
INTRODUCTION
Extranodal marginal zone lymphoma (EMZL) is a heterogeneous group of low-grade mature B-cell lymphomas that demonstrate different genetic alterations depending on the tumor origin. A number of chromosomal abnormalities have been described in EMZL, including trisomies 3, 12, and 18 and the specific chromosomal translocations t(11;18)(q21;q21), t(1;14)(p22;q32), t(3;14)(p14.1;q32), and t(14;18)(q32;q21). The frequency of chromosomal alterations is dependent on the primary tumor site, which suggests that genetic alterations are associated with different etiologies [1]. t(11;18)(q21;q21), involving the API2 and MALT1 genes, is most frequently found in EMZL from the lung (38-53%) and stomach (17-31%) and less commonly in tumors at other sites. t(14;18)(q32;q21), involving the IGH and MALT1 genes, is found in ocular, salivary and cutaneous marginal zone lymphoma (MZL) but not in EMZL from the G-I tract, lung, and thyroid [2]. t(3;14)(p14.1;q32), involving FOXP1, has been described in ocular MZL (25%), thyroid MZL (50%), and cutaneous MZL (10%), but has rarely been found in tumors at other sites [3]. t(1;14)(p22;q32) has been reported in intestinal MZL (13%) but not at other sites. Ocular MZL is the second most common type of EMZL and arises from the eyelid, conjunctiva, lacrimal gland and orbit. Overall, 30% of ocular MZL cases involve specific chromosomal translocations for which oncogenic activity is linked to antigen receptor-associated activation of nuclear factor (NF)-κB. In addition to chromosomal translocation, TNFAIP3, a negative regulator of the NF-κB pathway, was found to be inactivated via somatic deletion and/or mutation in ocular MZL in 12-37% of ocular MZL cases [4]. The fact that these translocations and somatic mutations have been identified in only a minority of ocular MZL cases appears to indicate that additional genomic alterations are involved in the development and progression of ocular MZL. Deep sequencing is currently the method of choice for cataloguing genomic changes in tumors. To obtain a comprehensive overview of the gene expression patterns and genomic alterations in ocular MZL, we produced a multidimensional genomic dataset based on data obtained from whole-genome sequencing (WGS), transcriptome sequencing, and targeted sequencing.
RESULTS
Somatic copy number of ocular MZL
To characterize the copy number and structural variations (SVs) of ocular MZL, we performed WGS on 10 matched pairs of tumor and normal samples (Supplementary Table 1). Tumors were sequenced to an average depth of 66x coverage, and matched germline samples were sequenced to 32x coverage (Supplementary Table 2-1). Our somatic copy number variation (CNV) calling [5] identified a total of 14 gain and 63 loss regions (Supplementary Table 3). The gain regions showed broad and low-amplitude changes, whereas the loss regions exhibited narrow and high-amplitude changes (Supplementary Figure 1). One significantly recurrent high-level loss region was identified (Supplementary Figure 2 and Supplementary Table 4). We confirmed the presence of previously identified common genetic alterations in ocular MZL. For example, the 6q23.3 region containing TNFAIP3, a negative regulator of the NF-κB signaling pathway, was significantly deleted across tumor samples (Figure 1A; FDR = 1.02E-03 using GISTIC): 50% of samples (5 of 10) demonstrated the deletion of 6q23.3, where 3 samples had a homozygous deletion (Supplementary Figure 3), and 2 had a heterozygous deletion. RNA-seq data for the same samples (Supplementary Table 2-2) showed a significantly lower average gene expression on 6q23.3 in the deletion samples (n=5) than in samples without the deletion (Figure 1B, P = 2.70E-02). We also found trisomies of chromosomes 3 and 18 in 50% (n=5) and 10% (n=1) of samples, respectively (Supplementary Table 3). The trisomy of chromosome 3 increased the mRNA expression of genes on this chromosome: the samples with this amplification (n=5) demonstrated significantly higher gene expression compared with samples without the amplification (Figure 1B).
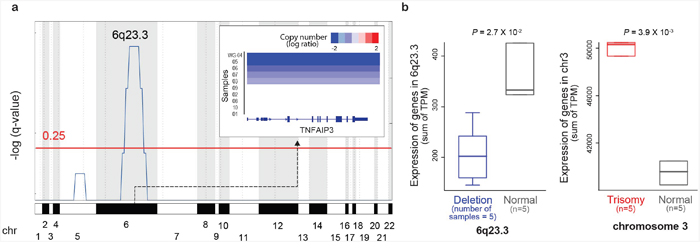
Figure 1: Copy number alterations in ocular MZL. a. GISTIC analysis of genomic regions with copy number loss.q-values (-log10 transformed) from GISTIC analysis (y-axis) are plotted across the genome (x-axis). Regions with q-values of <0.25 (red line) were considered significant. TNFAIP3 copy numbers in significantly deleted regions per sample are shown in the top right inset. b. The effects of copy number alternations on gene expression. The x-axis denotes samples grouped by copy number status, while the y-axis denotes expression in transcripts per million (TPM). P-values were derived from one-sided t-tests.
