
INTRODUCTION
Hypoxic-ischemic encephalopathy (HIE), caused by a lack of oxygen or blood flow to the brain around the time of birth, affects 1-2 in every 1000 live births in the UK [1] and far more in the developing world [2]. The consequences for babies and parents affected by HIE are devastating with a high proportion of survivors suffering from severe and long-lasting motor and cognitive impairments [3]. Neuroprotective therapy with hypothermia improves outcome and prevents death or neurological outcome in one in seven babies which represents a breakthrough in the care of these vulnerable neonates [4]. Although therapeutic hypothermia is not universally successful, it provides proof-of-concept that intervention following injury can offer significant benefit. However, in order to provide additional more effective therapies we urgently require a more thorough understanding of the underlying pathophysiology.
Brain injury develops in different phases following hypoxia-ischemia (HI) depending on the brain region and severity of the primary insult [5, 6]. Cell death after neonatal brain injury is characterized morphologically by a mixed necrotic, necroptotic, autophagic and apoptotic phenotype [7-10], with predominantly neurons vulnerable to insult in the near term brain. However, data from our lab and others strongly suggest that the common thread linking these diverse mechanisms is mitochondrial perturbation [11-13].
HI triggers multiple signaling events such as excitotoxicity, release of reactive oxygen species and increase in intracellular calcium [5, 6, 11, 12]. Bcl-2 family member Bax is activated and translocates to the mitochondria where it and family member Bak homo-oligomerise forming pores ultimately leading to mitochondrial outer membrane permeabilization (MOMP) [5]. MOMP allows the efflux of pro-apoptotic proteins such as cytochrome c and apoptosis inducing factor (AIF) into the cytosol [13-17] thereby initiating a cascade resulting in activation of caspases, degradation of DNA and ultimately cell death [14, 15, 18].
It is, however, still unclear how MOMP is initiated. Besides changes in pro- vs. anti-apoptotic Bcl-2 protein ratio, several other upstream regulators may be involved such as caspase-2 and c-Jun N-terminal kinase (JNK) [19, 20]. The tumor suppressor protein p53 (Trp53 mouse gene) is one such candidate recently implicated in mediating neuronal cell death observed after neonatal HI. Nuclear p53 triggers apoptosis via multiple pathways, for example cell cycle arrest, the regulation of autophagy [21], through transactivating pro-apoptotic and repressing anti-apoptotic genes [22]. In addition, p53 also has cytoplasmic actions at the mitochondrial level and can promote Bax-dependent MOMP [23, 24]. Nuclear export of p53 depends on mono-ubiquitination by the E3 ubiquitin-ligase Mdm2 and once at the mitochondria, p53 is deubiquitinated by the ubiquitin specific peptidase 7 (HAUSP) [25]. Cytosolic p53 is probably inactivated through binding to Bcl-XL and/or Mcl-1 [24, 26]. P53 is, however, liberated from Bcl-XL/Mcl-1 by high levels of p53 up-regulated modulator of apoptosis (PUMA) and can subsequently interact with Bax at the outer mitochondrial membrane.
There are several reasons to suspect that p53 is important in the context of Bax-dependent MOMP and cell death in the neonatal brain. Firstly, the expression of p53 and several of its downstream genes are induced in the brain after neonatal HI [27, 28]. Secondly, p53, PUMA and Noxa accumulate in the cytosol and p53 translocates to mitochondria after neonatal HI [28-30]. Thirdly, inhibiting NFκB (a regulator of p53) by blocking IκB-kinase with a Nemo binding domain TAT peptide reduces p53 levels and subsequently cytochrome c release and brain injury in a neonatal HI mouse model [29-31]. Finally, Pifithrin-μ (PFT-μ, inhibitor of the mitochondrial actions of p53) [32] blocks mitochondrial accumulation of p53, cytochrome c release and subsequent activation of caspase-3, substantially reducing neonatal brain injury [31]. However, PFT-μ also has effects on microglial activation [33] and heat shock proteins (HSP) [34] and we still lack convincing genetic proof that p53 is involved.
The aim of the present study is to apply a two-pronged approach to evaluate whether, firstly, a partial gene deficiency of Trp53, comparing Trp53 heterozygote mice with wild types, confer neuroprotection in a neonatal model of term HI and, secondly, conditional neuronal deletion of Trp53 (utilizing CaMK2αCre – Lox Trp53 mice [35]) affects the extent of HI brain injury in neonatal and juvenile mice.
