
INTRODUCTION
Ovarian cancer is the most lethal gynecologic malignancy, accounting for more than 150,000 deaths annually worldwide [1]. The reason for the poor prognosis of ovarian cancer is that the majority of patients present an advanced stage of this disease, characterized by metastasis to the peritoneal cavity. Importantly, intra-tumor heterogeneity has been reported for these types of tumors [2, 3]. Tumors are thought to originate from a single clonal state that then expands clonally, accompanied by genetic changes that give rise to functional differences, resulting in different stages and characteristics of neoplastic development [4, 5].
Copy number variation (CNV) is increasingly linked to the genetic and phenotypic diversity among cancers, and is frequently associated with the activation of oncogenic drivers or the deletion of tumor suppressors [6–9]. Using either conventional metaphase chromosome-based comparative genomic hybridization [10, 11] or array-based high-resolution genomic technology for identifying genome-wide CNVs in ovarian cancer [12–19], previous studies have identified regions of frequently increased copy number along 1q, 3q26, 7q32–q36, 8q24, 17q32 and 20q13, and reduced copy number along 1p36, 4q, 13q, 16q, 18q and Xq12. In addition, a number of high-level amplifications have been highlighted as predictive biomarkers, including those of CCNE1, RB1, MYC, ERBB2, PIK3CA, EVI1, AKT2, NOTCH3 and FGFR1. While studies of numerous patient cohorts have enabled the precise characterization of the genetic alterations that predict clinical outcomes [19, 20] or chemoresistance [12], as well as the precise comparison of the genetic alterations between primary and metastatic lesions [15] or histotype-specific ovarian cancers [21], there has been little effort to correlate clinical outcomes with the genetic alterations of ovarian cancer cell subclones characterized by distinct invasive/migratory capacities. Considering the high malignant potential and poor 5-year survival rate associated with this type of cancer, the mechanisms underlying advanced ovarian cancer should be elucidated through comparison of the genetic profiles of heterogeneous neoplastic subclones.
We previously established stable cellular subclones derived from the human epithelial ovarian cancer cell lines A2780 and SKOV3, which exhibit distinct invasive/migratory capacities [22]. A-H and S-H cells (A2780 and SKOV3 subclones with higher invasive/migratory capacities) exhibited enhanced proliferative, anti-apoptotic and anti-anoikic activity and reduced autophagic activity compared with A-L and S-L cells (A2780 and SKOV3 subclones with lower invasive/migratory capacities). In addition, A-H and S-H cells were significantly more resistant to cisplatin and Taxol in vitro and had higher capacities for tumor formation in vivo than A-L and S-L cells [22]. These two pairs of subclones with the same hereditary background served as models of intra-tumor heterogeneity. In the present study, we searched for differences in the genomic CNVs of the chosen subclones. Determining the genetic and molecular events leading to the distinct invasive/migratory capacities of these subclones will improve the accuracy of clinical interpretations and the effectiveness of therapeutics for advanced ovarian cancer.
RESULTS
Validation of the CNV data
We identified two pairs of subclones derived from the ovarian cancer cell lines A2780 and SKOV3 in our previous work [22]. A-H and S-H cells had higher invasive/migratory capacities than A-L and S-L cells, respectively. We also found that A-H and S-H cells showed enhanced proliferative and anti-apoptotic activities compared with A-L and S-L cells. Moreover, they had higher level of resistance to cisplatin and Taxol in vitro and tumor formation capacity in vivo [22]. Affymetrix CytoScan™ HD microarrays were used to investigate regions of DNA with copy number alterations for the four subclones. For validation of the array data, we selected several regions for quantitative PCR analysis of A-H versus A-L copy number and S-H versus S-L copy number.
In the A-H versus A-L validation, the relative gene copy numbers in regions of 11q12.2, 12p13.1, 12p12.1 and 19q13.32 of A-H were found to be amplified, whereas the relative gene copy numbers in regions of 4q25, 5q21.3, 5q22.2, 5q31.2, 5q33.3, 9q34.12, 9q34.3 and 9q22.33 of A-H revealed deletion, when the copy number of A-L was set as 1. In contrast, when the gene copy number of A-H was set as 1, the copy numbers in regions of 2q32.3, 2q32.2 and 15q25.1 of A-L were amplified. For S-H/S-L validation, regions of 11q12.1, 12p13.1, 12p12.1 and 19q13.32 of S-H were amplified and regions of 8p23.3 and 17p13.1 of S-H were deleted relative to S-L. In contrast, in S-L cells, regions of 2p14, 3p21.31, 10q24.32, 10q26.3, 15q11.2, 15q15.2 and 15q22.31 were amplified and regions of 8p12 and 8p11.23 were deleted relative to S-H (Supplementary Figure 1). The relative copy numbers agreed with the array data.
Copy number profiling of the heterogeneous invasive/migratory subclones
We compared the genomic DNA copy numbers of highly and minimally invasive/migratory subclones with a HapMap control set, to determine specific amplifications and deletions in cancer cell lines versus normal samples. The CNV profiles for the subclones are shown in Figure 1. The distributions of altered regions were quite different in the A2780- and SKOV3-derived subclones. In each cell line, a large number of chromosomal differences revealed some degree of genetic heterogeneity between A-H and A-L, S-H and S-L. Encouragingly, the majority of regions agreed with those previously published in studies of ovarian cancer [15, 17–19]. These included amplifications in 1q, 7q35-36, 17q and 20q and deletions in 4q, 5q, 13q, 16q and 18q, among others, in both A-H and A-L cells, as well as amplifications in 1q, 3q, 6p, 7q35-36, 8q, 12p and 20q and deletions in 1p36, 4q, 16q, 17p, 17q, 22q and Xq, among others, in both S-H and S-L cells. It was clear from our analysis that there were fewer copy number changes in the A2780-derived subclones than in the SKOV3-derived subclones. According to previous studies on histotype-specific CNVs in ovarian cancer [21, 23], ovarian serous cancer is characterized by 1q, 3q, 6p, 7q, 8q, 11q, 12p and 20q amplification and 1p36, 4q, 5q, 6q, 8p, 11p, 13q, 15q, 16q, 17, 18q, 22q and X deletion relative to other subtypes. Apparently, the SKOV3-derived subclones were more molecularly similar to ovarian serous cancer than were the A2780-derived subclones.