Recurrent structural variations associated with the disruption of TNFAIP3
We sought to discover five different types of SVs [6] (i.e., insertion, deletion, inversion, duplication, and translocation) in each sample (Supplementary Figures 4-5) and identified an average of 40 SVs per sample. Interestingly, we found 5 SV breakpoints in 3 samples (30%) in the first intron of IL20RA (Supplementary Figure 6), which encodes a subunit for the interleukin-20 receptor in the 6q23.3 region. The rearrangements included deletions, intra- and inter-chromosomal translocations, and complex SV (Supplementary Figure 7). For example, the WG-06 sample carried multiple rearrangements including 2 inter-chromosomal translocations with different chromosomes and 1 complex SV. As IL20RA does not appear to be expressed in lymphoid organs [7], the first intron of IL20RA might be a hotspot for structural genomic instability in ocular MZL. In particular, given that IL20RA lies upstream (specifically, 1 mb) of TNFAIP3, and that all samples with SVs (n=3) carry the homozygous deletion of TNFAIP3 (Figure 2), these SVs can be thought of as an important mechanism for the complete inactivation of TNFAIP3. The known balanced translocations in MALT lymphomas, such as t(11;18)(q21;q21) API2-MALT1, t(14;18)(q32;q21) IgH-MALT1, t(1;14)(p22;q32) BCL10-IgH, and t(3;14) (p14.1;q32) FOXP1-IgH were not found in our 10 samples. Additionally, no corresponding fusion transcripts were detected in the RNA-seq data.
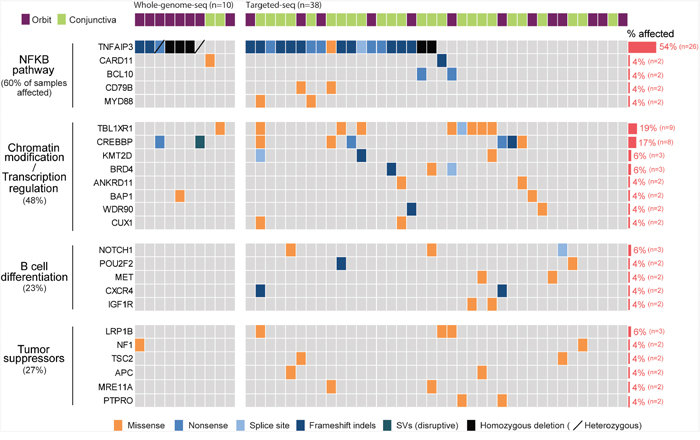
Figure 2: Landscape of somatic alternations in ocular MZL. Somatic alternations detected by whole genome (n=10) and targeted sequencing (n=38) are represented as a heatmap. Each column and row represents an affected individual and a gene, respectively. Genes altered in at least two patients were selected, and the fraction of affected individuals per gene is shown on the right. Mutated genes were clustered into four groups according to signaling pathway or molecular function.
Prevalence of the TNFAIP3 mutation in ocular MZL
We next called somatic single-nucleotide variants (SNVs) and identified an average of 22 non-silent substitutions per tumor sample (range, 12-44; Supplementary Figure 4 and Supplementary Table 5). In total, 70% (n=7) of the samples carried disruptive alterations in TNFAIP3 (i.e., nonsense, frameshift indels, or deletions), and the samples with such alterations were associated with NF-κB pathway overexpression (Supplementary Figure 8), which was in line with the notion that TNFAIP3 inactivation is implicated in the pathogenesis of ocular MZL via the constitutive activation of the NF-κB pathway [4]. To obtain a more comprehensive mutational landscape of ocular MZL, we further performed targeted deep sequencing of 430 cancer-related genes in 38 tumor samples (average sequencing depth of 400x per sample; Supplementary Table 2-1). Among a total of 48 tumor samples, 54% (n=26) demonstrated alterations in TNFAIP3 (Figure 2 and Supplementary Table 5). Our clonality analysis with the whole-genome-sequenced samples revealed that the TNFAIP3 mutations were all clonal (Supplementary Figure 9 and Supplementary Table 6-1). Together, these results indicated that alterations in TNFAIP3 are likely early driving events during lymphomagenesis. Other recurrent mutations activating the NF-κB pathway included CARD11 (4%), BCL10 (4%), CD79B (4%), and MYD88 (4%), all of which are known to function as a positive regulator of NF-κB pathway. The majority of the alterations in these genes appear to be activating mutations, including truncating mutations with potential gain-of-function in BCL10. Alterations in BCL10, CARD11, and MYD88 have been implicated in ocular MZL pathogenesis [8], and CD79B somatic changes in splenic MZL [9] have been reported. These mutations were mutually exclusive to each other while most of them coincided with TNFAIP3. A total of 55% of samples demonstrated gene alterations involving activation of the NF-κB pathway.
Frequent alterations in TBL1XR1
TBL1XR1, which is essential for transcriptional repression mediated by unliganded nuclear receptors (NRs) and other regulated transcription factors [10], was the most commonly mutated gene after TNFAIP3. Nine out of 48 samples (18%) harbored 13 TBL1XR1 mutations (Figure 2). The majority of the mutations (92%, n=12) were missense variants, and mutations in all variants were located within the WD40 domain, which has been known to be involved in binding nuclear receptor corepressor (NCoR) and histone deacetylase 3 (HDAC3) (Figure 3A) [10, 11]. All of these somatic mutations were heterozygotes as validated by Sanger sequencing (Supplementary Figure 10). Since many mutations are located near the top face of the domain and some of them are directly related to the DHSW tetrad, the core scaffold of WD40 domain (Figure 3B), they are expected to affect the binding of TBL1XR1 to the partner proteins [12]. Mutations in TBL1XR1 were significantly enriched in conjunctival ocular MZL (P=4.63E-02 by χ2 test), suggesting their role in its pathogenesis. Our clonality analysis revealed that the TBL1XR1 mutations were clonal (Supplementary Figure 9 and Supplementary Table 6-1).
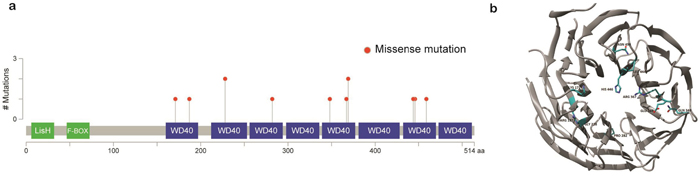
Figure 3: Schematic representation of mutations in TBL1XR1. a. Location of missense mutations in the TBL1XR1 protein. Frequency of missense mutations (y-axis) is shown by amino acid (x-axis). b. Protein structure of TBL1XR1 with mutations. Mutant residues are highlighted in cyan. Mutations are incorporated into TBL1XR1 model using swapaa tool of Chimera version 1.10.2 considering appropriate rotamer.