RESULTS
Trp53 hets are less injured at posterior levels of the brain following moderate (50 min) but not severe (60 min) of hypoxia-ischemia at postnatal day (PND) 9
At PND 9, mixed genotype litters of both sexes were randomly subjected to either a moderate 50 min or severe 60 min episode of HI. Loss of grey matter (MAP-2 staining) and white matter (MBP staining) was evaluated 7 days following HI.
Following moderate (50 min) HI, no differences in body weight were observed between genotypes at PND 9 and PND 16. Analysis of the infarct revealed that Trp53 hets (n=18; females n=8, males n=10) showed significantly less MAP-2 tissue loss in the posterior levels of the brain (Figure 1A, E) compared with Trp53 WT (n=21; females n=8, males n=13; 16.1 ± 3.5% vs 30.6 ± 4.0%, p<0.05). No differences were observed at the central or anterior levels of the brain. No differences were observed in volume tissue loss (Figure 1B) or loss of MBP staining (Figure 1C).
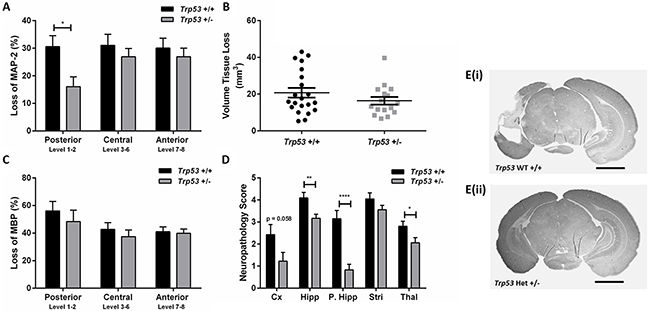
Figure 1: Brain injury assessment of Trp53 WT (+/+) and Het (+/-) mice 7 days following 50 min HI at PND 9. MAP-2 tissue loss A. volume tissue loss B. MBP loss C. and neuropathology scores D. were assessed at 8 levels of the brain in Trp53 WT (black bars; n=21) and Trp53 het (grey bars; n=16). MAP-2 stained images at posterior levels of the brain from Trp53 WT +/+ E(i). and Trp53 het +/- E(ii). Mean ± SEM. * p< 0.05, ** p < 0.01, **** p<0.0001. Cortex (Cx), Hippocampus (Hipp), posterior hippocampus (P. Hipp), Striatum (Stri), Thalamus (Thal). Scale bar represents 2 mm.
The atrophy and infarction (0 to 6) for each region of interest (cortex, hippocampus, striatum and thalamus) were assessed to obtain a neuropathology score of the left, injured, hemisphere (Figure 1D) [36]. Trp53 hets showed significantly less injury particularly in the posterior region of the hippocampus and the thalamus. A trend towards less injury was also seen in the cortex (p=0.058). No differences were observed in the striatum. In spite of the quite marked protection in the Trp53 het mice in the posterior part of the brain, we observed no difference in overall volume tissue loss as the central and anterior part of the brain including striatum and most parts of cerebral cortex were unaffected by p53 genotype.
Following a severe (60 min) duration of HI there were no differences between Trp53 WT (n=20; females n=11, males n=9) and Trp53 hets (n=29; females n=15, males n=14) when assessed at PND 16 for MAP-2 tissue loss (Figure 2A), volume tissue loss (Figure 2B) or MBP loss (Figure 2C).
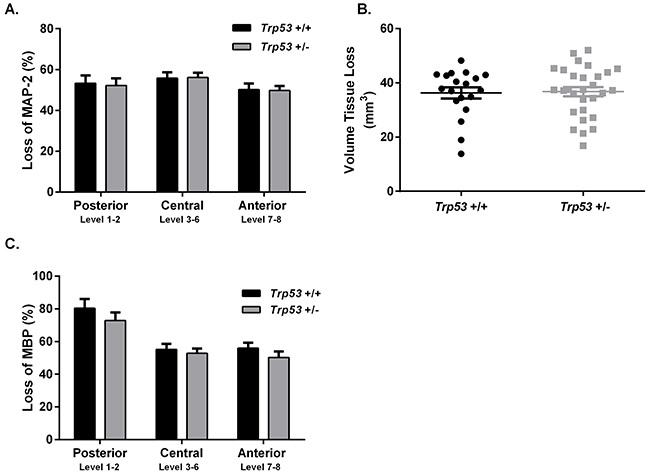
Figure 2: Brain injury assessment of Trp53 WT (+/+) and Het (+/-) mice 7 days following 60 min HI at P9. MAP-2 tissue loss A. volume tissue loss B. and MBP loss C. were assessed at 8 levels of the brain in Trp53 WT (black bars; n=20) and Trp53 het (grey bars; n=29). Mean ± SEM.