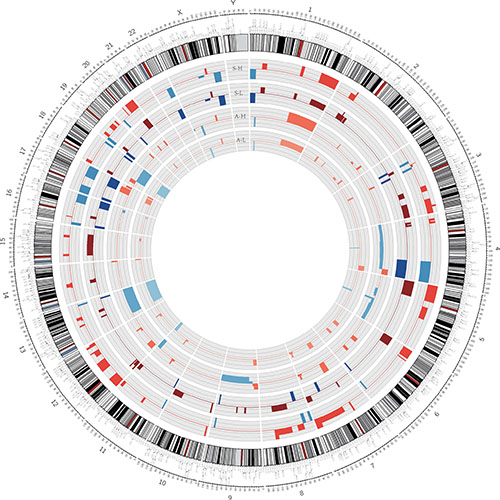
Figure 1: Genetic heterogeneity of the distinct highly and minimally invasive/migratory subclones. Circos plot of segmented CNVs in S-H/S-L and A-H/A-L cells. Colored bands expanding toward the center or the periphery of the diagram represent copy number losses or gains, respectively (red, gain; blue, loss).
In the comparison of A-H and A-L, while the CNVs of both A-H and A-L overlapped significantly with those identified in previous studies, large regions were different between the two subclones. Segments of gains in 9p22.3-9p22.2, 11q12.1-11q12.2, 12p13.31-12p11.23, 17q25.2, 18p11.21, 18q11.1-18q11.2, 19q13.32 and 20q13.33 appeared only in A-H (named “A-H-specific gain”), whereas segments of gains in 2p14, 2q32.1, 2q32.2-2q32.3, 15q25.1-15q25.2 and 15q25.3 were observed only in A-L (named “A-L-specific gain”). While we also observed losses in segments of A-H only (named “A-H-specific loss”), we did not find any chromosomal losses in A-L only. Segment locations and genes with CNVs, both shared and specific, are documented in Supplementary Table 4.
Functional analysis by DAVID [24, 25] indicated that the proteins encoded by the “A-H-specific gain” genes (245 genes) were involved in sensory perception, signal transduction, defense responses, nucleotide excision repair, DNA damage removal and cell proliferation. The proteins encoded by the “A-H-specific loss” genes (2313 genes) displayed significant enrichment for cell adhesion and extracellular structure organization. The loss of such genes may promote the epithelial-mesenchymal transition, whereby cancer cells lose their polarity and adhesive abilities and acquire migratory and invasive capacities that facilitate their detachment from the primary tumor and invasion through the basement membrane into the circulation to remote sites [26–28]. Thus, the loss of these genes could explain the higher invasive/migratory capacities of A-H cells. The “A-L-specific gain” genes (55 genes) pertained mainly to cell apoptosis and death. Copy number gains in these genes might contribute to the lower capacity for proliferation and invasion/migration and the higher rate of apoptosis of the A-L subclone (Supplementary Table 5).
In the S-H and S-L comparison, segments of gains that appeared only in S-H were distributed across 3p, 5q, 6p, 7q, 8p, 11, 12, 14q, 16p, 17, 19q and 20q (named “S-H-specific gain”), while segments of losses only in S-H were located on 8p23.3-8p23.2, 8p21.3-8p21.2, 8q24.12, 17p13.1-17p11.2 and Xq21.31 (named “S-H-specific loss”). S-L also harbored specific gains in 2p14, 2p16.1, 3p21.31, 3q25.31, 6p25.3, 8q24.12, 9p11.2, 9q21.11, 9q13, 10q24.32-10q26.3, 11q24.2, 15q11.2-15q25.2 and 16p11.2 and specific losses in 7q31.1, 8p12-8p11.23 and 8p11.23 (named “S-L-specific gain” and “S-L-specific loss,” respectively; Supplementary Table 6).
GO analysis of the genes within regions specific to S-H and S-L was also performed. The GO terms and KEGG enrichment results are listed in Supplementary Table 7. The genes with “S-H-specific gain” (1520 genes) included the Wnt/β-catenin signaling pathway members WNT2 and CAV1; CAV2, the product of which is likely to be involved in signal transduction, cellular growth and apoptosis; the cell-cycle-related gene CDKN1B; and cortactin binding protein 2 (CTTNBP2). The proteins encoded by these genes could function in microtubule polymerization, protein complex assembly and vasodilation. Other notable GO terms with P values < 0.05 include the following: cell-cell signaling, regulation of growth, nucleotide excision repair, DNA damage removal, negative regulation of apoptosis, negative regulation of programmed cell death, and negative regulation of cell adhesion. The identification of such pathways seems consistent with the high level of proliferation and low level of apoptosis in S-H cells. Moreover, KEGG analysis revealed significant enrichment of genes with products involved in Melanoma and Glioma (P = 0.013 and 0.039) and a trend for those involved in Prostate cancer and Non-small lung cancer (P = 0.059 and 0.098). In contrast, the proteins encoded by the “S-H-specific loss” genes (182 genes) appeared to be involved in apoptosis (hsa04210, P = 0.002), consistent with the low level of apoptosis among S-H cells. Major functional categories for “S-L-specific gains” (836 genes) included cell death and apoptosis, regulation of the cell cycle, and regulation of cell-matrix adhesion. “S-L-specific losses” (22 genes) were enriched for the regulation of protein binding. The annotations for specific gains/losses in S-H/S-L differed from those of A-H/A-L, demonstrating the heterogeneity of ovarian cancer cell lines. This also suggested that genetic alterations might contribute to functional differences in the two pairs of heterogeneous subclones.
By using matched highly and minimally invasive/migratory subclones, we could also detect regions that were altered in both subclones and further changed in one subclone. These included, for instance, amplifications that were shared between both high and low subclones relative to a normal baseline, and were further amplified in the high or low subclone compared to the other (Supplementary Table 8). For consistency, we focused our analysis on the above results. In the case of shared amplifications in A-H and A-L that were further amplified in A-H cells, we also observed enrichment of genes encoding proteins that were involved in sensory perception, the immune response and signal transduction. Genes amplified in both S-H and S-L and further amplified in S-H were involved in the immune response and growth, and KEGG analysis also revealed a trend in pathways in cancer. We also observed that ECM1 and AKT3, members of the PI3K/AKT/mTOR pathway, as well as RASSF5, RYR2 and IGFBP3, were amplified in both high and low subclones relative to normal cells, and also amplified in both A-H and S-H compared with A-L and S-L, consistent with our previous gene expression data [22].
Analysis of genes identified in CNV regions in both highly or both minimally invasive/migratory subclones
Although both pairs of subclones had cell-line-specific characteristics, comparison data between A-H and A-L overlapped to some extent with comparison data between S-H and S-L. There were 118 genes amplified in both “A-H-specific gains” and “S-H-specific gains,” and 27 genes amplified in both “A-L-specific gains” and “S-L-specific gains.” However, there was no gene deleted in both “A-H-specific losses” and “S-H-specific losses,” or in both “A-L-specific losses” and “S-L-specific losses” (Figure 2).