Alterations of other genes
Genes involved in chromatin modification and/or transcriptional regulation comprised the second most frequently mutated gene group in ocular MZL. cAMP response element binding protein (CREBBP), which belongs to the KAT3 family of histone/protein lysine acetyltransferases [13], was mutated in 17% of samples (n=8). KMT2D, which encodes a histone methyltransferase [13], was mutated in 6% of samples (n=3). The majority of the mutations in these genes were truncating variants that include the disruptive SV in CREBBP. This is in agreement with previous findings [14] that the loss-of-function of these genes is pathogenically involved in B-cell lymphoma. The other genes mutated in this group include ANKRD11, BAP1, WDR90, and CUX1. In total, 44% of cases carried mutations in this group, demonstrating that alterations to chromatin modifiers and/or transcription regulators also shape the mutational landscape of ocular MZL. Moreover, genes involved in B-cell differentiation or tumor suppression were also mutated. For example, several genes previously identified in other types of lymphomas, such as NOTCH1 [15], POU2F2 [16], CXCR4 [17], and IGF1R [18], were affected by mutations. Overall, 23% and 27% of samples harbored mutations in the B-cell differentiation and tumor suppressor groups, respectively. The specific impacts of some of the mutations on lymphomagenesis remain to be investigated.
Gene expression signature in ocular MZL
To better delineate the molecular pathogenesis of ocular MZL, we compared the gene expression profiles of ocular MZL samples (n=10) with normal marginal zone B cells (Figure 4; n=10) at the pathway level using single sample Gene Set Enrichment Analysis (ssGSEA). We identified 12 differentially expressed pathways in ocular MZL (p value <0.01), of which 11 were up-regulated, and the other was down-regulated (Supplementary Figure 11). The up-regulated pathways included those playing a crucial role in B-cell proliferation (e.g., NF-κB, CD40, and IL10) and NF-κB crosstalk pathways (e.g., EGFR_SMRTE, TNFR1, and STRESS), which are known to coordinately activate the NF-κB pathway [19, 20], highlighting the fact that aberrant activation of the NF-κB pathway is a hallmark of ocular MZL. In addition, apoptosis-suppressing pathways (HIVNEF, HSP27, and VIP), which confer apoptosis resistance in lymphocytes [21, 22], were over-expressed. The roles of other differentially expressed pathways in ocular MZL development remain to be elucidated. Enriched gene ontology (GO) terms in ocular MZL samples are displayed in Supplementary Figure 12.
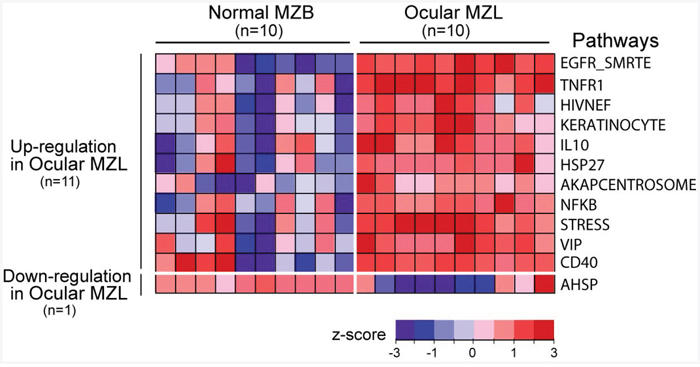
Figure 4: Gene expression signature in ocular MZL. Differentially expressed pathways between ocular MZL (n=10) and normal marginal zone B-cell samples (n=10) are shown (p-value <0.01 from a two-sided t-test). The pathway activity level calculated by ssGSEA is a transformed z -score and is shown as a heatmap.
Functional characterization of TBL1XR1
To verify the functional characterization of the mutations, we analyzed the interactions of TBL1XR1 mutants with NCoR complex components. These mutations were located in the WD40 domain, which is involved in interactions with NCoR and is the part of the corepressor complex that includes HDAC3, G-protein pathway suppressor 2 (GPS2), silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) and TBL1 [10]. NCoR controls the balance between transcriptional repression and activation through ubiquitination-mediated degradation. HDAC3 is responsible for the deacetylation of histone, which regulates transcriptional repression. TBL1XR1 is required for transcriptional activation, serving as a corepressor/coactivator exchange factor that regulates the degradation of corepressor components and the recruitment of coactivator complexes [11].
Using four mutants of highly conserved residues (G187R, L282P, H348Q, and S459N), we first analyzed the binding of corepressor complex proteins to TBL1XR1. Binding of NCoR and HDAC3 with disease-specific mutants of TBL1XR1 was increased compared with that of wild-type TBL1XR1 in a co-immunoprecipitation assay (Figure 5B). Expression of the NCoR protein in the total cell lysate was decreased in cells transfected with mutant TBL1XR1. The increased binding between NCoR and TBL1XR1 appears to facilitate the degradation of NCoR. However, there was no notable difference in HDAC3 expression levels in mutant TBL1XR1-expressing cells (Figure 5C). These results imply that these residues are not directly involved in protein binding, but have a role in maintaining local conformation. The fact that the mutations are not present at the residues in the protein-protein interaction (PPI) hotspot of WD40 domains [12] also supports our interpretation that mutants might induce PPI-enhancing conformational changes rather than abrogate PPI.