No effect of conditional knock-down of Trp53 on injury at PND 12
In order to reduce any influence on phenotype due to the global absence of p53, we generated a conditional Trp53 knockout mouse in which deletion of Trp53 was limited to neurons in the forebrain. We chose to combine Trp53-lox animals with mice expressing Cre recombinase under the control of the calcium/calmodulin-dependent protein kinase II alpha (CaMK2α) promoter. The CaMK2α promoter is reported to facilitate early, neuronal expression of the gene of interest [37]. PND12 mixed litters consisting of WT Lox Trp53+/+ (n=16; females n=7, males n=9) and KO Lox Trp53f/+ (n=24; males n=13, females n=11) were subjected to 50 min of HI. There was, however, no difference between genotypes in MAP-2 tissue loss overall or in the hippocampus (data not shown).
CaMK2α Cre activation is initially detected at PND 19
In order to investigate the regional and age-dependent expression of Cre recombinase activity which may have influenced the previous result, CaMK2α-Cre mice were bred with Rosa26 LacZ reporter mice. X-gal staining of β-galactosidase revealed no positive Cre recombinase activity (shown by blue stained cells) at PND 7 or PND 14. At PND 19, blue stained cells were present in the CA1 and CA2 regions of the hippocampus, with subtle staining seen in the cortex. By PND 28 strong staining was present throughout the hippocampus and cortex, at the level of approximately Bregma -2.12mm. This was consistent in pups generated from paternal (bred with a ROSA26 female) or maternal (bred with a ROSA26 male) Cre inheritance (Figure 3).
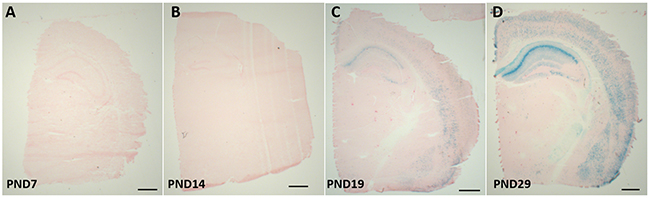
Figure 3: β-gal expression in the brains of CaMK2α Cre - Rosa26 reporter mice. CaMK2α Cre mice were bred with ROSA26 reporter mice; brains were collected at postnatal day (PND) 7 A. PND 14 B. PND 19 C. and PND29 D. B-gal staining is representative of cre-recombinase activity, which occurs from PND 19. Scale bar represents 1000 μm.
Trp53 messenger RNA expression in CaMK2α Cre-Trp53 Lox mice
As antibodies directed towards mouse p53 are unreliable for immunohistochemistry (data not shown), to confirm CaMK2α Cre-mediated deletion of Trp53 from neurons, in situ hybridization was conducted for Trp53 mRNA in WT Lox Trp53+/+ and KO Lox Trp53f/+ (Figure 4). Brains were assessed at PND 7, PND 12 and at 6 weeks of age. Trp53 mRNA expression was present at PND 7 and PND 12 in both genotypes (Figure 4A, 4B, 4E, 4F). At 6 weeks of age WT Lox Trp53+/+ (Figure 4C, 4D) exhibited Trp53 mRNA staining in the hippocampus, in contrast to KO Lox Trp53f/+ that showed sparse hippocampal Trp53 expression (Figure 4G, 4H). This staining was consistent with the regions where we observed Cre-recombinase activation, shown by x-gal staining (Figure 3D). Traditional Trp53 KO mice served as a negative control and no Trp53 mRNA staining was observed throughout the brain (Figure 4I, 4J).
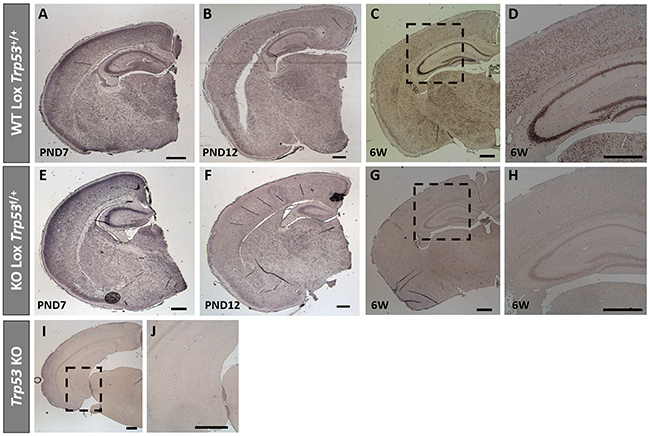
Figure 4: Trp53 in situ hybridization on brains from WT Lox Trp53+/+, KO Lox Trp53f/+ and Trp53 KO. In situ hybridization of Trp53 in WT Lox Trp53+/+ A-D. KO Lox Trp53f/+ E-H. and traditional Trp53 KO I, J. at postnatal day (PND) 7 A, E. PND 12 B, F. 6 weeks of age C, G. and [higher magnification D, H.]. Traditional Trp53 KO I. higher magnification J. shows no Trp53 staining. Scale bars represent 500 μm.