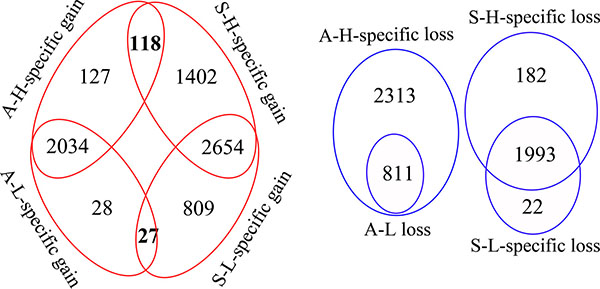
Figure 2: Diagrams indicating the overlapping genes with CNV between the distinct invasive/migratory subclones. The red circle indicates the number of amplified genes and the blue circle indicates the number of deleted genes. The red circle demonstrates that there were 245 (117+118) genes amplified as “A-H-specific gains” and 1520 (1402+118) genes amplified as “S-H-specific gains,” with 118 genes overlapping between them. There were 55 (28+27) genes amplified as “A-L-specific gains” and 836 (809+27) genes amplified as “S-L-specific gains,” with 27 genes overlapping between them. The blue circle demonstrates that there were 2313 genes deleted as “A-H-specific losses” and 182 genes deleted as “S-H-specific losses,” as well as 22 genes deleted as “S-L-specific losses”.
Functional analysis of genes within regions with specific gains for both A-H and S-H (118 genes) revealed enrichment mainly in genes with products involved in defense response (10 genes, P = 0.0035), nucleotide excision repair, DNA damage removal (3 genes, P = 0.0054), cell activation (6 genes, P = 0.0146), regulation of immune system processes (positive regulation of leukocyte activation, 4 genes, P = 0.0162; innate immune response activating cell surface receptor signaling pathway, 2 genes, P = 0.02; positive regulation of immune system process, 5 genes, P = 0.0317; T cell and lymphocyte costimulation, 2 genes, P = 0.0395), cell proliferation (7 genes, P = 0.0221) and G1/S transition of mitotic cell cycle (3 genes, P = 0.0322). Shared A-L- and S-L-specific gains (27 genes) were enriched for genes encoding proteins involved in cell apoptosis and death. The top annotations with P values < 0.05 were the following: apoptosis (5 genes, P = 0.0046), programmed cell death (5 genes, P = 0.0049), cell death (5 genes, P = 0.0082), and death (5 genes, P = 0.0088) (Figure 3, Supplementary Table 9).
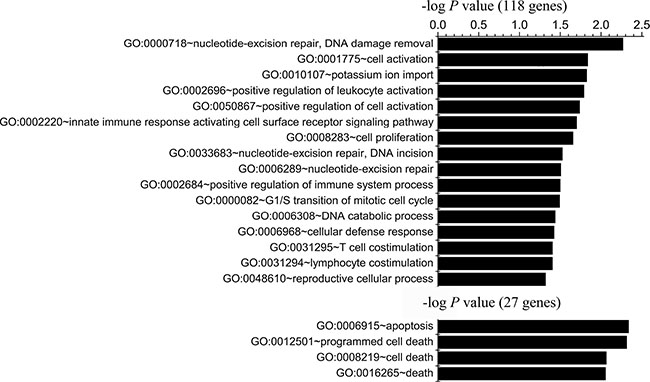
Figure 3: Functional enrichment analysis of genes identified in CNV regions in both highly or both minimally invasive/migratory subclones. Gene Ontology-based annotation was used for functional enrichment analysis of genes with shared A-H/S-H-specific gains (118 genes) and genes with shared A-L/S-L-specific gains (27 genes) through DAVID. The bar represents the -log of the P value for the significance of enrichment. Only annotations with significant P values < 0.05 are shown.
Assessment of candidate gene expression and patient outcomes
Because the primary purpose of this study was to identify genes that were distinctively altered in highly and minimally invasive/migratory subclones and had a functional effect on tumor metastasis, all genes that were exclusively amplified in both A-H and S-H (relative to A-L and S-L) were analyzed. In the gene ontology analysis, there were both A-H- and S-H-specific gains in a signature of immune genes, including defense response genes, immune system regulatory genes, and immune cell activation genes. These results were consistent with the results of a previous study of serous and endometrioid tumors mainly from the Australian Ovarian Cancer Study [29]. These investigators defined a subtype of high-grade ovarian cancer characterized by up-regulation of immune response genes, with worse survival (both overall survival (OS) and progression-free survival (PFS), P < 0.001) than endometrioid ovarian tumors or serous tumors of low grade, early stage and low malignant potential. Another previous study identified amplified regions mapping to 12p12.1, 19q12, 20q11.21 and 20q13.12 with significantly worse outcomes (OS, P = 0.0028; PFS, P < 0.001) [20]. For comparison to our data, the region of 12p12.1 amplified in both A-H- and S-H-specific gains overlapped with the regions of the previous study, indicating a worse outcome.
We also assessed these genes with respect to clinical outcomes using the TCGA database. We found that patients with altered copy numbers of defense response genes (KLRC4, CLEC1A, KLRC2, ABCC9, KLRC3, OLR1, KCNJ8, MS4A2, CLEC7A and CLEC1B) had a poorer OS than patients without such alterations. Patients with alterations of immune system and immune cell activation genes (BLOC1S3, KLRK1, MS4A2, CLEC7A and CD5), as well as cell activation genes (PLCZ1, BLOC1S3, MS4A1, KLRK1, CLEC7A and ERCC1) also had poor OS. However, the differences associated with these three categories of genes were not significant (P = 0.245, 0.151, 0.239, respectively). Amplification of nucleotide excision repair, DNA damage removal genes (DDB1, ERCC1 and ERCC2) was associated with trends for worse OS (P = 0.0662) and PFS (P = 0.0919). Both highly invasive/migratory subclones were also characterized by amplification of cell proliferation genes (BCAT1, PRPF19, IFLTD1, MS4A2, CD5, ERCC1 and ERCC2), which was associated with a significant difference in OS (P = 0.0283) but not PFS (P = 0.222), and by amplification of genes with products involved in the G1/S transition of the cell cycle (BCAT1, CDKN1B and MARK4), which was associated with a significant difference in OS (P = 0.00454) but not PFS (P = 0.148) (Supplementary Figure 2–7).
In the analysis of each gene with both A-H- and S-H-specific gains, TRAPPC6A, BLOC1S3, EXOC3L2, CKM, KLC3, ERCC2, PPP1R13L, CD3EAP, ERCC1, FOSB, RTN2, PPM1N, VASP and OPA3 amplifications were associated with significantly worse outcomes (OS: all P < 0.01; PFS: all P < 0.05). MARK4, BCL2L14, LRP6, MANSC1, DUSP16, CREBL2, GPR19, CDKN1B, ST8SIA1 and PPP1R37 amplifications were associated with significantly differences in OS but not PFS (OS: all P < 0.05). MS4A8, MS4A15 and MS4A10 amplifications were associated with significantly differences in PFS but not OS (PFS: all P < 0.05) (Supplementary Table 10).