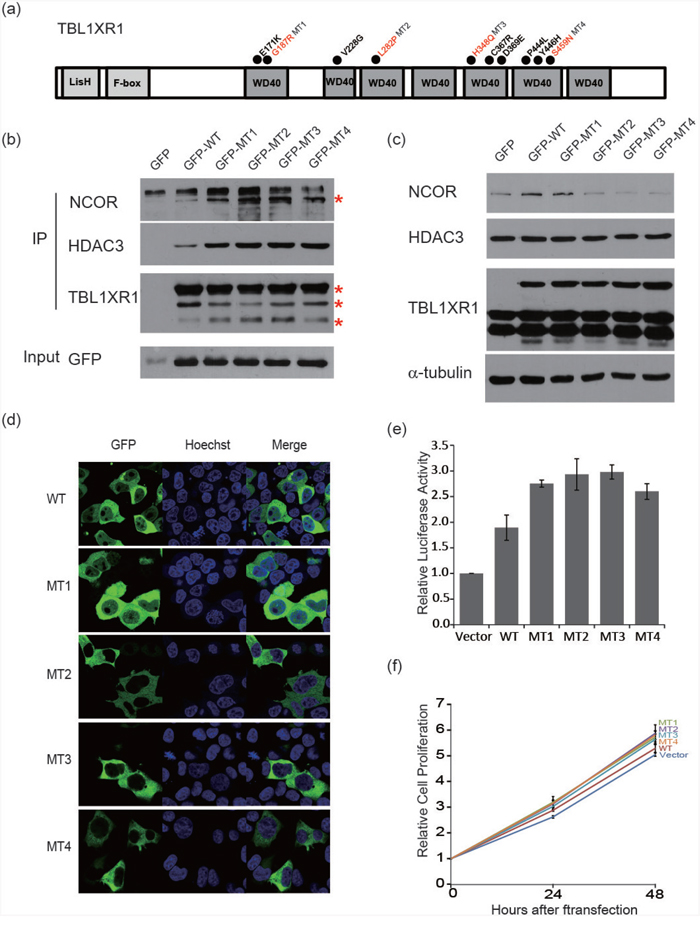
Figure 5: Functional characterization of TBL1XR1 WD40 domain alternations. a. Schematic diagram of the structure of TBL1XR1. Mutants occurred in the WD40 domain as indicated with circles. b. Co-immunoprecipitation of TBL1XR1 mutants. TBL1XR1 physically interacts with NCoR repressor complexes. Mutant TBL1XR1 interacts with NCoR and HDAC3 more frequently than wild-type TBL1XR1. c. Expression of NCoR protein levels in cells transfected with wild-type or mutant TBL1XR1. d. Fluorescence images of TBL1XR1 and Hoechst staining and merged images in 293T cells transfected with each TBL1XR1 mutant. e. NF-κB activity assay measuring the effects of each TBL1XR1 mutant. f. Cell proliferation comparison of TBL1XR1 mutants expressed in cells over 48 h using a CCK-8 assay.
Next, we analyzed the localization of wild-type and mutant TBL1XR1 (Figure 5D). Normally, wild-type TBL1XR1 is localized in both the nucleus and cytoplasm [23]. When TBL1XR1 is localized in the nucleus, cell growth is reduced, and apoptosis is increased [24]. TBL1XR1 mutants transfected into 293T cells demonstrated differential localization compared with the localization of wild-type TBL1XR1. Certain TBL1XR1 mutants (L282P, H348Q, and S459N) tended to localize in the cytoplasm, while another TBL1XR1 mutant (G187R) was located in both the cytoplasm and nucleus. These changes in the localization of mutant TBL1XR1 may lead to inhibition of the repressor complex and anti-apoptotic progression [23, 24].
The corepressor complex exerts its repressive effects via interactions with transcription factors, including NF-κB and JUN [10, 25]. Therefore, we examined the extent to which TBL1XR1 mutations affect the repressive function of this complex, by investigating the expression pattern of NF-κB and JUN target genes. We first examined gene expression similarities between the samples using microarray expression data for cell lines expressing the mutant (L282P and H348Q) and wild-type TBL1XR1, and cell line treated with TBL1XR1 siRNA (Figure 6A). The mutant samples clustered more closely with the wild-type than the siRNA knockdown sample, suggesting that L282P and H348Q are gain-of-function mutations. We performed gene set enrichment analysis (GSEA) and found that not only the mutant cell lines (Figure 6B) but also a patient sample harboring somatic missense variants (n=2) in TBL1XR1 (Figure 6C) demonstrated significantly up-regulated NF-κB and JUN target genes compared with wild-type TBL1XR1 samples. Based on the NF-κB luciferase reporter assay, we verified that expression of the TBL1XR1 mutants in BJAB (human Burkitt lymphoma) cells enhanced luciferase activity by approximately 1.5-fold compared with that of wild-type TBL1XR1 (Figure 5E). Overall, these results indicate that TBL1XR1 mutants can activate the transcription of some transcription factors such NF-κB and JUN by degrading the corepressor complex. Finally, we analyzed the effects of the TBL1XR1 mutants on cell proliferation. BJAB cells transfected with constructs expressing the TBL1XR1 mutants demonstrated a proliferation rate that was approximately 10-15% higher than cells expressing wild-type TBL1XR1 (Figure 5F).