Conditional knock-down of Trp53 was not protective against hypoxic-ischemic injury at PND 28
Based on the LacZ reporter staining and the in situ data showing that Trp53 expression was significantly reduced at PND 28, we induced 40 min of HI at PND 28 in mixed litters from Cre-Lox mice. No differences in brain injury were seen between WT Lox Trp53+/+ (n=8; females n=4, males n=4) and KO Lox Trp53f/+ (n=16; females n=8, males n=8) 7 days following HI. No differences were observed in loss of MAP-2 (total group or separated for sex), volume tissue loss or loss of MBP (Figure 5). We also performed neuropathology scoring of the different regions and found that brain injury in the juvenile brain (PND 28) was observed in the same regions (hippocampus, thalamus, striatum and cortex) as in the immature (PND 9 or PND 12) brain which agree with previous work [38]. However, we found no difference between genotypes following HI at PND 28.
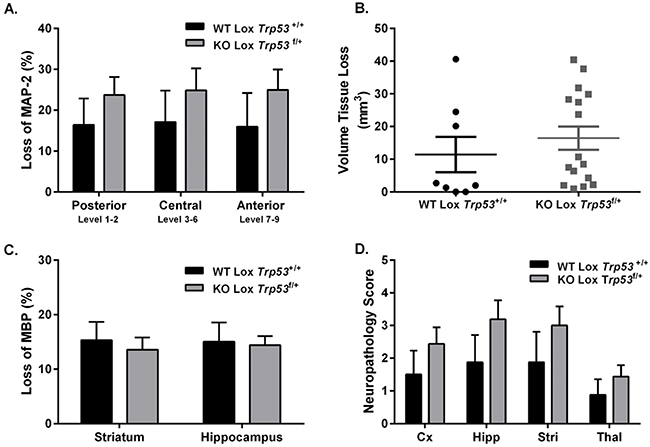
Figure 5: Brain injury assessment of WT Lox Trp53+/+ and KO Lox Trp53f/+ mice 7 days following 40 min HI at PND 28. MAP-2 tissue loss A. volume tissue loss B. MBP loss C. and neuropathology score D. were assessed in WT Lox Trp53+/+ (black bars; n=8) and KO Lox Trp53f/+ (grey bars; n=16). Mean ± SEM. Cortex (Cx), Hippocampus (Hipp), Striatum (Stri), Thalamus (Thal).
DISCUSSION
In summary, we found that heterozygote Trp53 gene deficiency conferred partial neuroprotection in the posterior part of the brain including the posterior part of the hippocampus, cerebral cortex and thalamus in response to a moderate insult of 50 min of HI but not following a more severe 60 min insult of HI in PND 9 mice. Brain injury was not affected in neonatal (PND 12) or juvenile (PND 28) mice by a conditional neuron-selective Trp53 gene deletion.
The moderate neuroprotection seen in the posterior part of the brain in neonatal Trp53 het mice contrasts with the profound tissue protection seen in adult mice. Adult Trp53 het mice develop ~80% smaller brain injury compared with WT controls after ischemia [39]. There are several possible explanations for the developmental difference in vulnerability dependent on p53. It was recently discovered that in the adult brain p53 interacts with cyclophilin D (CyD) and the complex induces opening of the mitochondrial permeability transition (MPT) pore leading to necrotic cell death [39]. Hence downregulation of p53 or blockage of CyD with cyclosporin A reduces ischemic injury in adults [40, 41]. However, in the setting of the immature brain the situation is different. Ischemic necrosis doesn’t seem to depend on CyD or MPT as cyclosporin A or CyD gene deficiency do not confer neuroprotection [14, 42]. Instead, RIP kinase-dependent necrosis is proposed to be critical for this mode of cell death [43]. Speculatively, partial p53 gene depletion is markedly protective in the adult brain as CyD dependent necrosis and apoptosis are both attenuated. In the immature brain, Bax-dependent apoptotic cell death is reduced in Trp53 het mice reducing vulnerability but the degree of protection is limited as necrosis is unaffected. It also could explain why p53 genotype did not influence brain injury after a severe insult of 60 min HI in the neonates, as necrosis becomes the predominant mode of cell death even in the immature brain under those conditions [7].