We then assessed the correlation between copy number and mRNA levels. The altered copy numbers of several genes, including BLOC1S3, ERCC1, DDB1, ERCC2, BCAT1, PRPF19, CDKN1B, MARK4, TRAPPC6A, KLC3, PPP1R13L, CD3EAP, RTN2, VASP, OPA3, LRP6, MANSC1, DUSP16, CREBL2, GPR19 and CDKN1B, correlated well with their mRNA levels in the TCGA database (Supplementary Table 10). We also detected the expression levels using RT-PCR and Western blot analysis of all the above genes in our distinct highly and minimally invasive/migratory subclones. We found that the mRNA and protein levels of DDB1, ERCC1, ERCC2, CKM, PRPF19, BCAT1, PPP1R13L, CDKN1B, CD3EAP and MARK4 were both significantly greater in A-H/S-H cells than in A-L/S-L cells (Supplementary Figure 8).
After integrating our analysis of CNV data with gene expression and patient outcome data, we identified 7 genes as strong candidates for therapeutic targeting in advanced ovarian cancer: DDB1, ERCC1, ERCC2, PRPF19, BCAT1, CDKN1B and MARK4 (Table 1). Relative copy numbers of these genes by quantitative PCR analysis of A-H versus A-L and S-H versus S-L were consistent with the array data (Supplementary Figure 9). Copy number gains correlated well with enhanced expression for DDB1, ERCC1, ERCC2, PRPF19, CDKN1B and MARK4, and with the same trend for BCAT1. Elevated ERCC1 and ERCC2 levels were consistently associated with worse outcomes (both OS and PFS, P < 0.01); CDKN1B and MARK4 amplification and increased expression were associated with significantly worse OS but not PFS (CDKN1B, OS: P = 0.0243, PFS: P = 0.345; MARK4, OS: P = 0.0034, PFS: P = 0.0905) (Figure 4). In addition, COX regression analysis revealed that ERCC1, ERCC2, CDKN1B and MARK4 were independent prognostic factors for OS. PRPF19, BCAT1 and DDB1 were identified according to the functional GO groups and their expression levels. Amplification of PRPF19 was not significantly associated with OS, but rather with a trend for better PFS (P = 0.0856). BCAT1 amplification was not associated with significantly worse outcomes, but was in the functional GO groups of cell proliferation and the G1/S transition of the cell cycle. Elevated DDB1 was not associated significantly with survival, but belonged to the group of DNA damage removal genes (Supplementary Figure 10). RT-PCR and Western blot analyses of our highly and minimally invasive/migratory subclones consistently confirmed the expression levels of the seven genes (Figure 5).
Table 1: Analyses of candidate genes in the TCGA ovarian cancer database (TCGA, Provisional) with the cBioPortal tool
Genes |
Location |
Correlation between copy number |
Survival (P value) |
||
|---|---|---|---|---|---|
Pearson |
Spearman |
OS |
PFS |
||
DDB1 |
11q12.2 |
0.565 |
0.523 |
0.89 |
0.961 |
ERCC1 |
19q13.32 |
0.547 |
0.626 |
0.00208 |
0.00778 |
ERCC2 |
19q13.32 |
0.63 |
0.696 |
0.00629 |
0.00778 |
PRPF19 |
11q12.2 |
0.478 |
0.461 |
0.451 |
0.0856 |
CDKN1B |
12p13.1 |
0.492 |
0.452 |
0.0243 |
0.345 |
BCAT1 |
12p12.1 |
0.42 |
0.262 |
0.152 |
0.729 |
MARK4 |
19q13.32 |
0.699 |
0.717 |
0.0034 |
0.0905 |
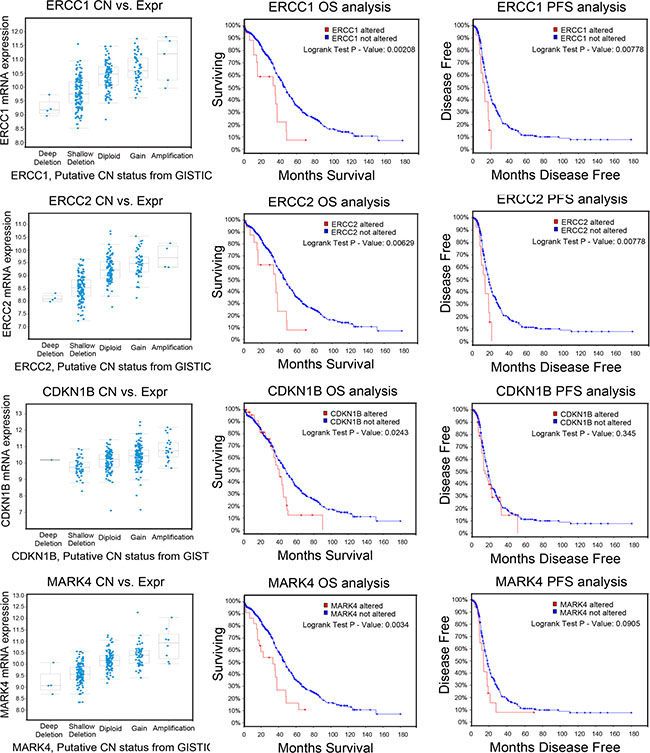
Figure 4: Reproduction of data from the TCGA database with the cBioPortal tool. The amplifications of candidate genes, ERCC1, ERCC2, CDKN1B and MARK4 correlated with gene expression and were associated with patient outcomes. Kaplan-Meier analysis of OS and PFS was performed with the copy number of the candidate gene as a categorical variable, so that the effects of genes with unaltered and altered copy numbers could be compared. The results of a Cox proportional hazards test, with residual disease as a copredictor, are shown as P values. The correlations between the copy numbers and the mRNA levels of the candidate genes, determined through RNA Seq analysis of the TCGA datasets, are also shown.
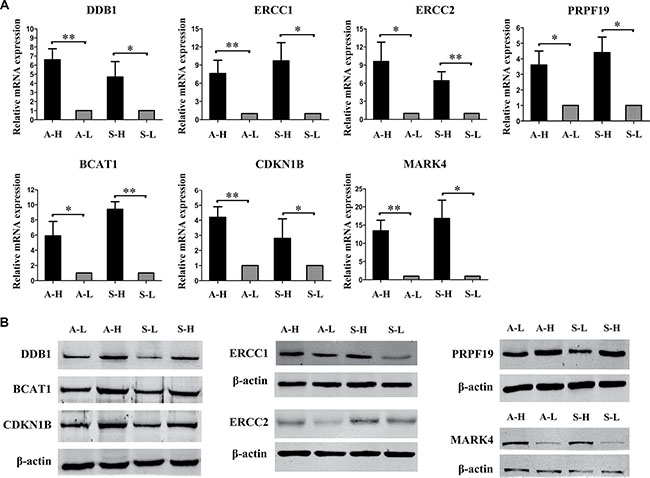
Figure 5: Expression of the candidate genes in highly and minimally invasive/migratory subclones. (A) RT-PCR and (B) Western blot analysis consistently confirmed that DDB1, ERCC1, ERCC2, PRPF19, BCAT1, CDKN1B and MARK4 expression were all greater in A-H/S-H cells than in A-L/S-L cells. Error bars represent the SEM, n = 3 (*P < 0.05. **P < 0.01).
