
Introduction
Triple negative breast cancer (TNBC) is a clinicopathological entity which includes breast tumors that do not express immunohistochemically detectable estrogen, progesterone and HER2 receptors [1]. They account for around 15% of all breast cancers, being more frequently expressed in young women and those from African and Hispanic descendants [1-3]. Genomic studies have revealed the heterogeneous nature of TNBC [4-6]. Indeed, this entity has been classified by gene expression analyses in several subgroups, including two basal-like (BL1, BL2), an immunomodulatory (IM), a mesenchymal (M), a mesenchymal stem-like (MSL) and a luminal/androgen receptor (LAR) subtype [5]. A similar study but using a smaller dataset described four groups that mimic the previous reported subtypes: luminal/androgen receptor (LAR), mesenchymal (MES), basal like/immune-suppressed (BLIS) and basal like/immune activated (BLIA) [7].
Although heterogeneous, most TNBC share common clinical features such as poor long term prognosis or a specific pattern of relapse, mainly during the first five years after diagnosis [8]. In addition, some data suggests that the IM and BLIA subgroups are associated with an enrichment of lymphocytes and are those with better outcome [5, 7]. These findings are in line with recent studies suggesting that TNBC with tumor-infiltrating lymphocytes (TILs) have better prognosis [9, 10].
Due to the lack of druggable known targets most patients with TNBC are treated with chemotherapy [11]. Chemotherapy regimens have clinical activity in some TNBC patients. The group of patients that obtain a pathological complete response (pCR) after neoadjuvant chemotherapy, have a better clinical outcome than those that do not achieve such response [12]. However, in the metastatic setting chemotherapy shows limited efficacy and most patients soon progress to these agents.
The poor clinical prognosis, the limited long term efficacy of chemotherapy and the absence of targeted therapies support research to identify new targets and develop novel therapies against this cancer. In this review we will discuss novel biological findings that are therapeutically exploitable as well as combinations of agents to efficiently augment and optimize existing therapies.
Novel therapeutic possibilities in TNBC
Molecular and functional studies have unraveled cellular functions that are important in the generation/progression of TNBC. On the bases of these studies, agents targeting distinct components of those functions have been developed. Figure 1 provides a schematic view of key components of such cellular functions for whom therapeutic agents have been developed. Some of them, already approved or under clinical evaluation, are shown.
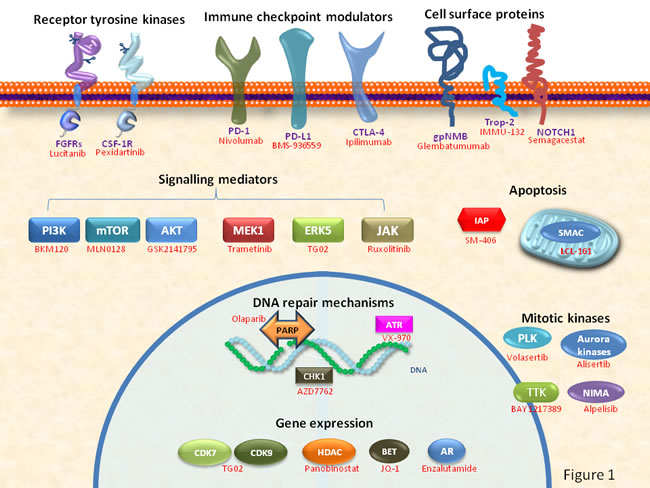
Figure 1: Schematic representation of cellular functions and key components used as potential drug targets for the therapy of TNBC. Examples of drugs already approved or under clinical development are shown in red.
DNA repair mechanisms
In TNBC alterations of the DNA repair machinery seem to be of significance, as there is an increased presence of somatic or acquired mutations in DNA repair genes, mainly BRCA1 or BRCA2 genes. Those genes code for key tumor-suppressor proteins that are important components of the homologous recombination DNA repair mechanism [13, 14] (Figure 2). In this context, chemotherapies that affect DNA like platinum compounds, and agents that act by inhibiting the PolyADP-Ribose Polymerase (PARP) protein have been evaluated in this subtype of breast cancer. Platinum chemotherapy causes DNA damage through induction of adducts or DNA crosslinking. These lesions are normally repaired through base excision repair mechanisms in which PARP1 plays an essential role. Platinum compounds have moderate activity in unselected TNBC tumors but are more active than taxanes particularly in tumors harboring BRCA mutations [15, 16].
Given the fact that in BRCA1/2 mutated tumors PARP inhibitors produce a synthetic lethality interaction [17, 18] (see Figure 2), these compounds are currently in clinical development in this indication. Recently, olaparib, an agent that targets several PARP isoforms including PARP1, PARP2 and PARP3, has been approved for the treatment of ovarian cancer in patients harboring deleterious mutations of the BRCA1/2 genes [19, 20]. In TNBC several phase II trials have evaluated various PARP inhibitors. Olaparib showed clinical activity when patients were selected for BRCA1 mutations, with response rates from 13-50% depending on the number of prior chemotherapies or the previous exposure to platinum-compounds [20, 21]. Of note, in unselected patients efficacy was very limited [22]. Velaparib is another inhibitor that targets PARP1 and PARP2. In a phase II trial clear signs of activity were observed with velaparib in combination with temozolomide in BRCA1 mutated patients [23]. Ongoing phase III studies with PARP inhibitors are shown in Table 1.
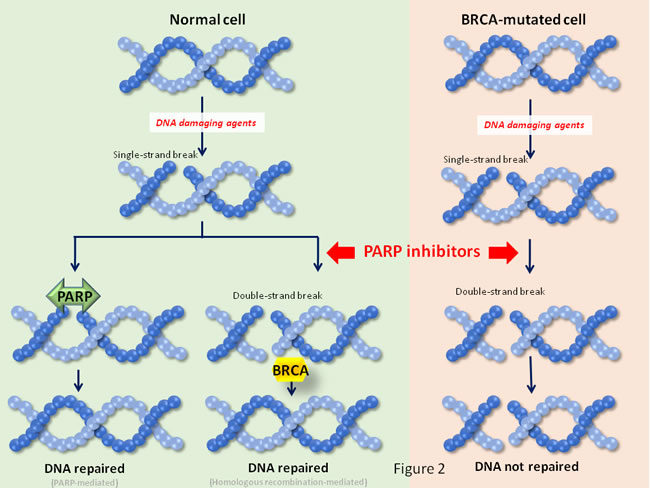
Figure 2: Synthetic lethality interactions for PARP inhibitors. In synthetic lethality two different pathways participate to sustain a cellular function. In the case shown in the figure, single strand DNA repair is accomplished by the action of PARP. In case the lesion progresses to a double strand break, it can be repaired by the action of BRCA1. In case one of the pathways is impaired (e. gr. by loss of function of BRCA1), the cellular function is still supported by the action of the other pathway. An alteration in a base, or a single strand break is usually repaired by the base excision repair mechanism, in which PARP proteins play an essential role. If inhibitors of PARP are used, then the single strand alteration may evolve to double strand breaks. The repair of lesions depends on the integrity of BRCA proteins. In patients whose tumors are defective in BRCA activity, the PARP inhibitors create DNA double strand breaks that are not repaired, leading to cell death.
Other druggable proteins that are involved in DNA repair include ATR or its effector kinase Chk1 [24]. Their inhibition has shown promising preclinical activity in TNBC that lack the expression of ERCC1 [25]. In this context, agents targeting these proteins are currently in clinical development (see Tables 2 and 3). Finally, agents that bind DNA like novel mithramycin analogs (mithralogs) or trabectedin have also shown activity in preclinical models, but their potential antitumoral activity in the clinical setting requires evaluation [26, 27].
Table 1: Summary of ongoing phase III studies in TNBC describing the new compound, indication and the study design.
Study number |
Indication |
New compound |
Control Arm |
Experimental Arm |
NCT02032277 |
Early Stage Triple Negative Breast Cancer |
ABT-888 (PARP inhibitor) |
Active Comparator: Arm A Veliparib + carboplatin + paclitaxel followed by doxorubicin/cyclophosphamide (AC) |
Placebo Comparator: Arm B Placebo + carboplatin + paclitaxel followed by AC. Arm C Placebo + placebo + paclitaxel followed by AC. |
NCT02555657 |
Metastatic Triple Negative Breast Cancer |
Pembrolizumab (MK-3475) (Anti-PD-1 Monoclonal Antibody) |
Experimental: Pembrolizumab Participants receive pembrolizumab 200 mg intravenously (IV) every 3 weeks (Q3W) for up to 35 administrations |
Active Comparator: Chemotherapy Participants receive capecitabine, eribulin, gemcitabine, or vinorelbine as therapy of physicians choice in accordance with local regulations and guidelines |
NCT02574455 |
Refractory/Relapsed Triple-Negative Breast Cancer |
Sacituzumab Govitecan IMMU-132 (ADC against Trop-2 linked to irinotecan metabolite SN-38) |
Experimental: IMMU-132 Sacituzumab govitecan (10 mg/kg on Days 1 and 8 of 21-day cycles) |
Active Comparator Treatment of Physician's: Eribulin (1.4 mg/m2 intravenously on Days 1 and 8 of a 21-day cycle). Capecitabine (1250 mg/m2 orally twice day for 2 weeks followed by a 1 week rest period given as a 21-day cycle). Gemcitabine (1250 mg/m2 intravenously on Days 1 and 8 of a 21-day cycle). Vinorelbine (30 mg/m2 weekly; IV injection over 6-10 min) |
NCT02425891 |
Previously Untreated Metastatic Triple Negative Breast Cancer |
Atezolizumab (MPDL3280A) (Anti-PD-1 Monoclonal Antibody) |
Active Comparator: Atezolizumab plus nab-paclitaxel |
Placebo Comparator: Placebo and nab-paclitaxel |
NCT00938652 |
ER-, PR-, and Her2-Negative Metastatic Breast Cancer |
BSI-201. Iniparib (PARP inhibitor) |
Active Comparator: Arm G/C gemcitabine/carboplatin on Days 1 and 8 of 21-day cycle(s) |
Experimental: Arm G/C/I gemcitabine/carboplatin on Days 1 and 8, plus iniparib on Days 1, 4, 8, and 11 of 21-day cycle(s) |
Abbreviations: ER, estrogen receptor; PR, progesterone receptor.