Whilst the ideal comparison to access the effect of genetic knockdown would be between Trp53 KO and WT pups, it is widely reported that traditional Trp53 KO mice develop malignancy throughout the body from a young age [44]. Also, whilst all our breeding paradigms were generating mixed genotype litters, only very few Trp53 KO (n=6) pups were born. These pups were subjected to 60 min HI and no difference between genotypes in injury was seen (data not shown). We therefore compared Trp53 het with Trp53 WT, which, in two studies in the adult mouse brain have shown that a partial knockdown of p53 is sufficient to protect the brain following focal ischemia [39, 45].
In order to reduce secondary phenotypic influences in conventional knock-out mice we also adopted the Cre-LoxP system to allow for conditional gene deletion in predominantly hippocampal and cortical neurons, regions that are vulnerable to near term hypoxic-ischemic injury. We chose the promoter CaMK2α [35] where the mRNA for CaMK2α is expressed in most brain regions at postnatal day 4-14 [46]. This significant increase in expression from second to third postnatal week is thought to correlate with active forebrain synaptogenesis [47]. However, according to our assessment of cre-recombinase activity, through breeding with a ROSA26 reporter mouse and staining for X-gal, CaMK2α cre-recombinase activation was only present from PND 19, consistent with Tsien et al., [37]. In addition, our in situ hybridization results also showed Trp53 expression was apparent until PND 12, which could explain the lack of neuroprotection in Cre-LoxP mice at PND 12. But it was surprising that HI brain injury was unaffected by conditional neuronal deletion of Trp53 also at PND 28 considering the marked cre-recombinase activation and loss of Trp53 gene expression at this age and the protective effect of conventional gene deletion in adults [39, 45].
This lack of effect suggests that selective gene deletion in neurons is insufficient to alter the extent of pan-cellular focal lesions produced in this HI model in mice at PND 9-60 [38, 48] and indeed, increasing evidence implies that p53 impacts glial function in development and neurodegeneration [49]. Enhanced expression of p53 in astrocytes has been reported 7 days following adult cerebral ischemia in rodents [50] and nitric oxide-induced cell death in astrocytes is mediated via p53 [51]. Overexpression of p53 has also been implicated in oligodendrocyte cell death in vitro and p53 expression is strongly associated with oligodendrocyte destruction in lesions of adult patients with multiple sclerosis [52, 53]. In addition to an essential role in cell death, p53 is also associated with oligodendrocyte precursor cell (OPC) development, where the optic nerve in Trp53 knockout mice at PND 7 showed an increase in OPC number but a decrease in the number of oligodendrocytes [54].
In models of demyelination, genetic knock down (Trp53 knockout mice) or pharmacological inhibition (PFT-α) of p53 resulted in preservation of myelinated fibres and increased oligodendrocyte survival [55]. This study also found decreased microglial activation and recruitment to the site of injury (corpus callosum) [55]. P53 expression in microglia has been shown to promote a pro-inflammatory phenotype that is damaging to neuronal synapses [56]. Numerous studies have also found that reduced expression of p53, such as in Trp53 knockout mice or following inhibition (PFT-μ), results in increased expression of anti-inflammatory genes and functions leading to tissue repair and phagocytosis, suggesting that in microglia, p53 modulates the activation state [33, 55, 57]. These studies indicate that the role of p53 in other cell types can significantly contribute to the progression of injury, and could explain the lack of protection following neuronal knockdown of Trp53. Additionally, this could have a greater impact in the adult and ageing brain.
Our findings strongly suggest that the CaM2Kα, from the T29-1 line, is not the ideal choice for neonatal experiments considering Cre, under the promoter of CamK2α, is not active till after PND 14. Whilst these experiments were being conducted the online cre repository was being characterized as a global resource [58]. As a consequence, it was recognized that Cre inherited from the maternal or paternal line can differ in expression, although we did not see this. In addition, it is important to recognize that Cre expression can differ between litter mates and other organs may be affected, all of which are important considerations when choosing a Cre line.
The partial neuroprotection we found in Trp53 het mice in response to moderate HI (50 min HI) is more subtle and limited to the posterior part of the brain in response to moderate degrees of HI when compared with our previous demonstration that PFT-μ reduces brain injury globally after neonatal HI [31]. The degree of neuroprotection exerted by PFT-μ was considerably more pronounced than by heterozygote Trp53 gene deficiency. This discrepancy could be due to that the degree of gene deletion was only partial (comparing Trp53 het vs. Trp53 WT) and that the cerebroprotective effect of PFT-μ is complex and not solely explained by its blockage of p53 interaction at the outer mitochondrial membrane. PFT-μ interacts with HSPs [34] and modulates the immuno-inflammatory response [33] which certainly could contribute to its neuroprotective efficacy in HI. In addition to PFT-μ, PFT-α has been used to inhibit the transcriptional activity of p53 which results in decreasing p53 DNA binding activity [59, 60]. Treatment with PFT-α given prior to [61], 1-6h following [62], or 6–9 days [63] following adult cerebral ischemia resulted in neuroprotection.