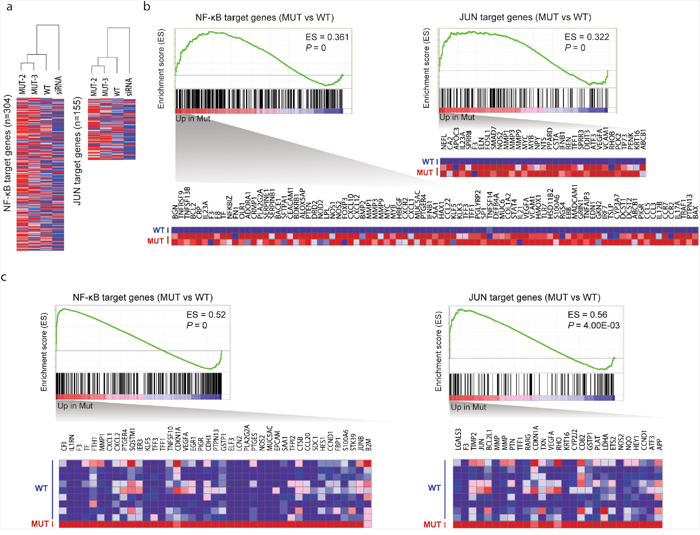
Figure 6: Effects of TBL1XR1 mutations on gene expression. a. Hierarchical clustering of gene expression microarray data. An unsupervised hierarchical clustering algorithm was used to group cell lines expressing mutant (L282P and H348Q), wild-type, and siRNA knockdown based on the expression patterns of NF-κB (n=304) and JUN target genes (n=155). b. GSEA of NF-κB and JUN target gene sets in mutant (n=2; L282P and H348Q) versus wild-type TBL1XR1 (n=1) based on microarray expression data. Enrichment scores (ES) and p-values are shown in the top right of the enrichment plot. Genes contributing to core enrichment are shown as a heatmap. c. GSEA of NF-κB and JUN target gene sets in mutant (n=1) versus wild-type TBL1XR1 (n=9) based on RNA-seq data.
Clinical impact of genomic alteration
All samples were thought to be negative for Chlamydophila psittaci (C. psittaci) based on the absence of WGS reads that aligned C. psittaci genome (Online Methods). Mutations in the MYD88, MET, and APC genes were found in tumor samples from 2 out of 11 patients who relapsed later, and none were found in patients without relapse (0/33) (p=0.054). Otherwise, mutations did not correlate with clinical parameters, including age, sex, international prognostic index, stage, performance status, LDH level, bone marrow involvement, or survival.
DISCUSSION
Here, we present the mutational landscape of ocular MZL and identify recurrent mutations in multiple genes involving altered signaling pathways. Commonly mutated genes are involved in the NF-kB signaling pathway, B-cell differentiation, and epigenetic modification. These findings are in line with those of previous studies although mutation frequency of each gene was variable according to the study [26, 27].
Ocular MZL arises on the chronic inflammatory background associated with infectious agents and autoimmune disease. Ongoing somatic hypermutation and biased usage of the IgVH genes in ocular MZL supports a role for antigen-driven lymphoproliferation and the clonal expansion of lymphoma cells [28]. C. psittaci infection is an etiologic agent that has been reported thus far, but this association has demonstrated geographical variability [29]. In our study, no chlamydia sequences were observed with whole-exome sequencing. NF-κB is a tightly regulated positive mediator of T- and B-cell development, proliferation, and survival. Constitutive activation of the NF-κB pathway plays an essential role in the neoplastic transformation of memory B cells and the progression of MZL [8]. While MZL derived from other sites, particularly the stomach and lung, involves chromosomal translocations of NF-κB signaling molecules that provoke antigen-independent MALT lymphoma growth, somatic mutation appears to be the major genetic event that activates NF-κB in ocular MZL. Our study confirms alterations in many genes belonging to different functional groups leading to the activation of NF-κB, where TNFAIP3 appears to be the major driver gene in terms of frequency and known functional impact. TNFAIP3 inhibits NF-κB activation by exerting dual ubiquitin-editing functions [30]. TNFAIP3-deleted mice with constitutive NF-κB activation demonstrate the overproduction of pro-inflammatory cytokines, severe multi-organ inflammation, enhanced proliferation upon activation, and finally develop autoimmune disease [31, 32]. However, TNFAIP3 inactivation may not be sufficient for the development of MZL and may require additional mutations. The alternative involvement of multiple genes, including CARD11, BCL10, CD79B, MYD88, and TNFRSF14, converging on the NF-κB pathway strongly supports their cooperation in the neoplastic transformation observed in ocular MZL. In addition, MZL involved the somatic mutation of several transcription factors that activate the NF-κB pathway. TBL1XR1, containing an F-box and the WD40 repeat domains, is a core component of NCoR and SMRT, which also contain HDAC3, transducin (beta)-like 1X (TBL1X), and GPS2 [33]. The F-box-like domain is essential for the recruitment of the 19S proteasome complex to nuclear receptor-regulated transcription units, and WD40 domains are important for binding with proteins such as NCoR [11]. To regulate the switch between corepression/coactivation, TBL1XR1 functions as an exchange cofactor via the recruitment of the ubiquitin conjugating/19S proteasome to the nuclear receptor corepressors NCoR and SMRT [34]. TBL1XR1 is regulated by post transcriptional modification, specifically phosphorylation and sumoylation, and is also required for the activation of the Wnt-β-catenin and NF-κB pathways [35, 36].
TBL1XR1 can become dysregulated by various mechanisms. In solid tumors, dysregulation commonly occurs through gene amplification. TBL1XR1 overexpression has been observed in diverse cancers [37, 38]. Conversely, hematopoietic neoplasms demonstrate abnormal regulation of TBL1XR1 through chromosome rearrangement and point mutations [39–41]. Recurrent mutations or deletions have been identified in 19% of primary central nervous system lymphoma cases, Sezary syndrome, and 2/9 ABC-type DLBCLs [39–42]. TBL1XR1 disrupts glucocorticoid receptor recruitment to the chromatin and results in glucocorticoid resistance in B-ALL [43]. Our study revealed that TBL1XR1 mutation at the WD40 domain facilitates binding with NCoR, leading to increased degradation of the NCoR complex, which results in activation of the NF-κB and c-Jun pathways.