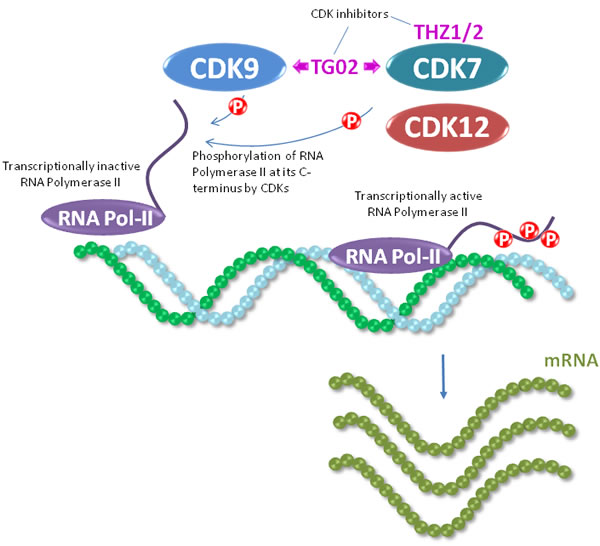
Figure 3: CDK7 and CDK9 act as regulators of RNA polymerase II, an enzyme critical in mRNA production which is required for protein synthesis. RNA polymerase II is recruited to gene promoters by transcription factors, and is phosphorylated in its C-terminal domain heptad repeats. At these heptad repeats, CDK7 phosphorylates RNA polymerase II on serine 5 during initiation of transcription, and CDK9 phosphorylates serine 2 during elongation of the transcripts. The activity of CDK7 and CDK9 can be targeted by the dual inhibitor TG02.
Targeting the mitotic process
TNBC is characterized by its high proliferation rate [1, 11, 28]. Recent studies have demonstrated that kinases involved in the formation of the mitotic spindle, such as TTK/Mps1, polo-like kinases (PLKs) or NIMA/NEK2, are overexpressed in this particular breast cancer subtype [29]. Agents against TTK or PLKs have shown antitumor activity in preclinical models and are currently under clinical development [30-33]. In the case of NIMA, knockdown of this gene in triple negative tumors induced cell cycle arrest followed by cell death [34]. Similarly, targeting aurora kinases A and B has shown an antitumoral effect in TNBC and these agents were synergistic with MEK inhibitors and agents targeting microtubules [35, 36].
Gene expression modulators
In a heterogeneous disease like TNBC targeting epigenomic components that can indirectly control several oncogenic functions represents an attractive approach. Bromodomain and extra terminal domain (BET) inhibitors and histone deacetylase inhibitors (HDACi) are two types of agents with an epigenetic mechanism of action that have preferentially been explored in TNBC. BET inhibitors belong to a new family of compounds that by inhibiting bromodomains can modify gene transcription [37]. These compounds have shown activity in several tumor types like neuroblastoma, where the expression of the transcription factor c-Myc was reduced; or in breast cancer by affecting key signaling elements involved in resistance to targeted agents [38, 39]. One of these compounds OTX015 is currently under early clinical development [40]. In preclinical models BET inhibitors have shown clinical activity in TNBC synergizing with chemotherapies used for the treatment of this disease [41, 42]. On the other side, HDACi have shown activity on TNBC tumor initiating cells [43, 44] and some of these agents are currently in phase II studies, mainly in combination with platinum-based therapies (see Table 2).
Through inhibition of the kinase activity of the cyclin-dependent kinases CDK7, CDK9 and CDK12 several agents have been reported to indirectly control transcription. These CDKs modulate the activity of the RNA polymerase II large subunit through phosphorylation at its C-terminal domain [45]. Such phosphorylation is required for initiation and elongation of transcription. A recent study has shown that CDK7 inhibition triggers cell death in preclinical models of TNBC, opening the door for the potential evaluation of CDK7 inhibitors in the clinical setting [46]. Moreover, the dual CDK7 and CDK9 inhibitory drug TG02 has shown antitumor effect in TNBC [47]. Interestingly, CDK12 has been associated with genomic stability [48], one of the characteristics of BRCA1/2 mutated TNBCs. In TNBC attenuation of CDK12 function increases the effect of PARP inhibitors [49].
As indicated above, the molecular classification of TNBC includes the luminal androgen receptor subtype, characterized by the expression of that receptor. In fact, approximately 10-15% of TNBCs express the AR [50, 51]. In this context several studies are exploring the role of targeting AR in this setting [52]. Single agent phase II studies have evaluated the clinical activity of anti-androgens in estrogen receptor negative or triple negative breast tumors. In these studies the clinical benefit rate defined as complete response, partial response and stable disease was around 40% at 4 months [53, 54]. Although the clinical activity of anti-androgens is not impressive, these data have stimulated the design of further studies in TNBC.
Inhibiting tyrosine kinase receptors and intracellular signaling nodes
In addition to the targeting of molecules involved in nuclear processes such as those defined above, targeting cell surface or intracellular signaling molecules, mainly kinases, that may favor a prooncogenic phenotype has also been explored in TNBC. With respect to cell surface receptors, studies on the fibroblast growth factor receptor (FGFR) family have shown that 9% and 4% of TNBC have FGFR1 and FGFR2 amplification, respectively [55, 56]. Inhibition of these receptors has been shown to reduce tumor growth in preclinical models, supporting the evaluation of agents against FGFR in the clinical setting [57, 58].
In the case of signaling routes, particular emphasis has been placed in the PI3K/mTOR/AKT pathway. Molecular studies indicated that this route is activated in 10-15% of TNBC, and such activation may be caused by mutations, loss of suppressor phosphatases like PTEN or INPP4B; or in a small proportion of patients, amplification of AKT [59, 60]. Of note, PIK3CA mutations seem to be more frequent in the mesenchymal subtype [28]. Recent studies showed the relevant role of PI3K/mTOR inhibitors in TNBC and their synergistic interaction with chemotherapy [61]. Interestingly, inhibition of this route produced only a cytostatic effect, but when combined with chemotherapy the association induced cell death [61].
Another kinase with a relevant role in TNBC that has been recently described is ERK5. Expression of this protein was associated with worse outcome; and agents inhibiting its function produced an anti-proliferative effect [47, 62]. Combination of ERK5 kinase inhibitors with chemotherapy was synergistic [47].
Targeting multiple key intracellular signaling nodes can be a better strategy than inhibiting single kinases [63]. In line with that idea novel multikinase inhibitors have shown preclinical activity mainly when targeting the PI3K/mTOR and the JAK/STAT pathways [64].
Finally, it should be mentioned that strategies aimed at inhibiting EGFR did not meet expectations. Clinical studies showed no signs of potential activity when combined with chemotherapy [65]. A similar lack of clear efficacy was observed for antiangiogenic therapies, with studies showing no benefit [66]. Even considering the lack of data to support these strategies in the clinical setting, some studies are currently exploring novel compounds against these targets in clinical trials, as can be seen in Table 2.
Immunologic agents: check point inhibitors and antibody drug conjugates (ADCs)
Strategies based on modulating antitumoral immune responses have demonstrated clinical efficacy in different oncological diseases. Among the various strategies tested, those using antibodies aimed at augmenting the cellular immune response against tumors, particularly with the use of checkpoint immunologics, and agents directed to cell surface proteins have progressed to the clinic.
With respect to agents aimed at modulating the cellular immune response, blockade of cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death protein-1 (PD-1) or its ligand (PD-L1) with antibodies has shown clinical benefit in different indications [67]. In breast cancer, expression of PD-L1 has been described in 20% of TNBC [68]. Breast tumors with presence of CTLA-4 have been linked with a worse outcome [69]. However, expression of these antigens cannot fully predict response to antibodies targeting them. A recent clinical study with the anti-PD-1 antibody pembroluzimab has shown evidence of clinical activity with a good safety profile [70]. In this context, some phase III studies are currently analyzing the antitumoral action of anti-PD-1 antibodies either as monotherapy or in combination with chemotherapy (see Table 1).
In addition to those strategies that have shown potential clinical efficacy, the immunomodulatory properties of the colony stimulating factor 1 (CSF-1) could be manipulated for treating TNBC. Knocking out CSF-1 in mice susceptible to breast cancer resulted in reduced macrophage recruitment to the tumor and delayed metastasis [71]. This may be a consequence of the immunosuppressive action of tumor-associated macrophages. In the clinical setting the ratio among neutrophils and lymphocytes predicts worse outcome when it is higher than a certain level, also suggesting the deleterious role of neutrophils on cancer [72]. In preclinical models the role of neutrophils in relation to lung colonization of metastases in breast cancer has recently been reported [73]. CSF-1-receptor inhibition increased CD8+ T cell tumor infiltration and enhanced antitumor response to paclitaxel in xenograft models [74]. Agents with this mechanism of action are currently in phase II studies (Table 2).