In summary, we have found that partial knockdown of Trp53 is protective following a moderate hypoxic-ischemic insult in posterior regions of the immature brain. More severe insults and conditional neuronal knockdown in the juvenile brain did not show any level of protection. Whilst p53 is a key mediator of neuronal cell death, acting upstream of Bax-dependent MOMP in the neonatal brain, further studies evaluating mediators of p53 activation could reveal new therapeutic targets.
MATERIALS AND METHODS
Ethics statement
All animal experimentation conducted in accordance with Department of Agriculture (Jordbruksverket, Sweden) and approved by the Gothenburg animal ethics committee (Application 52-2012).
Animals
Mice were bred in-house, kept in a 12 hour light/dark cycle, and fed standard laboratory chow diet and drinking water, ad libitum (Experimental Biomedicine, University of Gothenburg, Sweden). All mice were purchased from The Jackson Laboratory.
Traditional Trp53 Knockouts
Trp53 heterozygote (Het; +/-) mice (Stock #002101; B6.129S2-Trp53tm1Tyj/J) [44] were bred together or with wild types (WT; +/+ [generated from Trp53 het-het cages]) to produce mixed genotypes; Trp53 knockout (KO; -/-), Trp53 het and Trp53 WT. Trp53 knockout mice tend to develop tumors in early life and were therefore not used in these studies. Mixed litters (Trp53 het and Trp53 WT) were used for HI experiments as previously described in adult experiments on ischemia [39].
Cre-LoxP conditional knockouts
Neuronal specific knockdown of Trp53 was achieved utilizing Cre-LoxP technology. Briefly, LoxP mice containing the (floxed) Trp53 allele (Lox; Cre-/- Lox+/+, strain #008462; B6.129P2-Trp53tm1Brn/J) were bred with mice expressing Cre recombinase under the promoter of calcium/calmodulin-dependent protein kinase II alpha, of the T29-1 line (Cre; Cre+/+ Lox-/-, CaMK2α-Cre T29-1, Stock# 005359; B6.Cg-Tg(CaMK2α-cre)T29-1Stl/J). First generation (Cre+/+ Lox-/- x Cre-/- Lox+/+) breeding resulted in heterozygote Cre-Lox mice (Cre+/- Lox+/-). These were subsequently bred with Lox mice to produce a second generation of mixed genotypes [Het Cre-Lox (Cre+/- Lox+/-), Hom Lox (Cre-/- Lox+/+), Het Lox (Cre-/- Lox+/-) and neuronal specific KO of Trp53 (KO Lox Trp53f/+; Cre+/- Lox+/+). These KO Lox Trp53f/+ were bred with Lox mice to generate mixed litters: WT Lox Trp53+/+ (Cre-/- Lox+/+) and KO Lox Trp53f/+ (Cre+/- Lox+/+) that were used for HI experiments.
Cre-ROSA26 characterization
Currently the online cre portal (http://www.creportal.org, http://www.informatics.jax.org/allele/MGI:2177650) only contains information regarding cre recombinase activity at ages Embryonic (E) 10.5, E15.5, PND 7 and PND 56. To further characterize CaMK2α-cre T29-1 cre activity we bred with ROSA26 LacZ reporter mice (Rosa26+/+; strain #003474 (B6.129S4-Gt(ROSA)26Sortm1Sor/J) [58], which generated Cre+/- Rosa26+/-. Both male and female Cre mice were used to breed with Rosa26 reporter mice to further assess whether maternal or paternal cre inheritance was different [58]. At PND 7, PND 14, PND 19 and PND 29 brains were harvested and frozen at -80°C. Coronal sections were cut on a cryostat at 10 μm. β-galactosidase staining was done following manufacturer instructions (Cat number: K1465-01, Invitrogen, Sweden). Sections were visualized on a Zeiss Microscope (Model Axio Imager 2, Zeiss, Germany) and captured on an AxioCam MRm Camera (Zeiss, Germany) using Zen Software (Zeiss, Germany).