Chromatin modification has emerged as a critical mechanism in lymphomagenesis. As shown here, MZL possesses a broad spectrum of somatic mutations in key genes that are involved in epigenetic regulation. CREBBP function as transcriptional coactivators for a large number of DNA binding transcription factors involved in multiple signaling pathways. CREBBP is targeted by inactivating mutations and deletions in 39% of DLBCL and 41% of FL cases [44]. CREBBP regulates BCL-2 expression and the rescue of immature B cells from apoptosis [45]. In B-cell lymphoma, mutant CREBBP proteins are deficient in acetylating BCL6 and p53, leading to the constitutive activation of BCL6 and decreased p53 tumor suppressor activity [46]. Deletion of CREBBP down-regulates the transcription of MHC class II genes [47], which contributes to the immune evasion of malignant lymphoma cells. Although the target genes of CREBBP mutations have yet to be explored in MZL, inactivating mutations of CREBBP may contribute to lymphomagenesis through immune evasion and the regulation of apoptosis. KMT2D affects the methylation of lysine 4 on histone H3 (H3K4) and the expression of a set of genes, including those involved in the CD40, JAK-STAT, Toll-like receptor and B cell receptor signaling pathways [48]. KMT2D mutations was reported in 22% of ocular MZL in previous study analyzing FFPE samples by targeted deep sequencing [27]. BRD4 is a BET protein linked to the regulation of NF-κB transcriptional activity. BRD4 maintains nuclear NF-κB levels by preventing its ubiquitination and degradation. BAP1 and WDR9 are novel chromatin modifiers which associations with malignant lymphoma have not been reported.
Several genes involved in B-cell differentiation were mutated. CXCR4 is required for the generation of the earliest identifiable B-cell precursor populations in bone marrow and promotes B-cell egress from Peyer's patches [49]. The transcription factor Oct-2 functions in the terminal phase of B-cell differentiation, and its depletion blocks the in vivo differentiation of antibody-secreting plasma cells [50]. The Met/HGF signaling pathway regulates B-cell adhesion in the germinal center and functions in B cell differentiation [51]. Of note, the novel c-Met mutation was found only in ocular MZL that later relapsed. Together with MYD88 and APC mutations, the c-Met mutation may be a marker for relapse, which is an uncommon event in ocular MZL. Further studies employing a larger cohort will be needed to confirm this hypothesis.
Collectively, the genetic alterations associated with ocular MZL appear to predominantly involve genes that play a role in NF-κB signaling pathways, with alterations in genes regulating chromatin modification, transcriptional regulation, and B-cell differentiation. The major driver gene is TNFAIP3, which is inactivated by recurrent disruptive structural variations involving the 6q23.3 chromosomal region, along with TBL1XR1 and CREBBP. In addition to these major driver genes, our results expand the directory of mutated genes in ocular MZL, some of which are novel findings. The functions of minor mutated genes and their roles in the initiation and progression of ocular MZL remain to be explored in future studies.
MATERIALS AND METHODS
Samples and sequencing
Clinical information for the patients is summarized in Supplementary Table 1. For WGS, 10 tumor samples with matched blood samples were obtained, and for targeted deep sequencing, 38 formalin-fixed, paraffin-embedded (FFPE) MZL tumor tissue samples were utilized. All patient samples were obtained with informed consent at the Samsung Medical Center, Seoul, Korea, and the study was approved by the Institutional Review Board in accordance with the Declaration of Helsinki.
Whole-genome-seq, targeted-seq, and RNA-seq with bioinformatic analysis were performed as described in the Supplementary information. Through the whole exome sequencing of MZL patient samples, potential mutations were selected and validated by Sanger sequencing.
Functional assays with TBL1XR1 mutants
Cell culture and cell lines
The BJAB (Burkitt lymphoma) cell line and 293T cell line were maintained in RPMI-1640 and DMEM, respectively, containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 250 ng/ml amphotericin B at 37°C and 5% CO2. Cell lines were regularly tested for mycoplasma infection using the MycoAlert mycoplasma detection kit (Lonza).
Cloning of TBL1XR1
A cDNA clone encoding full-length human TBL1XR1 was isolated from a human placenta cDNA library by PCR amplification. A PCR-generated DNA fragment encoding TBL1XR1 was cloned into the pcDNA3-GFP vector to express proteins with a GFP tag at the N terminus. Mutants of TBL1XR1, specifically Gly187Arg (GGA to GTA), Leu282Pro (CTA to CCA), His348Glu (CAT to CAA) and Ser459Asn (AGT to AAT), were generated using the QuikChange kit (Stratagene). Mutant constructs were verified by sequencing.
Transfection and RNA interference
To overexpress TBL1XR1 in the BJAB cell line, plasmids expressing N-terminal GFP- tagged wild-type TBL1XR1 and TBL1XR1 mutants (Gly187Arg, Leu282Pro, His348Glu and Ser459Asn) were transfected into cells using the Nucleofector I device (Amaxa) with Nucleofector solution V and the program O-017.
Co-immunoprecipitation
For co-immunoprecipitation, whole cell lysates were clarified by centrifugation at 12,000×g for 10 min. Cell lysates were mixed with protein G magnetic beads (Thermo Scientific) and 2 μg of GFP antibody (Abcam) and incubated for 2 h at 4°C on a rotator. Immune complexes were collected using a magnet and washed two times with washing buffer. Whole-cell lysate and beads associated with protein complexes were separated by SDS-PAGE and immunoblotted with appropriate antibodies.
Immunoblotting assay
Antibodies used for protein blotting were TBL1XR1 (Novus), HDAC3 and NCoR (Abcam) and alpha-tubulin (Santa Cruz). Horseradish peroxidase (HRP)-conjugated secondary antibodies (Bio-Rad) were used to detect primary antibodies.