The second group of immunologic agents is based on the recognition of cell surface proteins by antibodies, or modified versions of antibodies carrying a cytotoxic drug. Two novel ADCs are in clinical development. IMMU-132 is an ADC against Trop-2 conjugated to the irinotecan metabolite SN-38 [75]. Trop-2 is a calcium signal transducer transmembrane protein that is expressed in normal tissue and overexpressed in many tumors [76]. IMMU-132 has shown promising signs of activity in early clinical studies [77]. IMMU-132 has received fast track designation from the FDA for metastatic TNBC (Table 1). Glembatumumab vedotin is an ADC against the glycoprotein non-metastatic B (gpNMB) expressed on the membrane of cancer cells. gpNMB was associated with worse outcome in breast cancer patients, particularly those with a triple negative phenotype [78]. In this case the antibody is linked to monomethyl auristatin E, a potent antimitotic agent [78]. Glembatumumab vedotin has demonstrated promising signs of activity in TNBCs overexpressing gpNMB and is now in clinical evaluation [79]. Other ADCs include those against the folate receptor (see Table 2).
Pathways involved in stemness
TNBC is a subtype of cancer where subpopulations of cells with stem-like characteristics are present. In this context, targeting signaling pathways involved in biological functions associated with the maintenance of the stem cell phenotype, has been explored preclinically. The JAK/STAT pathway is preferentially active in basal-like tumors [64, 80]. Inhibition of this pathway with kinase inhibitors produces tumor regression in preclinical models [64, 80]. Another pathway that participates in the maintenance of stem cell properties is the Notch signaling pathway [81]. Mutations at NOTCH1, NOTCH2 and NOTCH3 and rearrangement at NOTCH1 have been described, and inhibition of NOTCH cleavage with gamma-secretase inhibitors has shown activity in preclinical models [82, 83]. Some studies are currently evaluating this family of compounds in TNBC (see Tables 2 and 3).
Inducing cell death by targeting apoptosis
Direct induction of cell death by using agonists of cell death receptors or inhibitors of anti-apoptotic proteins can represent another antitumoral strategy in TNBC. Agents that act on inhibitors of apoptotic proteins (IAPs) have shown potential signs of activity in different preclinical models [84, 85]. LCL-161 is a second mitochondrial-derived activator of caspases (SMAC) mimetic that has shown clinical activity in TNBC in combination with paclitaxel in the neoadjuvant setting, when selecting patients with a specific gene signature [86]. Recently, two articles have described the role of PIM kinase inhibition as an effective antitumor strategy in basal-like tumors [87, 88].
Optimizing combinatorial strategies
Given the fact that TNBC is a heterogeneous disease, combined inhibition of relevant vulnerabilities seems to be a better approach than targeting individual events. This is in line with the clinical experience which demonstrated that targeting of individual routes often results in escape of inhibition of tumor growth by secondary activation of other protumorigenic routes. The development of novel drug combinations to improve management of patients with TNBC requires identification of tumors that most likely would respond to a given combination. In addition, and given the fact that TNBCs are sensitive to classical chemotherapies, the new drug is expected to be tested in combination with one of those chemotherapies. Below we will discuss drug combination strategies focusing on opportunities for clinical development and their limitations.
DNA-damaging agents
The finding that a number of TNBC present alterations in DNA repair mechanisms opened the possibility of exploiting this situation and fostered the development of PARP inhibitors. PARP inhibitors are synergistic with agents that induce double strand breaks. Among chemotherapies that produce double strand breaks, platinum compounds and temozolomide are the most used in the clinical setting, for glioblastoma and TNBC, respectively (Tables 2 and 3).
Table 2: List of ongoing phase II trials in TNBC as from clinicaltrials.gov, access Jun 1, 2016.
Study number |
New compound |
Study design |
Indication |
NCT01745367 |
Triple Negative Breast Cancer (TNBC) |
Tivozanib Hydrochloride (AV-951, an oral VEGF receptor tyrosine kinase inhibitor ) |
Active Comparator: Placebo in combination with paclitaxel Placebo orally once daily on a 3 weeks on/1 week off schedule with 90 mg/m2 of paclitaxel administered intravenously 3 weeks on (Day 1, Day 8 and Day 15)/1 week off (4 weeks = 1 Cycle). Experimental: Tivozanib Hydrochloride in combination with paclitaxel 1.5 mg tivozanib hydrochloride orally once daily on a 3 weeks on/1 week off schedule with 90 mg/m2 of paclitaxel administered intravenously 3 weeks on (Day 1, Day 8 and Day 15)/1 week off (4 weeks = 1 Cycle). |
NCT02393794 |
Triple Negative Breast Cancer |
Romidepsin (histone deacetylase inhibitor) |
Experimental: Romidepsin (8mg/m2) + Cisplatin (75mg/m2) Romidepsin 8mg/m2 IV on days 2 & 9 of each 21 day cycle Cisplatin 75mg/m2 IV on day 1 of each 21 day cycle Experimental: Romidepsin (10mg/m2) + Cisplatin (75mg/m2) Romidepsin 10mg/m2 IV on days 2 & 9 of each 21 day cycle Cisplatin 75mg/m2 IV on day 1 of each 21 day cycle Experimental: Romidepsin (12mg/m2) + Cisplatin (75mg/m2) Romidepsin 12mg/m2 IV on days 2 & 9 of each 21 day cycle Cisplatin 75mg/m2 IV on day 1 of each 21 day cycle Experimental: Romidepsin Dose Expansion Romidepsin maximum tolerated dose (MTD) from Phase I IV on days 2 & 9 of each 21 day cycle Cisplatin 75mg/m2 IV on day 1 of each 21 day cycle |
NCT02161679 |
Triple Negative Breast Cancer |
IMMU-132 Antibody drug conjugate |
Experimental: IMMU-132 Active Comparator: IMMU-132 plus Carboplatin |
NCT01997333 |
gpNMB Over-Expressing, Triple Negative Breast Cancer (METRIC) |
CDX-011 glembatumumab vedotin (antibody-drug conjugate) |
Active Comparator: Capecitabine Capecitabine will be administered on Days 1 through 14 of each 21 day cycle. Experimental: Drug: CDX-011 CDX-011 administered as an intravenous infusion on Day 1 of each 21 day cycle. |
NCT02593175 |
Localized Triple-Negative Breast Cancer (TNBC) |
Panitumumab (anti-EGFR mAb) |
Experimental: Panitumumab + Carboplatin + Paclitaxel One week before Cycle 1 participants receive a single dose of Panitumumab 1000 mg by vein. About 1 week after the first Panitumumab dose, participants have an image-guided core biopsy and/or a fine needle aspiration (FNA) to remove breast tissue. Participants then receive the study drug combination for 4 cycles. Each cycle is 21 days. On Day 1, 8, and 15 of each cycle, participants receive Panitumumab 2.5 mg by vein and Paclitaxel 80 mg/m2 by vein. On Day 1 of each cycle Carboplatin AUC 6 received by vein. |
NCT01307891 |
Metastatic, Triple Negative Breast Cancer |
Abraxane ( formulation of paclitaxel) |
Experimental: Combination Abraxane and Tigatuzumab Patients will receive Abraxane at 100 mg/m2 X 3 doses on Days 1, 8, and 15 at 28-day intervals in combination with tigatuzumab to be administered as a 10 mg/kg loading dose followed by 5 mg/kg for the first cycle and then every other week on Days 1 and 15 for subsequent cycles. Patients will be evaluated for response every 8 weeks. Patients with disease progression will be taken off the study. Experimental: Abraxane alone Patients will receive Abraxane at 100 mg/m2 weekly X 3 doses on Days 1, 8, and 15 at 28-day intervals. Treatment may continue without interruption in patients with complete response (CR), partial response (PR), or stable disease (SD) until there is progression of the disease or unacceptable toxicity. Patients will have the option to crossover to the combination arm based upon the pre-clinical data. |
NCT02402764 |
Metastatic Triple Negative Breast Cancer |
Selinexor (KPT-330) is a first in class SINE™ XPO1 antagonist) |
Experimental: Selinexor Treatment |
NCT02368691 |
Androgen Receptor-Positive Triple Negative Breast Cancer (AR+ TNBC) |
GTx-024 (selective androgen receptor modulator) |
Experimental: GTx-024 GTx-024 capsules, 18 mg PO once-daily for up to 12 months |
NCT01617668 |
Triple Negative Breast Cancer |
LCL161 ( is a small molecular antagonist of the inhibitor of apoptosis (IAP)) |
Experimental: Paclitaxel with LCL161 Active Comparator: Paclitaxel without LCL161 |
NCT01176669 |
Triple Negative Breast Cancer |
Apatinib (VEGFR2 TK inhibitor). |
Drug: Apatinib Apatinib was administratered at 750 mg/d in Phase IIa. The actual average dose intensity delivered was 525 mg/d due to toxicities. So, in Phase IIb, the starting dose of apatinib will be 500mg/d. Two dose reductions will be allowed to 375 and then 250 mg/d. |
NCT00813956 |
Triple Negative Breast Cancer |
BSI-201 (Parp inhibitor) |
Experimental: standard chemotherapy plus BSI-201 |
NCT02000882 |
TNBC BC Brain Met |
BKM120 ( PI3K inhibitor) |
