B, Yan JJ, Özhan G, Moro E, Argenton F, Brand M, Weidinger G, Antos CL. Simplet/Fam53b is required for Wnt signal transduction by regulating β-catenin nuclear localization. Development. 2014; 141: 3529-39.
39. Kotiyal S, Bhattacharya S. Breast cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res Commun. 2014; 453: 112-6.
40. Demirkan B. The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment. J Clin Med. 2013; 2: 264-82.
41. Agnelli L, Forcato M, Ferrari F, Tuana G, Todoerti K, Walker BA, Morgan GJ, Lombardi L, Bicciato S, Neri A. The reconstruction of transcriptional networks reveals critical genes with implications for clinical outcome of multiple myeloma. Clin Cancer Res. 2011; 17: 7402-12.
42. Mori M, Vignaroli G, Cau Y, Dinić J, Hill R, Rossi M, Colecchia D, Pešić M, Link W, Chiariello M, Ottmann C, Botta M. Discovery of 14-3-3 protein-protein interaction inhibitors that sensitize multidrug-resistant cancer cells to doxorubicin and the Akt inhibitor GSK690693. ChemMedChem. 2014; 9: 973-83
43. Han B, Xie H, Chen Q, Zhang JT. Sensitizing hormone-refractory prostate cancer cells to drug treatment by targeting 14-3-3sigma. Mol Cancer Ther. 2006; 5: 903-12.
44. Nakamura Y, Oshima K, Naoi Y, Nakayama T, Kim SJ, Shimazu K, Shimomura A, Maruyama N, Tamaki Y, Noguchi S. 14-3-3σ expression is associated with poor pathological complete response to neoadjuvant chemotherapy in human breast cancers. Breast Cancer Res Treat. 2012; 134: 229-36.
45. Li J, Humphreys K, Heikkinen T, Aittomäki K, Blomqvist C, Pharoah PD, Dunning AM, Ahmed S, Hooning MJ, Martens JW, van den Ouweland AM, Alfredsson L, Palotie A, et al. A combined analysis of genome-wide association studies in breast cancer. Breast Cancer Res Treat. 2011; 126: 717-27.
46. Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claud e J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013; 45: 353,61, 361e1-2.
47. Broeks A, Schmidt MK, Sherman ME, Couch FJ, Hopper JL, Dite GS, Apicella C, Smith LD, Hammet F, Southey MC, Van ‘t Veer LJ, de Groot R, Smit VT, et al. Low penetrance breast cancer susceptibility loci are associated with specific breast tumor subtypes: findings from the Breast Cancer Association Consortium. Hum Mol Genet. 2011; 20: 3289-303.
48. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. JSTOR. 1995; 57: 239-300.
49. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486: 346-52.
50. Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28: 1353-8.
51. Guo X, Long J, Zeng C, Michailidou K, Ghoussaini M, Bolla MK, Wang Q, Milne RL, Shu XO, Cai Q, Beesley J, Kar SP, Andrulis IL, et al. Fine-scale mapping of the 4q24 locus identifies two independent loci associated with breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2015; 24: 1680-91.
52. Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010; 123: 725-31.
53. Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, Majumder MM, Malani D, Murumägi A, Knowles J, Porkka K, Heckman C, Kallioniemi O, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep. 2014; 4: 5193.