DISCUSSION
Studies performed by others [30–32] and in our lab [33–35] have demonstrated that there is heterogeneity among ovarian cancers. We used the limiting-dilution method to isolate and establish heterogeneous subclones of the ovarian cancer cell lines A2780 and SKOV3 [22]. Among these subclones A-H/S-H and A-L/S-L cells exhibited the highest and lowest invasive/migratory capacities, respectively. A-H and S-H cells displayed enhanced proliferation and anti-apoptotic activity, as well as significant resistance to cisplatin and Taxol in vitro, and a higher capacity for tumor formation in vivo, compared with A-L and S-L cells, respectively. These studies demonstrated that we successfully produced a model consisting of clones with distinct invasive/migratory capacities and the same hereditary background. We hypothesized that heterogeneity may preferentially evolve due to initial alterations in DNA, followed by alterations in chromosomal copy numbers. In the present study, we used array-based technology to systematically analyze CNVs in the heterogeneous invasive/migratory models. We also integrated our data with clinical findings to determine the relationship among CNVs, gene expression and patient outcomes, and thus identified a number of genes that may be good therapeutic targets.
Several studies have described the copy number aberrance in ovarian cancer, and our data agreed well with these previous CNV analyses. However, it was clear that the A2780-derived subclones differed extensively from the SKOV3-derived subclones. This may have contributed to cell-line-specific characteristics and the heterogeneity of these ovarian cancer cell lines. The A2780 cell line is probably of endometrioid origin, while the SKOV3 cell line is of clear cell carcinoma origin [36]. The different ovarian cancer histotypes practically represent genetic disparities. Moreover, such heterogeneity could also be due to the cellular phenotype differences between the two cell lines. A2780 cells exhibit an epithelial/intermediate (round) phenotype accompanied with robust expression of epithelial markers, while SKOV3 cells display a typical mesenchymal (spindle-like) morphology accompanied with predominant expression of mesenchymal markers [37]. Accordingly, some of the differences observed in the CNV patterns between the two cell lines might be cellular phenotype-related. In this sense in our results, for example, the A-H-specific loss genes were significantly enriched in cell adhesion and extracellular structure, which cold promote epithelial-to-mesenchymal transition, indicating a mesenchymal-like change in A-H cells.
We performed functional enrichment analysis of our data set to identify genes of specific annotations that were distinctively altered in A-H and A-L, S-H and S-L pairs. It is possible that different mechanisms were responsible for the two distinct cell lines. We observed that “A-H-specific gains” were enriched in signal transduction through G-protein coupled receptors, which involve in many biological functions, such as the sensing of taste, light and odor, chemotaxis, and inflammatory and immune responses [38, 39]. In addition, the loss of genes involved in cell-cell adhesion in A-H cells might have contributed to their high level of invasion/migration. On the other hand, the “S-H-specific gains” were distinctly enriched in microtubule polymerization and vasodilation, as well as regulation of growth and negative regulation of apoptosis/programmed cell death/cell adhesion, which may have promoted cell proliferation and metastasis. These findings strongly suggest that distinct genetic pathways caused the distinct invasive/migratory capacities of the A2780- and SKOV3-derived subclones.
It was also clear from our analysis that some of the same pathways promoted the higher invasive/migratory capacities of A-H and S-H cells, including defense responses, nucleotide excision repair, DNA damage removal, positive regulation of immune system processes, cell activation, cell proliferation and the G1/S transition of the cell cycle. An immune signature has also been shown to be involved in pathological inflammatory conditions and cancer [40]. The expression of inflammation-related programs due to the activation of oncogenes induces the formation of an inflammatory microenvironment, which in turn promotes carcinogenesis [41]. The enrichment of the GO categories of nucleotide excision repair, DNA damage removal and cell proliferation was also consistent with the functional attributes of the A-H and S-H subclones. In contrast, A-L and S-L both had amplicons enriched mainly in cell apoptosis and death, demonstrating a relationship between clonal genomic changes and the low invasive/migratory capacities.
GO analysis allowed us to group the affected genes according to their biological processes, providing new insights into tumor metastasis. Our proof-of-principle screen indicated that several genes had strong prognostic significance for advanced ovarian tumors. A previous study indicated that the expression of immune response genes was elevated and survival was significantly lower in patients with high-grade serous and endometrioid tumors than in those with low-grade/low-malignant-potential tumors [29]. While the amplification of groups of defense response genes in the TCGA database, as well as immune system and immune cell activation genes, was associated with worse OS in our study, the differences were not significant. Immune responses have long been considered important events in tumor progression. Thus, it is necessary to further explore the behavior of ovarian cancer in the context of immune responses.
The amplification of the DNA damage repair genes DDB1, ERCC1 and ERCC2 was associated with worse outcomes in our study. DDB1, encoded by a gene located at chromosome 11q12.2, is an important positive regulator of nucleotide excision repair [42] and is responsible for resistance to platinum-based agents [43]. ERCC1/2, which are encoded by genes located at chromosome 19q13.32, are the two major components of the nucleotide excision repair process, particularly for DNA damage caused by chemotherapeutic agents [44, 45]. Studies have suggested the use of ERCC1/2 as molecular predictors of clinical resistance to platinum-based chemotherapy [46]. These conclusions may help to explain our previous data showing that A-H/S-H cells exhibited greater resistance to cisplatin and Taxol than A-L/S-L cells. In addition, our data revealed that ERCC1/2 amplification was associated with significantly worse OS and PFS. These results indicate that DDB1, ERCC1 and ERCC2 could be used as predictors of chemoresistance, and that ERCC1/2 could predict poor clinical outcomes.
Besides immune signature and DNA damage repair genes, genes encoding proteins involved in cell proliferation and G1/S transition were notable for their association with worse OS. Among the genes herein, PRPF19, BCAT1, CDKN1B, and MARK4 could be considered as potential candidates for therapeutic intervention. PRPF19 (pre-mRNA-processing factor 19) is a U-box-containing E3 ubiquitin ligase and is involved in the DNA damage response [47]. PRPF19 expression was found to be greater in gastric cancer tissues and/or metastatic lymph nodes than in peri-cancerous tissues [48]. Another report indicated that PRPF19 expression was elevated in most hepatocellular carcinoma tissues and cell lines, and its overexpression correlated positively with vascular invasion and tumor capsule breakthrough, probably through the p38 mitogen-activated protein kinase/twist1 pathway [49]. To date, PRPF19 has not been studied in ovarian cancer; however, these previous studies imply that the gain of PRPF19 is a critical event during the progression of cancer, making it a promising target for malignancies with aberrant PRPF19 expression.