Experimental: BKM120 plus Capecitabine BKM120 will be administered at a dose of 100 mg orally (PO) daily. Capecitabine will be administered at a dose of 1000 mg/m2 orally (PO) twice a day (rounded down to the nearest 500 mg pill) 14 days on and 7 days off. |
NCT00951054 |
Advanced, Metastatic Triple Negative Breast Cancer |
NK012 ( is an SN-38-releasing polymeric micelle) |
|
NCT02435680 |
Advanced Triple Negative Breast Cancer (TNBC) |
MCS110 ( a monoclonal antibody with potent neutralizing activity against macrophage colony-stimulating factor for the treatment of tumor-induced osteolysis) |
Experimental: Arm 1: MCS110+carboplatin+gemcitabine MCS110+carboplatin+gemcitabine Active Comparator: Arm 2: carboplatin+gemcitabine carboplatin+gemcitabine |
NCT01964924 |
Metastatic Triple-Negative Breast Cancer |
GSK2141795 (Akt Inhibitor) |
Experimental: Treatment (trametinib, Akt inhibitor GSK2141795) PART 1: Patients receive trametinib PO QD on days 1-28. Courses repeat every 28 days in the absence of disease progression or unacceptable toxicity. Patients who experience disease progression continue to Part 2. PART 2: Patients receive trametinib as in Part 1 and also receive Akt inhibitor GSK2141795 PO QD on days 1-28. Courses repeat every 28 days in the absence of disease progression or unacceptable toxicity. |
NCT01639248 |
Previously Treated Locally Advanced + Metastatic TNBC |
ENMD-2076 ( Aurora + Angiogenic Kinase Inhibitor) |
Experimental: ENMD-2076 Treatment ENMD-2076 |
NCT02447003 |
Metastatic Triple-Negative Breast Cancer |
Pembrolizumab (MK-3475) |
Experimental: Pembrolizumab Participants receive pembrolizumab, 200 mg intravenously (IV) on Day 1 of each 3-week cycle (Q3W) for up to 24 months |
NCT02203513 |
BRCA1/2 Mutation Associated Breast or Ovarian Cancer, Triple Negative Breast Cancer, High Grade Serous Ovarian Cancer, and Metastatic Castrate-Resistant Prostate Cancer |
LY2606368 (Chk1/2 Inhibitor) |
Experimental: 1 Women with gBRCAm associated breast or ovarian cancer Experimental: 2 HGSOC at low genetic risk Experimental: 3 TNBC at low genetic risk |
NCT02162719 |
Metastatic Triple-Negative Breast Cancer |
Ipatasertib (GDC-0068) (Akt inhibitor) |
Experimental: Arm 1 Paclitaxel + Ipatasertib Active Comparator: Arm 2 Paclitaxel + Placebo |
NCT01045304 |
Metastatic Triple Negative Breast Cancer |
SAR240550 (BSI-201) is a PARP1 inhibitor. |
Experimental: Gencitabine + iniparib twice weekly Gemcitabine, 1000 mg/m² IV over 30 minutes and carboplatin, area under the curve (AUC) = 2, IV over 60 minutes, both on Days 1 and 8 of 3-week cycles. Iniparib, 5.6 mg/kg IV over 60 minutes on Days 1, 4, 8 and 11 of 3-week cycles Experimental: Gencitabine + iniparib weekly Gemcitabine, 1000 mg/m² IV over 30 minutes and carboplatin, area under the curve (AUC) = 2, IV over 60 minutes, both on Days 1 and 8 of 3-week cycles. Iniparib, 11.2 mg/kg IV over 60 minutes on Days 1 and 8 of 3-week cycles |
NCT01953536 |
Triple Negative Breast Cancer |
Vintafolide (ADC against the folate receptor linked with vinblastine) |
Experimental: Vintafolide Participants receive intravenous (IV) vintafolide 2.5 mg on Days 1, 3, 5, 15, 17, and 19 of a 28-day cycle. Experimental: Vintafolide + Paclitaxel Participants receive IV vintafolide 2.5 mg on Days 1, 3, 5, 15, 17, and 19 of one 28-day cycle and receive IV paclitaxel on Days 1, 8, 15, and 22 of a 28-day cycle. Active Comparator: Paclitaxel Participants receive IV paclitaxel on Days 1, 8, 15, and 22 of a 28-day cycle. |
NCT01074970 |
Triple Negative Breast Cancer |
Rucaparin (PARP Inhibitor) |
Active Comparator: Arm A: Cisplatin Monotherapy Cisplatin 75 mg/m2 IV infusion over 60 minutes, D1 every 21 days for 4 cycles Active Comparator: Arm B: Combination Therapy Rucaparib 24mg C1,30mg C2-4, D1,2,3 every 21 days for 4 cycles Cisplatin 75 mg/m2 IV infusion over 60 minutes, D1 every 21 days for 4 cycles |
NCT02301988 |
Early Stage Triple Negative Breast Cancer |
Ipatasertib (GDC-0068) (AKT inhibitor) |
Experimental: Arm 1: Paclitaxel + Ipatasertib Placebo Comparator: Arm 2: Paclitaxel + Placebo |
NCT01204125 |
Triple Negative Breast Cancer |
Iniparib (SAR2405550 -BSI-201) (Parp inhibitor) |
Experimental: SAR240550 twice weekly/ paclitaxel weekly SAR240550 will be administered at the dose of 5.6mg/kg as a 60-min intravenous (IV) infusion. Patients will receive SAR240550 infusions twice weekly (day 1 and day 4; total dose of 11.2mg/kg per week) and paclitaxel weekly as a 60-min IV infusion (day 1; dose of 80mg/m2). Experimental: SAR240550 weekly/ paclitaxel weekly SAR240550 will be administered at the dose of 11.2 mg/kg as a 60-min intravenous (IV) infusion. Patients will receive SAR240550 infusions once weekly (day 1; total dose of 11.2mg/kg per week) and paclitaxel weekly as a 60-min IV infusion (day 1; dose of 80mg/m2). Active Comparator: Paclitaxel alone Paclitaxel will be administered at the dose of 80mg/m2 as a 60-min IV infusion. Patients will receive weekly (day 1) paclitaxel infusions. |
NCT02299635 |
Advanced Breast Cancer With Or Without Notch Alterations |
PF-03084014 A selective gamma secretase (GS) inhibitor |
Experimental: PF-03084014 PF-03084014 will be administered orally, continuously, twice daily at 150 mg, but the dose can be reduced to 100 mg or 80 mg. |
NCT01629615 |
Triple Negative Metastatic Breast Cancer |
BKM120 (PI3K Inhibitor) |
Drug: BKM120 BKM120 oral capsules. 100 mg daily in cycles of 28 days, until disease progression |
NCT01421472 |
HER2-negative Breast Cancer |
MM-121 (SAR256212) , is an investigational human monoclonal antibody |
Experimental: MM-121 (SAR256212) + paclitaxel Active Comparator: Paclitaxel |
NCT01698281 |
Refractory in Triple Negative Breast Cancer |
AEZS-108 zoptarelin doxorubicin (an LHRH agonist linked to doxorubicin) |
Experimental: AEZS-108 AEZS-108 (267 mg/m2, 2-hour IV infusion every Day 1 of a 21-day (3-week) cycle Active Comparator: Standard single agent cytotoxic chemotherapy commercially available SSCC (doses below the recommended package insert at the discretion of treating oncologist), on a 21-day cycle (although weekly administration is allowed; note: pegylated liposomal doxorubicin will be administered on a 28-day cycle). |
NCT02423603 |
Triple-Negative Advanced or Metastatic Breast Cancer |
AZD5363 (serine/threonine AKT/PKB, protein kinase B kinase inhibitor) |
Active Comparator: Paclitaxel + AZD5363 Patients receive Paclitaxel on Day 1, Day 8 and Day 15 plus AZD5363/Placebo on Days 2-5, Days 9-12, and Days 16-19. Upon Paclitaxel withdrawal, Patient receive AZD5363/Placebo on Days 2-5, Days 9-12, Days 16-19 and Days 23-27. Placebo Comparator: Paclitaxel + Placebo Patients receive Paclitaxel on Day 1, Day 8 and Day 15. plus AZD5363/Placebo on Days 2-5, Days 9-12, and Days 16-19. Upon Paclitaxel withdrawal, Patient receive AZD5363/Placebo on Days 2-5, Days 9-12, Days 16-19 and Days 23-27. |
NCT00540358 |
Triple Negative Metastatic Breast Cancer |
BSI-201 iniparib (Parp Inhibitor) |
Active Comparator: Arm G/C Standard chemotherapy with gemcitabine/carboplatin on Days 1 and 8 of 21-day cycle(s) Experimental: Arm G/C/I Standard chemotherapy with gemcitabine/carboplatin on Days 1 and 8, plus iniparib on Days 1, 4, 8, and 11 of 21-day cycle(s) |
NCT01818999 |
Metastatic Breast Cancer |
Ixabepilone |
Experimental: arm one IXABEPILONE and STEREOTACTIC BODY RADIATION THERAPY (SBRT) |
NCT01127763 |
Breast Cancer |
RAD001 (mTOR inhibitor) |
Experimental: RAD001+carboplatin Carboplatin (starting dose was initially AUC 6, later decreased to AUC 5, then AUC 4) every 3 weeks as IV infusion and RAD001 as 5 mg pill each day until disease progression or unacceptable toxicity. |
NCT00827567 |
Triple Negative Metastatic Breast Cancer |
RAD001 (mTOR inhibitor) |
Experimental: RAD 001 RAD001-10 mg by mouth once everyday |
NCT01319539 |
Stage I, Stage II, or Stage III Breast Cancer |
MK2206 (AKT inhibitor) |
Experimental: Treatment (Akt inhibitor MK2206) Patients receive Akt inhibitor MK2206 PO on days -9 and -2, and undergo segmental resection or total mastectomy on day 0. |
NCT01234402 |
Previously Treated Breast Cancer Patients |
IMC-18F1 (Anti-VEGFR) |
Experimental: Ramucirumab DP + Capecitabine Cycles repeat until disease progression, the development of unacceptable toxicity, noncompliance, or withdrawal of consent by the patient. Experimental: IMC-18F1 + Capecitabine Cycles repeat until disease progression, the development of unacceptable toxicity, noncompliance, or withdrawal of consent by the patient. Active Comparator: Capecitabine Crossover Study |
NCT01234402 |
Previously Treated Breast Cancer Patients |
Ramucirumab (Anti-VEGFR2) |
Experimental: Ramucirumab DP + Capecitabine Cycles repeat until disease progression, the development of unacceptable toxicity, noncompliance, or withdrawal of consent by the patient. Experimental: IMC-18F1 + Capecitabine Cycles repeat until disease progression, the development of unacceptable toxicity, noncompliance, or withdrawal of consent by the patient. Active Comparator: Capecitabine Crossover Study |
Abbreviations: VEGF, Vascular Endothelial Growth Factor; TK, tyrosine kinase.