Immunofluorescence analysis
293T cells were transiently transfected with GFP-tagged TBL1XR1 expression plasmids using Lipofectamine 3000 transfection reagent (Life Technologies). Then, the nuclei were stained with Nucblue (Hoechst dye, Thermo Scientific). Images were obtained using a confocal laser scanning microscope (CLSM-780, Zeiss).
Cell growth assays and NF-κB activity assays
At 48 h after transfection, BJAB cells were seeded into 96-well plates in triplicate at a density of 7.5×103 cells/well with 100 μl of RPMI-1640 containing 10% FBS and antibiotics. Cell growth was analyzed using Cell Counting Kit-8 (Dojindo) according to the manufacturer’s instructions. For the luciferase assay, cells were co-transfected with three NF-κB binding site (3xκBL) luciferase vectors and hRL (Renilla luciferase). Then, 5×105 cells were cultured in 24-well plates; after 24 h, the cells were lysed, and firefly and Renilla luciferase activities were measured with a Glomax 20/20 luminometer (Promega) using a dual luciferase reporter assay system (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity.
Gene expression analysis
Gene expression analysis using Agilent’s Gene Expression Hybridization Kit (GPL13497) was performed for cell lines expressing the mutant (L282P and H348Q) and wild-type TBL1XR1, and cell line treated with TBL1XR1 siRNA.
ACKNOWLEDGMENTS
This work was supported by grants from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C3414 to Y.H.K., HI15C3224, HI13C2096 to W.Y.P. and HI14C2331 to H.Y.Y.), and by a grant from NRF(National Research Foundation of Korea) funded by the Korean Government (NRF-2016H1A2A1907072 to H.J).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Streubel B, Simonitsch-Klupp I, Müllauer L, Lamprecht A, Huber D, Siebert R, Stolte M, Trautinger F, Lukas J, Püspök A, Formanek M, Assanasen T, Müller-Hermelink HK, et al. Variable frequencies of MALT lymphoma-associated genetic aberrations in MALT lymphomas of different sites. Leukemia. 2004;18:1722-1726.
2. Streubel B, Lamprecht A, Dierlamm J, Cerroni L, Stolte M, Ott G, Raderer M, Chott A. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood. 2003; 101:2335-2339.
3. Streubel B, Vinatzer U, Lamprecht A, Raderer M, Chott A. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia. 2005; 19:652-658.
4. Bi Y, Zeng N, Chanudet E, Huang Y, Hamoudi RA, Liu H, Dong G, Watkins AJ, Ley SC, Zou L, Chen R, Zhu X, Du MQ. A20 inactivation in ocular adnexal MALT lymphoma. Haematologica. 2012;97:926-930.
5. Xi R, Lee S, Xia Y, Kim TM, Park PJ. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res. 2016;44:6274-6286.
6. Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, Hsieh CH, Zhang C, Ren X, Protopopov A, Chin L, Kucherlapati R, Lee C, Park PJ. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153:919-929.
7. Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, Eagan M, Foster D, Haldeman BA, Hammond A, Haugen H, Jelinek L, Kelly JD, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell. 2001;104:9-19.
8. Yan Q, Wang M, Moody S, Xue X, Huang Y, Bi Y, Du MQ. Distinct involvement of NF-kappaB regulators by somatic mutation in ocular adnexal malt lymphoma. Br J Haematol. 2013;160:851-854.
9. Troen G, Warsame A, Delabie J. CD79B and MYD88 Mutations in Splenic Marginal Zone Lymphoma. ISRN Oncol. 2013;2013:252318. doi: 10.1155/2013/252318.
10. Li JY, Daniels G, Wang J, Zhang X. TBL1XR1 in physiological and pathological states. Am J Clin Exp Urol. 2015;3:13-23.
11. Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003;22:1336-1346.
12. Wang Y, Hu XJ, Zou XD, Wu XH, Ye ZQ, Wu YD. WDSPdb: a database for WD40-repeat proteins. Nucleic Acids Res. 2015;43:D339-344.
13. Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148-1159.
14. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298-303.
15. Vaqué JP, Martínez N, Batlle-López A, Pérez C, Montes-Moreno S, Sánchez-Beato M, Piris MA. B-cell lymphoma mutations: improving diagnostics and enabling targeted therapies. Haematologica. 2014;99:222-231.
16. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, Fox H, Jacobi K, Saiya-Cork K, Bixby D, Lebovic D, Roulston D, Shedden K, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123:1487-1498.
17. Schmidt J, Federmann B, Schindler N, Steinhilber J, Bonzheim I, Fend F, Quintanilla-Martinez L. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169:795-803.
18. Yaktapour N, Übelhart R, Schüler J, Aumann K, Dierks C, Burger M, Pfeifer D, Jumaa H, Veelken H, Brummer T, Zirlik K. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood. 2013;122:1621-1633.
19. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695-708.
20. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
21. Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645-652.
22. Delgado M, Ganea D. VIP and PACAP inhibit activation induced apoptosis in T lymphocytes. Ann N Y Acad Sci. 2000;921:55-67.
23. Daniels G, Li Y, Gellert LL, Zhou A, Melamed J, Wu X, Zhang X, Zhang D, Meruelo D, Logan SK, Basch R, Lee P. TBLR1 as an androgen receptor (AR) coactivator selectively activates AR target genes to inhibit prostate cancer growth. Endocr Relat Cancer. 2014;21:127-142.
24. Daniels G, Zhang X, Zhong X, Santiago L, Wang LH, Wu X, Zhang JY, Liang F, Li X, Neubert TA, Steinke L, Shen Y, Basch R, et al. Cytoplasmic, full length and novel cleaved variant, TBLR1 reduces apoptosis in prostate cancer under androgen deprivation. Oncotarget. 2016;7:39556-39571. doi: 10.18632/oncotarget.9005.
25. Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009;23:681-693.
26. Cani AK, Soliman M, Hovelson DH, Liu CJ, McDaniel AS, Haller MJ, Bratley JV, Rahrig SE, Li Q, Briceño CA, Tomlins SA, Rao RC. Comprehensive genomic profiling of orbital and ocular adnexal lymphomas identifies frequent alterations in MYD88 and chromatin modifiers: new routes to targeted therapies. Mod Pathol. 2016;29:685-697
27. Johansson P, Klein-Hitpass L, Grabellus F, Arnold G, Klapper W, Pförtner R, Dührsen U, Eckstein A, Dürig J, Küppers R. Recurrent mutations in NF-κB pathway components, KMT2D, and NOTCH1/2 in ocular adnexal MALT-type marginal zone lymphomas. Oncotarget. 2016;7:62627-39. doi: 10.18632/oncotarget.11548.
28. Coupland SE, Foss HD, Anagnostopoulos I, Hummel M, Stein H. Immunoglobulin VH gene expression among extranodal marginal zone B-cell lymphomas of the ocular adnexa. Invest Ophthalmol Vis Sci. 1999;40:555-562.
29. Chanudet E, Zhou Y, Bacon CM, Wotherspoon AC, Müller-Hermelink HK, Adam P, Dong HY, de Jong D, Li Y, Wei R, Gong X, Wu Q, Ranaldi R, et al. Chlamydia psittaci is variably associated with ocular adnexal MALT lymphoma in different geographical regions. J Pathol. 2006;209:344-351.
30. Heyninck K, Beyaert R. A20 inhibits NF-kappaB activation by dual ubiquitin-editing functions. Trends Biochem Sci. 2005;30:1-4.
31. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350-2354.
32. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wójtowicz E, Soberon V, Schenten D, Mack B, Reutelshöfer M, Beyaert R, Amann K, van Loo G, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011;117:2227-2236.
33. Zhang J, Kalkum M, Chait BT, Roeder RG. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611-623.
34. Choi HK, Choi KC, Yoo JY, Song M, Ko SJ, Kim CH, Ahn JH, Chun KH, Yook JI, Yoon HG. Reversible SUMOylation of TBL1-TBLR1 regulates beta-catenin-mediated Wnt signaling. Mol Cell. 2011;43:203-216.
35. Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511-526.
36. Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG. TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell. 2008;29:755-766.
37. Chen SP, Yang Q, Wang CJ, Zhang LJ, Fang Y, Lei FY, Wu S, Song LB, Guo X, Guo L. Transducin beta-like 1 X-linked receptor 1 suppresses cisplatin sensitivity in nasopharyngeal carcinoma via activation of NF-kB pathway. Mol Cancer. 2014;13:195.
38. Liu Y, Sun W, Zhang K, Zheng H, Ma Y, Lin D, Zhang X, Feng L, Lei W, Zhang Z, Guo S, Han N, Tong W, et al. Identification of genes differentially expressed in human primary lung squamous cell carcinoma. Lung Cancer. 2007;56:307-317.
39. Andersson E, Eldfors S, Edgren H, Ellonen P, Väkevä L, Ranki A, Mustjoki S. Novel TBL1XR1, EPHA7 and SLFN12 mutations in a Sezary syndrome patient discovered by whole exome sequencing. Exp Dermatol. 2014;23:366-368.
40. Bruno A, Boisselier B, Labreche K, Marie Y, Polivka M, Jouvet A, Adam C, Figarella-Branger D, Miquel C, Eimer S, Houillier C, Soussain C, Mokhtari K, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. 2014;5:5065-5075. doi: 10.18632/oncotarget.2080.
41. Scott DW, Mungall KL, Ben-Neriah S, Rogic S, Morin RD, Slack GW, Tan KL, Chan FC, Lim RS, Connors JM, Marra MA, Mungall AJ, Steidl C, et al. TBL1XR1/TP63: a novel recurrent gene fusion in B-cell non-Hodgkin lymphoma. Blood. 2012;119:4949-4952.
42. Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, Maingonnat C, Jais JP, Peyrouze P, Figeac M, Molina TJ, Desmots F, Fest T, et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res. 2016;22:2919-2928.
43. Jones CL, Bhatla T, Blum R, Wang J, Paugh SW, Wen X, Bourgeois W, Bitterman DS, Raetz EA, Morrison DJ, Teachey DT, Evans WE, Garabedian MJ, et al. Loss of TBL1XR1 disrupts glucocorticoid receptor recruitment to chromatin and results in glucocorticoid resistance in a B-lymphoblastic leukemia model. J Biol Chem. 2014;289:20502-20515.
44. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189-195.
45. Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546-5556.
46. Giebler HA, Lemasson I, Nyborg JK. p53 recruitment of CREB binding protein mediated through phosphorylated CREB: a novel pathway of tumor suppressor regulation. Mol Cell Biol. 2000;20:4849-4858.
47. Fontes JD, Kanazawa S, Jean D, Peterlin BM. Interactions between the class II transactivator and CREB binding protein increase transcription of major histocompatibility complex class II genes. Mol Cell Biol. 1999;19:941-947.
48. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, Ge K, Dalla-Favera R, Pasqualucci L. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21:1190-1198.
49. Schmidt TH, Bannard O, Gray EE, Cyster JG. CXCR4 promotes B cell egress from Peyer's patches. J Exp Med. 2013;210:1099-1107.
50. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15:160-171.
51. Skibinski G, Skibinska A, James K. The role of hepatocyte growth factor and its receptor c-met in interactions between lymphocytes and stromal cells in secondary human lymphoid organs. Immunology. 2001;102:506-514.