BCAT1 is highly overexpressed in serous ovarian cancers [50]. Knockdown of BCAT1 dramatically reduced the rates of cell proliferation, migration and invasion, and silencing of BCAT1 was reported to suppress ovarian tumorigenesis and induce the expression of several tumor suppressors. Additionally, survival was prolonged when BCAT1 was suppressed in a xenograft model of advanced epithelial ovarian cancer [51]. Thus, this transaminase could be considered a novel malignancy biomarker and a putative therapeutic target.
p27 protein is encoded by the cyclin-dependent kinase inhibitor 1B (CDKN1B) gene, mapped to chromosome 12p13. Polymorphisms in CDKN1B may be associated with reduced susceptibility to cancer, particularly ovarian cancer [52]. In a global online biomarker validation platform developed to mine all available microarray data in 1287 ovarian cancer patients, CDKN1B expression was found to be associated with survival [53]. We also detected amplification and overexpression of CDKN1B in both A-H and S-H cells; thus, it will be necessary to examine the pathological processes of CDKN1B CNV in advanced-stage ovarian cancer.
The MARK4 gene encodes a member of the microtubule affinity-regulating kinase family, and is inseparably linked with many human diseases, including cancer, diet-induced obesity, type 2 diabetes, and neurodegenerative disorders. MARK4 was found to bind with a number of proteins linked to cell motility, clearly suggesting its involvement in the promotion of the cytoskeleton. It is also involved in the stimulation of cell migration and metastasis in breast and lung cancers [54–56]. To date, MARK4 has been found to contribute to the development of hepatocellular carcinomas [57], gliomas [58], prostate cancer [59] and breast cancer [60]. However, no research on MARK4 has been performed for ovarian cancer, so further study is required regarding its potential involvement in the metastasis of ovarian carcinomas.
In summary, we have systematically analyzed the functions of the altered genes in ovarian cancer cell line models with heterogeneous invasive/migratory capacities. Integrating the data on copy number diversity with clinical outcomes and mRNA/protein expression facilitated the search for potential therapeutic targets. Therefore, we predict that clone-specific functional and genetic profiling will be a helpful method of identifying new molecular pathways underlying cancer and new biomarkers for clinical applications. This will not only reveal the heterogeneity in ovarian cancers of different developmental stages, but also elucidate other factors that account for cancer deaths, such as genes that promote chemoresistance.
MATERIALS AND METHODS
Cell culture
The human ovarian cancer cell lines A2780 and SKOV3 were purchased from the Cell Support Center, Institute of Basic Medical Science, Chinese Academy of Medical Sciences. A2780 was established from ovarian tumor tissue obtained from an untreated patient in the UK [61, 62]. SKOV3 cells, which were isolated from the ascites of a patient with ovarian adenocarcinoma, are resistant to several cytotoxic drugs, including diphtheria toxin, cis-platinum and Adriamycin [63]. A2780 and SKOV3 are the most popular cell line models for ovarian cancer studies in the literature. Their highly and minimally invasive/migratory subclones were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Hyclone, Logan, Utah, USA) supplemented with 10% heat-inactivated FBS (Gibco, Carlsbad, CA, USA), 100 μg/mL streptomycin and 100 IU/mL penicillin at 37°C in a humid incubator containing 5% CO2. The cells were subcultured when they reached approximately 80% confluence.
CNV assay
Genomic DNA was extracted from the cultured cells with a Gentra Puregene kit (Qiagen, Hilden, Germany). The DNA samples were genotyped with a CytoScan™ HD array (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s instructions. This array contains 2,696,550 CNV markers, including 743,304 genotypable SNP probes and 1,953,246 non-polymorphic probes. The data were visualized and then analyzed in the Affymetrix Chromosome Analysis Suite (ChAS) software package [64].
Validation of gene CNVs
We selected a subset of regions identified as varying in copy number between A-H and A-L, S-H and S-L. Genomic DNA was extracted from cultured cells with a Gentra Puregene kit (Qiagen) according to the manufacturer’s instructions. The primers listed in Supplementary Table 1 were used to determine the relative gene copy numbers between A-H and A-L, S-H and S-L. PCR was conducted on a CFX 96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) with a SYBR Green PCR Kit (Takara Bio Inc., Otsu, Japan). The following PCR protocol was used: 95°C for 10 min, followed by 40 cycles of amplification at 95°C for 15 s and 60°C for 1 min. Dissociation curve analyses were conducted to confirm the specificity of the PCR products. The gene copy numbers were normalized against the levels of RNase P-2 and analyzed by the comparative Ct method (ΔΔCt). The mean values of the fold changes obtained in three independent experiments were calculated.
Gene copy numbers in ovarian cancer samples and clinical outcomes
We obtained copy number data for 603 high-grade serous ovarian cancer samples from The Cancer Genome Atlas (TCGA) [65]. The data in the TCGA ovarian serous cystadenocarcinoma database were analyzed with the cBioPortal online analytical tool (http://www.cbioportal.org/public-portal/) in the “TCGA, provisional” category. The clinical copy number profiles were summarized through “OncoPrint.” The “Plot” function was used to generate copy number status/mRNA expression (RNA Seq V2 RSEM) (log2) correlation plots. The gene expression values were compared with matching copy number values by means of Spearman and Pearson correlations. The “Survival” function was used to plot Kaplan-Meier curves [66, 67]. Multivariate analysis for independent prognostic factors was performed using COX regression model.
RNA isolation and real-time PCR assay for mRNA expression
Total RNA was extracted with the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was then synthesized with a QuantScript Reverse Transcription Kit (Tiangen, China). The primers are listed in Supplementary Table 2. Real-time PCR was performed on a CFX 96 Real-Time PCR Detection System with a SYBR Green PCR Kit. The analysis of each sample was repeated at least three times.
Western blot
Cells were prepared in cold radioimmunoprecipitation assay lysis buffer (Applygen technology, China) containing freshly added 0.01% protease inhibitor (Sigma, Louis, MO, USA) for 30 min. The solutions were centrifuged at 12000 × g for 10 min at 4°C, and the supernatants were collected. Approximately 50 μg of total protein was separated on a 10–15% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. After being blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 for 2 h at room temperature, the membranes were incubated with primary antibodies overnight at 4°C, followed by horseradish peroxidase-conjugated secondary antibodies (Santa Cruz, CA, USA) for 2 h at room temperature. The signals were visualized with a SuperEnhanced Chemiluminescence Detection Kit (Applygen technology, China). Details about the primary antibodies are presented in Supplementary Table 3. Each assay was performed at least three times.