The adequate use of PARP inhibitors in combination with other compounds provoking DNA damage relies on the identification of tumors that may be sensitive to the combination. In this respect, mutations in BRCA1 and BRCA2 genes have been reported to be the principal predictors of response to PARP inhibitors. Similarly, molecular alterations in other DNA-repair genes, including ATM, Fanconi’s anemia genes or CHEK2, predict response to PARP inhibitors in prostate cancer [89].
The principal mechanism of resistance to PARP inhibitors includes the presence of mutations at the BRCA1/2 genes that restore their function [90, 91]. The presence of alternative splicing of the BRCA gene has also been suggested as a potential mechanism of resistance [92].
BET and CDK inhibitors
Association of BET inhibitors has shown to be synergistic with chemotherapy in preclinical models of lymphoma and TNBC [37, 42, 93]. Recent data in hematological malignancies suggest that WNT signaling and rewiring of transcriptional programs are potentially associated with primary and secondary resistance to BET inhibitors [94]. Preclinical data suggest that inhibition of CDK12 can increase the effect of PARP inhibitors [49]. The CDK7/9 inhibitor TG02 was shown to synergize with chemotherapy, especially with taxotere, in TNBC cellular models [47].
Kinase inhibitors
As mentioned before, a wide range of these agents have been evaluated in combination. The reason behind this is the complementary antitumoral action when combining several kinase inhibitors. In addition, such combinations may be more effective by inhibiting mechanisms of resistance that were developed by activation of secondary loops [95]. In this context the main limitation for targeted combinations, beyond the lack of efficacy in the clinical setting, is related to the overlapping of toxicities, mainly when different kinase inhibitors are combined. For example, preclinical data suggested the synergistic antitumoral effect when combining PI3K and MEK inhibitors [96, 97]. However early phase studies evaluating PI3K and MEK inhibitors in combination showed poor tolerability [98]. The combinations of drugs targeting both pathways seemed to have greater activity in KRAS mutated tumors, mainly ovarian tumors, but the high grade of toxicity observed, limited the clinical development [98].
An alternative approach for the clinical development of these compounds could be to combine these agents with standard chemotherapy. This is for instance the case for the association of PI3K inhibitors with taxanes. While targeting the PI3K/AKT/mTOR pathway by itself is only cytostatic, combination of PI3K inhibitors with agents targeting mitosis produces a cytotoxic effect in preclinical models [61]. That circumstance could translate into an increase in efficacy in the clinical setting. In fact, such strategy is now being evaluated in some clinical studies (Tables 2 and 3). A similar approach has been taken for the clinical development of spindle checkpoint kinase inhibitors that are under evaluation in combination with taxanes (Tables 2 and 3). Finally, combinations of PI3K inhibitors with anti-androgens or PARP inhibitors are under evaluation, as described in other parts of this review.
Table 3: List of ongoing phase I studies in TNBC, as from clinicaltrials.gov, access Jun 1, 2016.
Study number |
Indication |
New drug |
Treatment in combination |
NCT02622074 |
Triple Negative Breast Cancer |
Pembrolizumab (MK-3475) A humanized monoclonal immunoglobulin (Ig) G4 antibody directed against human cell surface receptor PD-1 |
Nab-paclitaxel Anthracycline Cyclophosphamide Carboplatin |
NCT00707707 |
Metastatic Triple Negative Breast Cancer |
AZD2281/Olaparib (inhibitor of PARP1/2) |
Paclitaxel |
NCT01884285 |
Triple Negative Breast Cancer Advanced Castrate-resistant Prostate Cancer (CRPC); Squamous Non-Small Cell Lung Cancer (sqNSCLC); |
AZD8186 (inhibitor of PI3Kβ and PI3Kδ) |
|
NCT01238952 |
Solid Tumors With Dose Expansion in Triple Negative Breast Cancer |
NK012 (SN-38-releasing polymeric micelle) |
Carboplatin |
NCT01624441 |
Metastatic Triple-Negative Breast Cancer |
Dinaciclib ( CDK inhibitor) |
Epirubicin Hydrochloride |
NCT02474173 |
Advanced Triple Negative Breast Cancer |
AT13387 (HSP90 Inhibitor) |
Paclitaxel |
NCT01939418 |
Metastatic Triple Negative Breast Cancer |
RAD001 (mTOR inhibitor) |
|
NCT01238133 |
Metastatic Triple Negative Breast Cancer |
RO4929097 (Gamma-Secretase/Notch Signalling Pathway Inhibitor) |
|
NCT01618136 |
Advanced Solid Tumors or With B-cell Malignancies oradvanced solid tumors |
E7449 Poly(ADP-Ribose) Polymerase (PARP) Inhibitor |
Temozolomide (TMZ) or With Carboplatin and Paclitaxel |
NCT02627430 |
Metastatic Advanced Solid Tumor or Recurrent Ovarian, Fallopian Tube, Primary Peritoneal, or Triple Negative Breast Cancer |
Talazoparib AT13387 (HSP90 Inhibitor) |
|
NCT02583542 |
Advanced Cancers (TORCMEK) |
AZD2014 mTOR serine/threonine kinase (TORC1/2) inhibitor |
Selumetinib |
NCT01623349 |
Recurrent Triple Negative Breast Cancer or High Grade Serous Ovarian Cancer |
BKM120 (oral PI3kinase Inhibitor) |
Olaparib |
NCT01623349 |
Recurrent Triple Negative Breast Cancer or High Grade Serous Ovarian Cancer |
BYL719 (Oral PI3kinase Inhibitor) |
Olaparib |
NCT01445418 |
Ovarian Cancer |
AZD2281 (PARP1/2 inhibitor) |
Carboplatin |
NCT02071862 |
Solid tumors |
CB-839 (Glutaminase Inhibitor) |
|
NCT02259114 |
Advanced Solid Tumors |
OTX015, a Small Molecule Inhibitor of the Bromodomain and Extra-Terminal (BET) Proteins |
|
NCT01698281 |
Refractory in Triple Negative Breast Cancer |
AEZS-108 zoptarelin doxorubicin an LHRH agonist linked to doxorubicin in women with platinum refractory |
|
NCT01876251 |
Advanced Breast Cancer |
PF-03084014 (gamma secretase inhibitor) |
Docetaxel |
NCT02632448 |
Advanced or Metastatic Cancer |
LY2880070 |
Gemcitabine |
NCT02476955 |
Breast Cancer Triple Negative |
ARQ 092 (Akt Inhibitor) |
Carboplatin Plus Paclitaxel |
NCT02027376 |
Triple Negative (TN) Advanced Breast Cancer (ABC) Patients (EDALINE) |
LDE225 (Hedgehog inhibitor) |
Docetaxel |
NCT01596751 |
Metastatic Breast Cancer |
PLX 3397 (multi-kinase inhibitor including CSF1R) |
Eribulin |
NCT01837095 |
Metastatic Breast Cancer |
POL6326 (CXCR4 inhibitor) |
Eribulin |
NCT00754312 |
Newly DiagnosedBreast Cancer |
SNDX-275 (Histone deacetylase inhibitor) |
|
NCT02154776 |
HR+, HER2-negative Post-menopausal Women With Advanced Breast Cancer |
LEE011 (CDK4/6 inhibitor) |
Buparlisib and Letrozole |
NCT01467310 |
Triple Negative Breast Cancer |
GSK1120212 (MEK1/2 allosteric inhibitor) |
|
NCT02158507 |
Metastatic Triple Negative Breast Cancer |
Veliparib (ABT-888) (PARP inhibitor) |
Lapatinib (Tykerb) |
Abbreviations: HR, hormone receptor.
Immunologic agents
Predicting which patients may respond to an immune-based antitumoral strategy remains one of the aspects that still requires much research. In the case of ADCs, the first step in their use is to define the degree of expression of the target that would confer therapeutic advantage. In this respect, preclinical data obtained in other tumors indicates that overexpression of the target is usually accompanied with sensitivity. The use of novel techniques like positron emission tomography imaging of radioactively labeled antibodies (immunoPET) could help selecting those patients that will have a clinical response [99].