Genotyping
The genotype of WT, Het and KO mice were determined by polymerase chain reaction (PCR) of genomic DNA extracted from tail clips by RedExtract-N-AMP Tissue PCR Kit (Sigma-Aldrich, USA). All primers were ordered from Eurofins MWG GmbH (Ebersberg, Germany). For traditional Trp53 transgenic mice; each PCR reaction (20μl) contained 2 μl (<250 ng DNA) and was made following KAPA Taq DNA Polymerase manufacturer’s instructions (KAPA BioSystems, USA) and primer concentrations of 0.08 μM IMR7777, 1μM IMR7778, 1.17 μM IMR8306. For Cre-Lox and Rosa26 genotyping: each PCR reaction (10 μl) contained 1 μl (50 – 100 ng) of genomic DNA, 5 μl of REDExtract-N-Amp PCR Reaction mix and all primers were used at a concentration of 1 μM. The PCR products were separated on a 1.5% agarose/0.5 x Tris-borate-ethylenediaminetetraacetic acid gel containing SYBR® Safe DNA gel stain (Invitrogen, Life Technologies Sweden). Primer details, PCR programs and expected results are listed in Table 1.
Table 1: Primers and PCR programs used for genotyping transgenic mice
Gene |
Sequence 5’ → 3’ |
Concentration (μM) |
PCR Program |
Result |
||
|---|---|---|---|---|---|---|
P53 |
IMR7777 |
WT Forward |
ACA GCG TGG TGG TAC CTT AT |
0.08 |
94°C for 3 min 35 cycles: [94°C for 30 sec; 66°C for 60 sec; 72°C for 90 sec] 72°C for 2 minutes. |
WT: 450 bp Het: 450bp and 650bp Mutant: 650 bp |
IMR8306 |
Mutant Forward |
CTA TCA GGA CAT AGC GTT GG |
1.17 |
|||
IMR7778 |
Common |
TAT ACT CAG AGC CGG CCT |
1 |
|||
Cre |
IMR1084 |
Transgene |
GCG GTC TGG CAG TAA AAA CTA TC |
1 |
94°C for 5 min 35 cycles: [94°C for 30 sec; 52°C for 60 sec; 72°C for 60 sec] 72°C for 2 min |
Transgene: ~100bp Internal positive control: 324 bp |
IMR1085 |
Transgene |
GTG AAA CAG CAT TGC TGT CAC TT |
1 |
|||
IMR7338 |
Internal positive control Forward |
CTA GGC CAC AGA ATT GAA AGA TCT |
1 |
|||
IMR7339 |
Internal positive control Reverse |
GTA GGT GGA AAT TCT AGC ATC ATC C |
1 |
|||
P53 Lox |
IMR8543 |
Forward |
GGT TAA ACC CAG CTT GAC CA |
1 |
94°C for 5 min 35 cycles: [94°C for 30 sec; 56°C for 60 sec; 72°C for 60 sec] 72°C for 5 min |
WT: 270 bp Het: 270 bp and 390 bp Mutant: 390 bp |
IMR8544 |
Reverse |
GGA GGC AGA GAC AGT TGG AG |
1 |
|||
Rosa26 |
IMR8545 |
WT Reverse |
GGA GCG GGA GAA ATG GAT ATG |
1 |
94°C for 3 min 35 cycles: [94°C for 30 sec; 65°C for 60 sec; 72°C for 60 sec] 72°C for 2 min |
WT: ~650 bp Het: 340 bp and ~650 bp Mutant: 340 bp |
IMR8052 |
Mutant Reverse |
GCG AAG AGT TTG TCC TCA ACC |
1 |
|||
IMR8545 |
Common |
AAA GTC GCT CTG AGT TGT TAT |
1 |
In situ hybridization
Mouse Trp53 complementary DNA was cloned from mouse brain total RNA by reverse transcription–polymerase chain reaction (Multiscribe; Applied Biosystems), as recommended by the manufacturer. Second-round amplification was performed with REDTaq (Sigma-Aldrich, Dorset, United Kingdom) and 0.5 μmol/L forward (5′-GGC-AAC-TAT-GGC-TTC-CAC-C-3′) and reverse (5′-CTC-CGT-CAT-GTG-CTG-TGA-C-3′) primers (based on accession number NM_001127233) using the following cycling parameters: 95°C for 5 minutes, 1 cycle; 95°C for 30 seconds; 65°C for 30 seconds; 72°C for 1 minute, 30 cycles; 72°C for 5 minutes. Each complementary DNA product was directionally cloned into TOPO II plasmid (Life Technologies, Carlsbad, CA), which incorporates T7 and SP6 RNA polymerase promoters flanking the cloning region defined by the HindIII or Xba 1 restriction enzyme site. Plasmids were sequenced, linearized, and transcribed with T7 (antisense) or SP6 (sense) RNA polymerases (Sigma) to yield digoxigenin-dUTP–labelled riboprobes in accordance with the manufacturer’s protocol (Ambion, Life Technologies). Transcription was performed for 24 hours at 37°C. The template complementary DNA was digested away by RNase-free DNase (2 U/μL, 15 minutes), and the riboprobes were purified (Megaclear Purification and Filtration System; Ambion, UK) and quantified by spectrophotometry and electrophoresis.