Statistical analysis
Statistical analysis of the data was performed with SPSS 17.0 software (SPSS, Chicago, Illinois, USA). The data were expressed as the mean values ± standard errors of the mean. The significance of differences in values was evaluated through analysis of variance or an unpaired two-tailed Student’s t-test. A P value < 0.05 was considered to indicate a significant difference. All experiments were repeated at least three times.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This study was supported by funds from the National Natural Science Foundation of China (program nos. 81502254, 81372780 and 81572576).
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer Journal international du cancer. 2015; 136:E359–386.
2. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012; 366:883–892.
3. McAlpine JN, Eisenkop SM, Spirtos NM. Tumor heterogeneity in ovarian cancer as demonstrated by in vitro chemoresistance assays. Gynecologic oncology. 2008; 110:360–364.
4. Heppner GH. Tumor heterogeneity. Cancer research. 1984; 44:2259–2265.
5. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012; 481:306–313.
6. Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010; 464:704–712.
7. Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nature reviews Genetics. 2007; 8:639–646.
8. Bollig-Fischer A, Michelhaugh SK, Wijesinghe P, Dyson G, Kruger A, Palanisamy N, Choi L, Alosh B, Ali-Fehmi R, Mittal S. Cytogenomic profiling of breast cancer brain metastases reveals potential for repurposing targeted therapeutics. Oncotarget. 2015; 6:14614–14624. doi: 10.18632/oncotarget.3786.
9. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615.
10. Sonoda G, Palazzo J, du Manoir S, Godwin AK, Feder M, Yakushiji M, Testa JR. Comparative genomic hybridization detects frequent overrepresentation of chromosomal material from 3q26, 8q24, and 20q13 in human ovarian carcinomas. Genes, chromosomes & cancer. 1997; 20:320–328.
11. Gray JW, Suzuki S, Kuo WL, Polikoff D, Deavers M, Smith-McCune K, Berchuck A, Pinkel D, Albertson D, Mills GB. Specific keynote: genome copy number abnormalities in ovarian cancer. Gynecologic oncology. 2003; 88:S16–21; discussion S22–14.
12. Etemadmoghadam D, deFazio A, Beroukhim R, Mermel C, George J, Getz G, Tothill R, Okamoto A, Raeder MB, Harnett P, Lade S, Akslen LA, Tinker AV, et al. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clinical cancer research. 2009; 15:1417–1427.
13. Kim SW, Kim JW, Kim YT, Kim JH, Kim S, Yoon BS, Nam EJ, Kim HY. Analysis of chromosomal changes in serous ovarian carcinoma using high-resolution array comparative genomic hybridization: Potential predictive markers of chemoresistant disease. Genes, chromosomes & cancer. 2007; 46:1–9.
14. Gorringe KL, Jacobs S, Thompson ER, Sridhar A, Qiu W, Choong DY, Campbell IG. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clinical cancer research. 2007; 13:4731–4739.
15. Malek JA, Mery E, Mahmoud YA, Al-Azwani EK, Roger L, Huang R, Jouve E, Lis R, Thiery JP, Querleu D, Rafii A. Copy number variation analysis of matched ovarian primary tumors and peritoneal metastasis. PloS one. 2011; 6:e28561.
16. Gorringe KL, George J, Anglesio MS, Ramakrishna M, Etemadmoghadam D, Cowin P, Sridhar A, Williams LH, Boyle SE, Yanaihara N, Okamoto A, Urashima M, Smyth GK, et al. Copy number analysis identifies novel interactions between genomic loci in ovarian cancer. PloS one. 2010; 5.
17. Wrzeszczynski KO, Varadan V, Byrnes J, Lum E, Kamalakaran S, Levine DA, Dimitrova N, Zhang MQ, Lucito R. Identification of tumor suppressors and oncogenes from genomic and epigenetic features in ovarian cancer. PloS one. 2011; 6:e28503.
18. Lee JY, Yoon JK, Kim B, Kim S, Kim MA, Lim H, Bang D, Song YS. Tumor evolution and intratumor heterogeneity of an epithelial ovarian cancer investigated using next-generation sequencing. BMC cancer. 2015; 15:85.
19. Wang ZC, Birkbak NJ, Culhane AC, Drapkin R, Fatima A, Tian R, Schwede M, Alsop K, Daniels KE, Piao H, Liu J, Etemadmoghadam D, Miron A, et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clinical cancer research. 2012; 18:5806–5815.
20. Engler DA, Gupta S, Growdon WB, Drapkin RI, Nitta M, Sergent PA, Allred SF, Gross J, Deavers MT, Kuo WL, Karlan BY, Rueda BR, Orsulic S, et al. Genome wide DNA copy number analysis of serous type ovarian carcinomas identifies genetic markers predictive of clinical outcome. PloS one. 2012; 7:e30996.
21. Huang RY, Chen GB, Matsumura N, Lai HC, Mori S, Li J, Wong MK, Konishi I, Thiery JP, Goh L. Histotype-specific copy-number alterations in ovarian cancer. BMC medical genomics. 2012; 5:47.
22. Bai H, Li H, Li W, Gui T, Yang J, Cao D, Shen K. The PI3K/AKT/mTOR pathway is a potential predictor of distinct invasive and migratory capacities in human ovarian cancer cell lines. Oncotarget. 2015; 6:25520–32. doi: 10.18632/oncotarget.4550.
23. Ramakrishna M, Williams LH, Boyle SE, Bearfoot JL, Sridhar A, Speed TP, Gorringe KL, Campbell IG. Identification of candidate growth promoting genes in ovarian cancer through integrated copy number and expression analysis. PloS one. 2010; 5:e9983.
24. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009; 37:1–13.
25. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009; 4:44–57.
26. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009; 119:1420–1428.
27. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. Journal of cellular physiology. 2007; 213:374–383.
28. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature reviews Cancer. 2002; 2:442–454.
29. Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, Traficante N, Fereday S, Hung JA, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clinical cancer research. 2008; 14:5198–5208.
30. Boesch M, Zeimet AG, Reimer D, Schmidt S, Gastl G, Parson W, Spoeck F, Hatina J, Wolf D, Sopper S. The side population of ovarian cancer cells defines a heterogeneous compartment exhibiting stem cell characteristics. Oncotarget. 2014; 5:7027–7039. doi: 10.18632/oncotarget.2053.
31. Davis SJ, Sheppard KE, Pearson RB, Campbell IG, Gorringe KL, Simpson KJ. Functional analysis of genes in regions commonly amplified in high-grade serous and endometrioid ovarian cancer. Clinical cancer research. 2013; 19:1411–1421.
32. Khalique L, Ayhan A, Weale ME, Jacobs IJ, Ramus SJ, Gayther SA. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. The Journal of pathology. 2007; 211:286–295.
33. Gui T, Bai H, Zeng J, Zhong Z, Cao D, Cui Q, Chen J, Yang J, Shen K. Tumor heterogeneity in the recurrence of epithelial ovarian cancer demonstrated by polycomb group proteins. OncoTargets and therapy. 2014; 7:1705–1716.