One of the potential mechanism of resistance to ADCs, as happens with antibodies, is associated with the limited drug distribution within the tumor due to their high molecular weight or tumor vascularization [100]. Similarly, elimination of chemotherapy from the intracellular compartment to the extracellular matrix could be another mechanism of resistance [101]. In this context, it has been suggested that chemotherapy can augment the activity of ADCs by destroying tumoral tissue, therefore facilitating the penetration of ADCs in different tumoral areas [102]. In the same line, new ADCs are designed to increase the payload ratio of chemotherapy linked with each antibody [103], so the amount of drug that reaches the tumor is higher. Another attractive possibility could be the use of ADCs targeting the stroma in combination with antitumor ADCs.
How to boost the anticancer properties of immune checkpoint modulators is an intensive area of research. In this case, the presence of various immune modulators in the tumor microenvironment appears to have an important role. Expression of checkpoint regulators PD-1, PD-L1 or CTLA-4 appears to be necessary but not sufficient to explain sensitivity to blocking antibodies against them. Some other cellular components of the tumor stroma, such as tumor-associated macrophages, myeloid-derived suppressor cells, or Treg lymphocytes may create an immunosuppressive environment that could prevent a strong antitumoral response by cytotoxic T lymphocytes. Some studies have suggested that the kinase inhibitor ibrutinib, that inhibits the BTK and ITK kinases, when added together with PD-L1 inhibitors, can boost the immune response by modulating the balance among T lymphocytes [104]. In several tumor types, combination of PD-L1 inhibitors with anti-VEGF strategies is under clinical evaluation [105]. However, combinations should be explored with caution: for instance the association of vemurafenib to ipilimumab in melanoma showed an unacceptable hepatotoxicity limiting therefore their clinical development [106].
Table 4: Potential combinations for new agents in TNBC.
Function |
Family of compounds |
Target/compound |
Potential Combination |
Development stage/rational |
Cell division |
Mitotic kinase inhibitors |
Aurora kinase inhibitors PLK inhibitors TTK inhibitors |
Chemotherapies. Taxane-based and platinum compounds |
Clinical stage |
Intracellular Signaling |
PI3K/mTOR inhibitors |
PI3K mTOR |
Anti-androgen receptors |
Overcoming mechanisms of resistance/Clinical stage |
Intracellular Signaling |
FGFR inhibitors |
Nintedanib, ponatinib, dovitinib |
Clinical stage |
|
Intracellular Signaling |
Erk5 |
TG02 |
Taxanes |
Increase cell death |
Intracellular Signaling |
JAK/STAT |
EC70124 |
Platinum compounds |
Increase cell death/preclinical |
DNA damage |
PARP inhibitors |
PARP Inhibitors: ABT-888, BSI-201 (Iniparib) |
DNA damaging agents in BRCA mutated tumors |
Increase cell death/ Clinical stage |
DNA damage |
ATR and Chek inhibitors |
ATR Chek1 (LY2606368) |
DNA damaging agents in ERCC1 and BRCA mutated tumors |
Increase cell death/Clinical stage |
Immunologic agents |
Check point inhibitors |
Anti-PD-1 antibodies: Pembrolizumab (MK-3475), Atezolizumab (MPDL3280A). |
Taxanes |
Clinical stage |
Immunologic agents |
ADCs |
Sacituzumab Govitecan IMMU-132, Glembatumumab vedotin |
Chemotherapy |
Augment drug penetration/Clinical stage |
Immunologic agents |
Ab against macrophage colony-stimulating factor |
MCS110 |
Carboplatin |
Clinical stage |
Targeting apoptosis |
second mitochondrial-derived activator of caspases (SMAC) mimetic |
LCL161 |
Taxanes |
Clinical stage |
Gene expression modulators |
BET inhibitors |
OTX015 |
Taxanes and DNA damaging agents |
Induces cell cycle arrest/Clinical stage |
Gene expression modulators |
CDK7, CDK9 inhibitors |
TG02, THZ1 |
Taxanes |
Increase cell death |
Gene expression modulators |
CDK12 inhibitors |
THZ1 |
PARP inhibitors |
Preclinical stage |
Gene expression modulators |
HDCi |
Romidepsin |
DNA damaging agents (platinum-based) |
Clinical stage |
PI3K and AR inhibitors
Given the fact that the LAR subtype is the one more enriched with mutations at the PIK3CA gene [28], studies evaluating the combination of anti-androgens with PI3K inhibitors are currently ongoing.
Conclusions
A wide range of targeted agents against different oncogenic and non-oncogenic vulnerabilities are currently under evaluation in TNBC. Unfortunately, classification of breast cancer by transcriptomic profiles has not contributed substantially to the identification of new targets or to the stratification of patients for new compounds. Indeed, the role of these subtypes in the clonal evolution of cancer tumors is under evaluation at this moment, with preliminary results not supporting a major role [107].
PARP inhibitors are currently in the late stages of drug development in TNBC patients with mutations at BRCA genes, suggesting that these compounds could be incorporated to the therapeutic armamentarium to fight this disease in the near future. Other families of agents with promising activity are novel ADCs and immune checkpoint inhibitors, both with ongoing phase III studies. On the other hand, the major limitation for all these compounds is the identification of biomarkers of response or companion diagnostics.
How to integrate these agents for treatment combinations is challenging and should always try to avoid the overlapping of toxicities. A proper evaluation of the selected combination in preclinical models should be mandatory before clinical testing. Finally, given the fact that chemotherapy is the standard of care in TNBC, the development of novel combinations will probably contemplate the use of the compounds together with a chemotherapeutic agent.
Funding sources
Instituto de Salud Carlos III (PI16/0112), ACEPAIN; Diputación de Albacete and CRIS Cancer Foundation (to AO). BAE (Beca Ampliación de Estudios) to AO for his stay at Yale University, CT, USA. Ministry of Economy and Competitiveness of Spain (BFU2015-71371-R), the Instituto de Salud Carlos III through the Spanish Cancer Centers Network Program (RD12/0036/0003) and CIBERONC, the scientific foundation of the AECC and the CRIS Foundation (to AP). The work carried out in our laboratories receive support from the European Community through the regional development funding program (FEDER).
Conflicts of interest
No conflict of interest to declare.
References
1. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z,Hernandez-Boussard T,Livasy C,Cowan D,Dressler L,Akslen LA,Ragaz J,Gown AMet al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10: 5367-5374.
2. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K,Karaca G,Troester MA,Tse CK,Edmiston S,Deming SL,Geradts J,Cheang MCet al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. Jama. 2006; 295: 2492-2502.
3. Amirikia KC, Mills P, Bush J, Newman LA. Higher population-based incidence rates of triple-negative breast cancer among young African-American women : Implications for breast cancer screening recommendations. Cancer. 2011;117: 2747-2753.
4. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F,Valero V,Lehmann BD,Pietenpol JA,Hortobagyi GN,Symmans WF,Ueno NT. Differential Response to Neoadjuvant Chemotherapy Among 7 Triple-Negative Breast Cancer Molecular Subtypes. Clin Cancer Res. 2013;19: 5533-5540.
5. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y,Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121: 2750-2767.
6. Vaca-Paniagua F, Alvarez-Gomez RM, Maldonado-Martinez HA, Perez-Plasencia C, Fragoso-Ontiveros V, Lasa-Gonsebatt F,Herrera LA,Cantú D,Bargallo-Rocha E,Mohar A,Durand G,Forey N,Voegele C,et al. Revealing the Molecular Portrait of Triple Negative Breast Tumors in an Understudied Population through Omics Analysis of Formalin-Fixed and Paraffin-Embedded Tissues. PLoS ONE. 2010;10: e0126762.
7. Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA,Savage MI,Osborne CK,Hilsenbeck SG,Chang JC,Mills GB,Lau CC,Brown PHl. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21: 1688-1698.
8. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA,Lickley LA,Rawlinson E,Sun P,Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007; 13: 4429-4434.
9. Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN,Martino S,Wang M,Jones VE,Saphner TJ,Wolff AC,Wood WC, Davidson NE,et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancers From Two Phase III Randomized Adjuvant Breast Cancer Trials: ECOG 2197 and ECOG 1199. J Clin Onco. 2014;32:2959-66l.
10. Ocana A, Diez-Gonzalez L, Adrover E,Fernandez-Aramburo A, Pandiella A, Amir E. Tumor-infiltrating lymphocytes in breast cancer: ready for prime time? J Clin Oncol. 2015;33: 1298-1299.
11. Ocana A, Pandiella A. Identifying breast cancer druggable oncogenic alterations: leasons learned and future options. Clin Cancer Res. 2008;14:961-70
12. Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA,Symmans WF,Gonzalez-Angulo AM,Hennessy B,Green M,Cristofanilli M,Hortobagyi GN,Pusztai L. J Clin Oncol. 2008; 26: 1275-1281.
13.Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N,Trudel M,Akslen LA. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst. 2003; 95: 1482-1485.
14. Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004; 4: 814-819.
15. Tutt A, Ellis P, Kilburn L, Gillett C, Pinder S, Abraham J, Barrett S, Barrett-Lee P, Chan S, Cheang M, Dowsett M, Fox L, Gazinska P,et al. The TNT trial: a randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). San Antonio Breast Cancer Symposium, 2014: abstr S3-01.
16. Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ,Rugo HS,Liu MC,Stearns V,Come SE,Timms KM,Hartman AR,Borger DR,et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2015;33: 1902-1909.
17. Ocana A, Pandiella A. Novel Synthetic Lethality Approaches for Drug Combinations and Early Drug Development. Curr Cancer Drug Targets. 2015;24:316-24.
18. Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R,Rayter S,Tutt AN,Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. Embo J. 2008; 27: 1368-1377.
19. Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T,Sridhara R,Lee E,Tzou A,Philip R,Chiu HJ,Ricks TK,Palmby T,et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015;21: 4257-4261.
20. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J,Mitchell G,Fried G,Stemmer SM,Hubert A,Rosengarten O,Steiner M,Loman N,et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33: 244-250.
21. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN,Friedlander M,Arun B,Loman N,Schmutzler RK,Wardley A,Mitchell G,Earl H,et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376: 235-244.
22. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K,Hirte H,Huntsman D,Clemons M,Gilks B,Yerushalmi R,Macpherson E,Carmichael J,et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12: 852-861.
23. Isakoff SJ,Puhalla S,Domchek SM,Friedlander M,Kaufman B,Robson M,Telli ML,Diéras V,Han HS,Garber JE,Johnson EF,Maag D,Qin Q,et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer.J Clin Oncol. 2010;28 (suppl; abstr 1019). .
24. D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362: 1909-1919.
25. Mohni KN, Kavanaugh GM, Cortez D. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014;74: 2835-2845.
26. Pandiella A, Moris F, Ocana A, Nunez LE, Montero JC. Antitumoral activity of the mithralog EC-8042 in triple negative breast cancer linked to cell cycle arrest in G2. Oncotarget. 2015;6: 32856-32867. doi: 10.18632/oncotarget.5942.
27. Blum JL, Goncalves A, Efrat N, Debled M, Conte P, Richards PD,Richards D,Lardelli P,Nieto A,Cullell-Young M,Delaloge S. A phase II trial of trabectedin in triple-negative and HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2016;155:295-302.
28. Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232: 142-150.
29. Ocana A, Perez-Pena J, Diez-Gonzalez L, Sanchez-Corrales V, Templeton A, Seruga B,Amir E,Pandiella A. Transcriptomic analyses identify association between mitotic kinases, PDZ-binding kinase and BUB1, and clinical outcome in breast cancer. Breast Cancer Res Treat. 2016;156: 1-8.
30. Maire V, Baldeyron C, Richardson M, Tesson B, Vincent-Salomon A, Gravier E,Marty-Prouvost B,De Koning L,Rigaill G,Dumont A,Gentien D,Barillot E,Roman-Roman S,et all. TTK/hMPS1 is an attractive therapeutic target for triple-negative breast cancer. PLoS ONE. 2013;8: e63712.
31. Maia AR, de Man J, Boon U, Janssen A, Song JY, Omerzu M,Sterrenburg JG,Prinsen MB,Willemsen-Seegers N,de Roos JA,van Doornmalen AM,Uitdehaag JC,Kops GJ,et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann Oncol. 2015;26:2180-92.
32. Koch A, Maia A, Janssen A, Medema RH. Molecular basis underlying resistance to Mps1/TTK inhibitors. Oncogene. 2016;35:2518-28..
33. Maire V, Nemati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G,Gravier E,Marty-Prouvost B,De Koning L,Lang G,Gentien D,Dumont A,Barillot E,et al.. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013;73: 813-823.
34. Cappello P, Blaser H, Gorrini C, Lin DC, Elia AJ, Wakeham A,Haider S,Boutros PC,Mason JM,Miller NA,Youngson B,Done SJ,Mak TW. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene. 2014;33: 2375-2384.
35. Kai K, Kondo K, Wang X, Xie X, Pitner MK, Reyes ME,Torres-Adorno AM,Masuda H,Hortobagyi GN,Bartholomeusz C,Saya H,Tripathy D,Sen S,et al. Antitumor Activity of KW-2450 against Triple-Negative Breast Cancer by Inhibiting Aurora A and B Kinases. Mol Cancer Ther. 2015;14: 2687-2699.
36. Bush TL, Payton M, Heller S, Chung G, Hanestad K, Rottman JB,Loberg R,Friberg G,Kendall RL,Saffran D,Radinsky R. AMG 900, a small-molecule inhibitor of aurora kinases, potentiates the activity of microtubule-targeting agents in human metastatic breast cancer models. Mol Cancer Ther. 2013;12: 2356-2366.
37. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54: 728-736.
38. Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L,Zhang Y,Liu B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015;6: 5501-5516. doi: 10.18632/oncotarget.3551
39. Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM,Parsons R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell. 2015;27: 837-851.
40. Odore E, Lokiec F, Cvitkovic E, Bekradda M, Herait P, Bourdel F,Kahatt C,Raffoux E,Stathis A,Thieblemont C,Quesnel B,Cunningham D,Riveiro ME,et al.Phase I Population Pharmacokinetic Assessment of the Oral Bromodomain Inhibitor OTX015 in Patients with Haematologic Malignancies. Clin Pharmacokinet. 2016;55:397-405.
41. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM,Janiszewska M,Huh SJ,Liang Y,Ryan J,Doherty E,Mohammed H,Guo H. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529: 413-417.
42. Perez-Peña J, Serrano-Heras G, Montero JC, Corrales-Sanchez V, Pandiella A, Ocana A.In silico analyses guides selection of BET inhibitors for triple-negative breast cancer treatment.Mol Cancer Ther. 2016; 15 (8): 1823-33.
43. Merino VF, Nguyen N, Jin K, Sadik H, Cho S, Korangath P,Han L,Foster YM,Zhou XC,Zhang Z,Connolly RM,Stearns V,Ali SZ,et al. Combined Treatment with Epigenetic, Differentiating, and Chemotherapeutic Agents Cooperatively Targets Tumor-Initiating Cells in Triple-Negative Breast Cancer. Cancer Res. 2016;76:2013-24
44. Schech A, Kazi A, Yu S, Shah P, Sabnis G. Histone Deacetylase Inhibitor Entinostat Inhibits Tumor-Initiating Cells in Triple-Negative Breast Cancer Cells. Mol Cancer The. 2015;r 14: 1848-1857.
45. Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15: 122.
46. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S,Yuzugullu H,Von T,Li H,Lin Z,Stover DG,Lim E,Wang ZC,et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell. 2015;163: 174-186.
47. Ortiz-Ruiz MJ, Alvarez-Fernandez S, Parrott T, Zaknoen S, Burrows FJ, Ocana A,Pandiella A,Esparís-Ogando A. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget. 2014;5: 11308-11318. doi: 10.18632/oncotarget.2324.
48. Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z,Cimermancic P,Ule J,Peterlin BM. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25: 2158-2172.
49. Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R,Campbell J,Sims D,Rafiq R,Hooper S,Chen L,Kozarewa I,Assiotis I,et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;s 74: 287-297.
50. Niemeier LA, Dabbs DJ, Beriwal S, Striebel JM, Bhargava R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod Pathol. 2010;23: 205-212.
51. Vera-Badillo FE, Templeton AJ, de Gouveia P, Diaz-Padilla I, Bedard PL, Al-Mubarak M,Seruga B,Tannock IF,Ocana A,Amir E. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106: djt319.
52. Proverbs-Singh T, Feldman JL, Morris MJ, Autio KA, Traina TA. Targeting the androgen receptor in prostate and breast cancer: several new agents in development. Endocr Relat Cancer. 2015;22: R87-R106.
53. Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA,Blackwell K,Rugo H,Nabell L,Forero A,Stearns V,Doane AS,Danso M,et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013;19: 5505-5512.
54. Traina TA, O’Shaughnessy JO, Nanda R,Schwartzberg L, Abramson V, Cortes J, Peterson A, Tudor IC, Blaney M, Steinberg JL, Kelly C, Trudeau M, Awada A,et al.Stage 1 results from MDV3100-11: a 2-stage study of enzalutamide, an androgen receptor inhibitor, in advanced AR+ triple-negative breast cancer. 37th Annual San Antonio Breast Cancer Symposium. December 2014. Abstract P5-19-09.
55. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010; 10: 116-129.
56. Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R,Geyer FC,van Kouwenhove M,Kreike B,Mackay A,Ashworth A,van de Vijver MJ,Reis-Filho JS. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010; 29: 2013-2023.
57. Sharpe R, Pearson A, Herrera-Abreu MT, Johnson D, Mackay A, Welti JC,Natrajan R,Reynolds AR,Reis-Filho JS,Ashworth A. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin Cancer Res. 2011;17: 5275-5286.
58. NCT02202746. A Study to Assess the Safety and Efficacy of the VEGFR-FGFR Inhibitor, Lucitanib, Given to Patients With FGF Aberrant Metastatic Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT02202746. Accessed Dec 15, 2015.
59. Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M,Lebigot I,Djelti F,Tourdès A,Gestraud P,Hupé P,Barillot E,Cruzalegui F,et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008; 10: R101.
60. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM,Lawrence MS,Sivachenko AY,Sougnez C,Zou L,Cortes ML,Fernandez-Lopez JC,Peng S,et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486: 405-409.
61. Montero JC, Esparis-Ogando A, Re-Louhau MF, Seoane S, Abad M, Calero R,Ocaña A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene. 2014;33: 148-156.
62. Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS ONE. 2009; 4: e5565.
63. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002; 298: 1912-1934.
64. Cuenca-Lopez MD, Serrano-Heras G, Montero JC, Corrales-Sanchez V, Gomez-Juarez M, Gascon-Escribano MJ,Morales JC,Voisin V,Núñez LE,Morís F,Bader GD,Pandiella A,Ocaña A. Antitumor activity of the novel multi-kinase inhibitor EC-70124 in triple negative breast cancer. Oncotarget. 2015;6:27923-37. doi: 10.18632/oncotarget.4736.
65. Baselga J, Gomez P, Greil R,Braga S,Climent MA,Wardley AM,Kaufman B,Stemmer SM,Pêgo A,Chan A,Goeminne JC,Graas MP,Kennedy MJ,et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013; 31: 2586-2592.
66. Bell R, Brown J, Parmar M,Toi M,Suter T,Steger GG,Pivot X,Mackey J,Jackisch C,Dent R,Hall P,Xu N,Morales L,et al. Final efficacy and updated safety results of the randomized phase III BEATRICE trial evaluating adjuvant bevacizumab-containing therapy in triple-negative early breast cancer. Ann Oncol. 2016.
67. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol. 2015;33: 23-35.
68. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S,Su X,Wang Y,Gonzalez-Angulo AM,Akcakanat A,Chawla A,Curran M,Hwu P,et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014; 2: 361-370
69. Yu H, Yang J, Jiao S, Li Y, Zhang W, Wang J. Cytotoxic T lymphocyte antigen 4 expression in human breast cancer: implications for prognosis. Cancer Immunol Immunother 2015; 64: 853-860..
70. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R,Pusztai L,Pathiraja K,Aktan G,Cheng JD,Karantza V,Buisseret L. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016;34:2460-7
71. Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression.J Mammary Gland Biol Neoplasia. 2002; 7: 147-162.
72. Templeton AJ, McNamara MG, Seruga B, Vera-Badillo FE, Aneja P, Ocana A,Leibowitz-Amit R,Sonpavde G,Knox JJ,Tran B,Tannock IF,Amir E.Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106: dju124.
73. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528: 413-417
74. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF,Gallagher WM,Wadhwani N,Keil SD,Junaid SA,Rugo HS,Hwang ES,Jirström K,et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011; 1: 54-67.
75. Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ, Jr., Vahdat LT,Thomas SS,Govindan SV,Maliakal PP,Wegener WA,Hamburger SA,Sharkey RM,Goldenberg DM. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin Cancer Res. 2015; 21: 3870-3878.
76. Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer. 2015;6: 84-105. doi: 10.18632/genesandcancer.40.
77. Bardia A, Starodub A, Moroose RL, Mayer IA, Diamond JR, Chuang E,Govindan SV,Sharkey RM,Maliakal P,Wegener WA,Hamburger SA, Ocean AJ, Goldenberg DM, et al. MMU-132, a new antibody-drug conjugate against Trop-2, as a novel therapeutic for patients with relapsed/refractory, metastatic, triple-negative breast cancer: results from phase I/II clinical trial (NCT01631552). 37th Annual San Antonio Breast Cancer Symposium. December 2014. Abstract P5-19-27
78. Rose AA, Grosset AA, Dong Z, Russo C, Macdonald PA, Bertos NR,St-Pierre Y,Simantov R,Hallett M,Park M,Gaboury L,Siegel PM. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin Cancer Res. 2916;16: 2147-2156.
79. Yardley DA, Weaver R, Melisko ME, Saleh MN, Arena FP, Forero A,Cigler T,Stopeck A,Citrin D,Oliff I,Bechhold R,Loutfi R,Garcia AA,et al EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB-Expressing Breast Cancer. J Clin Oncol. 2015; 33: 1609-1619
80. Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR,Bloushtain-Qimron N,Kim JJ,Choudhury SA,Maruyama R,Wu Z,Gönen M,Mulvey LA,et al .The JAK2/STAT3 signaling pathway is required for growth of CD44CD24 stem cell-like breast cancer cells in human tumors. The Journal of clinical investigation. 2011; 121: 2723-2735..
81. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR,Bundred NJ,Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70: 709-718.
82. Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C,Yuan J,Ware C,MacLean J,Garrett-Engele PWKluk M,Laskey J,Haines BB,et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014; 4: 1154-1167.
83. Wang K, Zhang Q, Li D,Ching K,Zhang C,Zheng X,Ozeck M,Shi S,Li X,Wang H,Rejto P,Christensen J,Olson P. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a gamma-secretase inhibitor. Clin Cancer Res. 2015;21: 1487-1496.
84. Weisberg E, Ray A, Barrett R,Nelson E,Christie AL,Porter D,Straub C,Zawel L,Daley JF,Lazo-Kallanian S,Stone R,Galinsky I,Frank D,et al. Smac mimetics: implications for enhancement of targeted therapies in leukemia. Leukemia. 2010;24: 2100-2109.
85. Zhang T, Li Y, Zou P,Yu JY,McEachern D,Wang S,Sun D. Physiologically based pharmacokinetic and pharmacodynamic modeling of an antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in a mouse xenograft model of human breast cancer. Biopharm Drug Dispos. 2013;34: 348-359.
86. PartonM, BardiaA, KummelS, EstevezL, HuangC, Cortes CastanJ, Ruiz BorregoM, TelliM, LluchA, LopezR, BeckT, Ismail-KhanR, ChenS, et al. A phase II, open-label, neoadjuvant, randomized study of LCL161 with paclitaxel in patients with triple-negative breast cancer (TNBC). J Clin Oncol. 33, 2015 (suppl; abstr 1014).
87. Horiuchi D, Camarda R, Zhou AY,Yau C,Momcilovic O,Balakrishnan S,Corella AN,Eyob H,Kessenbrock K,Lawson DA,Marsh LA,Anderton BN.Rohrberg J,et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat Med. 2016; 22: 1321-1329.
88. Braso-Maristany F, Filosto S, Catchpole S,Marlow R,Quist J,Francesch-Domenech E,Plumb DA,Zakka L,Gazinska P,Liccardi G,Meier P,Gris-Oliver A,Cheang MC,et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med. 2016; 22: 1303-1313.
89. Mateo J, Carreira S, Sandhu S,Miranda S,Mossop H,Perez-Lopez R,Nava Rodrigues D,Robinson D,Omlin A,Tunariu N,Boysen G,Porta N,Flohr P,et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373: 1697-1708.
90. Edwards SL, Brough R, Lord CJ,Natrajan R,Vatcheva R,Levine DA,Boyd J,Reis-Filho JS,Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008; 451: 1111-1115.
91. Sakai W, Swisher EM, Karlan BY,Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008; 451: 1116-1120.
92. Colombo M, Blok MJ, Whiley P,Santamariña M,Gutiérrez-Enríquez S,Romero A,Garre P,Becker A,Smith LD,De Vecchi G,Brandão RD,Tserpelis D,Brown M, et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: a report from the ENIGMA consortium. Hum Mol Genet. 2014;23: 3666-3680.
93. Boi M, Gaudio E, Bonetti P,Kwee I,Bernasconi E,Tarantelli C,Rinaldi A,Testoni M,Cascione L,Ponzoni M,Mensah AA,Stathis A,Stussi G,et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin Cancer Res. 2015;21: 1628-1638.
94. Rathert P, Roth M, Neumann T,Muerdter F,Roe JS,Muhar M,Deswal S,Cerny-Reiterer S,Peter B,Jude J,Hoffmann T,Boryń ŁM,Axelsson E,et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525: 543-547.
95. Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16: 280-296.
96. Hoeflich KP, O’Brien C, Boyd Z,Cavet G,Guerrero S,Jung K,Januario T,Savage H,Punnoose E,Truong T,Zhou W,Berry L,Murray L. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009; 15: 4649-4664.
97. Craig DW, O’Shaughnessy JA, Kiefer JA,Aldrich J,Sinari S,Moses TM,Wong S,Dinh J,Christoforides A,Blum JL,Aitelli CL,Osborne CR,Izatt T,et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;12: 104-116.
98. Bedard PL, Tabernero J, Janku F,Wainberg ZA,Paz-Ares L,Vansteenkiste J,Van Cutsem E,Pérez-García J,Stathis A,Britten CD,Le N,Carter K,Demanse D. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21: 730-738.
99. Lamberts LE, Williams SP, Terwisscha van Scheltinga AG,Lub-de Hooge MN, Schröder CP, Gietema JA, Brouwers AH, de Vries EG.. Antibody positron emission tomography imaging in anticancer drug development. J Clin Oncol. 2015;33: 1491-1504.
100. Tan Q, Saggar JK, Yu M,Wang M,Tannock IF. Mechanisms of Drug Resistance Related to the Microenvironment of Solid Tumors and Possible Strategies to Inhibit Them. Cancer J. 2015;21: 254-262.
101. Yu M, Ocana A, Tannock IF. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013;32: 211-227.
102. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006; 6: 583-592.
103.http://www.tarveda.com/pentarin.html.
104. Sagiv-Barfi I, Kohrt HE, Czerwinski DKNg PP,Chang BY,Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112: E966-972.
105. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14: 561-584.
106. Ribas A, Hodi FS, Callahan M,Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368: 1365-1366.
107. Nguyen LV,Pellacani D,Lefort S,Kannan N,Osako T,Makarem M,Cox CL,Kennedy W,Beer P,Carles A,Moksa M,Bilenky M,Balani S,et al. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature. 2015;528: 267-271.