34. Zeng J, Ruan J, Luo L, Shi J, Cui Q, Yang J, Shen K. Molecular portraits of heterogeneity related to cancer stem cells in human ovarian cancer. International journal of gynecological cancer. 2014; 24:29–35.
35. Gui T, Cao D, Yang J, Shen K. Tumor heterogeneity has important consequences for personalized medicine in ovarian cancer. Histology and histopathology. 2015; 30:173–181.
36. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature communications. 2013; 4:2126.
37. Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, Murtaza M, van IWF, Heine AA, Smid M, Koudijs MJ, Brenton JD, Berns EM, et al. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PloS one. 2014; 9:e103988.
38. Nathans J, Hogness DS. Isolation, sequence analysis, and intron-exon arrangement of the gene encoding bovine rhodopsin. Cell. 1983; 34:807–814.
39. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001; 357:539–545.
40. Li HS, Watowich SS. Innate immune regulation by STAT-mediated transcriptional mechanisms. Immunological reviews. 2014; 261:84–101.
41. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Current opinion in immunology. 2010; 22:231–237.
42. Li J, Wang QE, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, Wani AA. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer research. 2006; 66:8590–8597.
43. Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell death & disease. 2014; 5:e1257.
44. Boccard SG, Marand SV, Geraci S, Pycroft L, Berger FR, Pelletier LA. Inhibition of DNA-repair genes Ercc1 and Mgmt enhances temozolomide efficacy in gliomas treatment: a pre-clinical study. Oncotarget. 2015; 6:29456–29468. doi: 10.18632/oncotarget.4909.
45. Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, Stahel R, Sabatier L, Pignon JP, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. The New England journal of medicine. 2006; 355:983–991.
46. Yin M, Yan J, Martinez-Balibrea E, Graziano F, Lenz HJ, Kim HJ, Robert J, Im SA, Wang WS, Etienne-Grimaldi MC, Wei Q. ERCC1 and ERCC2 polymorphisms predict clinical outcomes of oxaliplatin-based chemotherapies in gastric and colorectal cancer: a systemic review and meta-analysis. Clinical cancer research. 2011; 17:1632–1640.
47. Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, Zou L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Molecular cell. 2014; 53:235–246.
48. Zhou J, Wang W, Xie Y, Zhao Y, Chen X, Xu W, Wang Y, Guan Z. Proteomics-Based Identification and Analysis of Proteins Associated with Helicobacter pylori in Gastric Cancer. PloS one. 2016; 11:e0146521.
49. Yin J, Wang L, Zhu JM, Yu Q, Xue RY, Fang Y, Zhang YA, Chen YJ, Liu TT, Dong L, Shen XZ. Prp19 facilitates invasion of hepatocellular carcinoma via p38 mitogen-activated protein kinase/twist1 pathway. Oncotarget. 2016; 7:21939–21951. doi: 10.18632/oncotarget.7877.
50. Ju W, Yoo BC, Kim IJ, Kim JW, Kim SC, Lee HP. Identification of genes with differential expression in chemoresistant epithelial ovarian cancer using high-density oligonucleotide microarrays. Oncology research. 2009; 18:47–56.
51. Wang ZQ, Faddaoui A, Bachvarova M, Plante M, Gregoire J, Renaud MC, Sebastianelli A, Guillemette C, Gobeil S, Macdonald E, Vanderhyden B, Bachvarov D. BCAT1 expression associates with ovarian cancer progression: possible implications in altered disease metabolism. Oncotarget. 2015; 6:31522–31543. doi: 10.18632/oncotarget.5159.
52. Lu Y, Gao K, Zhang M, Zhou A, Zhou X, Guan Z, Shi X, Ge S. Genetic Association Between CDKN1B rs2066827 Polymorphism and Susceptibility to Cancer. Medicine. 2015; 94:e1217.
53. Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocrine-related cancer. 2012; 19:197–208.
54. Naz F, Anjum F, Islam A, Ahmad F, Hassan MI. Microtubule affinity-regulating kinase 4: structure, function, and regulation. Cell biochemistry and biophysics. 2013; 67:485–499.
55. Trinczek B, Brajenovic M, Ebneth A, Drewes G. MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. The Journal of biological chemistry. 2004; 279:5915–5923.
56. Pardo OE, Castellano L, Munro CE, Hu Y, Mauri F, Krell J, Lara R, Pinho FG, Choudhury T, Frampton AE, Pellegrino L, Pshezhetskiy D, Wang Y, et al. miR-515–5p controls cancer cell migration through MARK4 regulation. EMBO reports. 2016; 17:570–584.
57. Kato T, Satoh S, Okabe H, Kitahara O, Ono K, Kihara C, Tanaka T, Tsunoda T, Yamaoka Y, Nakamura Y, Furukawa Y. Isolation of a novel human gene, MARKL1, homologous to MARK3 and its involvement in hepatocellular carcinogenesis. Neoplasia. 2001; 3:4–9.
58. Beghini A, Magnani I, Roversi G, Piepoli T, Di Terlizzi S, Moroni RF, Pollo B, Fuhrman Conti AM, Cowell JK, Finocchiaro G, Larizza L. The neural progenitor-restricted isoform of the MARK4 gene in 19q13.2 is upregulated in human gliomas and overexpressed in a subset of glioblastoma cell lines. Oncogene. 2003; 22:2581–2591.
59. Jenardhanan P, Mannu J, Mathur PP. The structural analysis of MARK4 and the exploration of specific inhibitors for the MARK family: a computational approach to obstruct the role of MARK4 in prostate cancer progression. Molecular bioSystems. 2014; 10:1845–1868.
60. Gabrovska PN, Smith RA, Haupt LM, Griffiths LR. Investigation of two Wnt signalling pathway single nucleotide polymorphisms in a breast cancer-affected Australian population. Twin research and human genetics. 2011; 14:562–567.
61. Hamilton TC, Young RC, Ozols RF. Experimental model systems of ovarian cancer: applications to the design and evaluation of new treatment approaches. Seminars in oncology. 1984; 11:285–298.
62. Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer research. 1987; 47:414–418.
63. Morimoto H, Yonehara S, Bonavida B. Overcoming tumor necrosis factor and drug resistance of human tumor cell lines by combination treatment with anti-Fas antibody and drugs or toxins. Cancer research. 1993; 53:2591–2596.
64. Uddin M, Thiruvahindrapuram B, Walker S, Wang Z, Hu P, Lamoureux S, Wei J, MacDonald JR, Pellecchia G, Lu C, Lionel AC, Gazzellone MJ, McLaughlin JR, et al. A high-resolution copy-number variation resource for clinical and population genetics. Genetics in medicine. 2015; 17:747–752.
65. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615.
66. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:pl1.
67. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–404.