In situ hybridization procedure
WT Lox Trp53+/+ and KO Lox Trp53f/+ pups were killed at PND 7, PND 12 and at 6 weeks of age, perfused with 0.9% physiological saline, followed by 5% buffered formalin. Brains were then paraffin embedded and 6 μm sections were cut at the level of the hippocampus. The in situ hybridization procedure was conducted as previously described [64], with overnight incubation occurring at 56°C and the 2× saline-sodium citrate washes occurring for 30 minutes at 55°C. Adjacent slides incubated with the sense probe and hybridization buffer alone were used as negative controls for specificity. Sections were visualized on a Leica Microscope (Model DM6000B, Leica Microsystems, UK) and captured on a MBF Bioscience Camera (Model Cx9000, MBF Bioscience, USA) using Stereo Investigator software (v5.65, MBF Bioscience, USA).
Induction of hypoxia-ischemia in neonatal and juvenile mice
Traditional Trp53 knockout mice at PND 9 and Cre-Lox mice at PND 12 and PND 28 were subjected to unilateral HI, according to the Rice–Vannucci model as previously described [36, 38, 65]. Briefly, the left common carotid artery was isolated and ligated. Pups recovered for 1 h in the parent cage. The litters were then placed in a chamber with a humidified hypoxic gas mixture (10% oxygen in nitrogen, 36°C) for PND 9 mice, 50 min (moderate hypoxia) or 60 min (severe hypoxia), PND 12 for 50 min, and PND 28 mice for 40 min. After hypoxic exposure, pups were returned to their biological dams until the conclusion of the experiment (7d post-HI). Mortality rates from the end of HI to +7 days post-HI were low: PND 9; 50 min and 60 min = 0; PND 12 for 50 min HI = 1/41 and PND 28 40 min HI = 2/28. The genotypes of these mice pups was not determined.
Brain injury assessment
Pups were killed, tail samples were collected for genotyping and mice were perfused intracardially with 0.9% physiological saline followed by 5% buffered formaldehyde (Histolab; Histolab AB, Sweden) as previously described [14, 66]. Paraffin-embedded tissue blocks were serially cut into 7 μm coronal sections on Superfrost® plus slides (Thermo Scientific, Sweden) with every 50th section stained. Microtubule associated protein-2 (MAP-2), a neuronal marker expressed in neurons and dendrites, and myelin basic protein (MBP), a major constituent of the myelin sheath of oligodendrocytes where loss of staining indicated infracted areas and MBP associated white matter loss respectively as previously described [14, 66]. Images were taken on a Nikon Optiphot-2 (Nikon, Japan) using an AVT Dolphin camera (Model 1 F145g, Nikon Germany). Positive stained areas were measured using the Olympus Micro Image analysis software system (V4.0, Olympus Optical, Tokyo, Japan).
Loss of MAP-2 and MBP staining was determined by subtracting the MAP-2 or MBP-positive area in the ipsilateral (injured) hemisphere from the contralateral (non-injured) hemisphere, and values expressed as a percentage of tissue loss from the contralateral hemisphere. Total volume loss was calculated according to the Cavalieri Principle; V = ΣA x P x T, where V = total volume, ΣA = sum of the areas, P = the inverse of the sections sampling fraction and T = section thickness. MBP staining was measured in the subcortical white matter region, at one level of the brain (approximately at the level of Bregma -2.12 mm) [67].
Neuropathology was evaluated by an assessor blinded to genotype. A modified semi-quantitative scoring scale [36, 68] was used to assess injury in the cortex, hippocampus, posterior hippocampus, striatum and thalamus. Each region was graded for degree of atrophy 0 to 3 (0 being no atrophy) and cellular infarction and necrosis 0 to 3 (0 indicating no injury), resulting to each region receiving a score between 0 – 6.
Data analysis
Data are expressed as mean ± SEM. All statistical analysis was performed on GraphPad Prism 6 Software (GraphPad Software, San Diego, USA), with statistical significance set at p< 0.05. Loss of MAP-2, loss of MBP, volume tissue loss and neuropathology score were assessed by a student’s t-test. Loss of MAP-2, separated for sex was assessed by a One-Way ANOVA for each region. If significant, a Tukey’s post-hoc analysis was conducted.
ACKNOWLEDGMENTS
We gratefully acknowledge Pernilla Svedin and Anna-Lena Leverin for their expert technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
We gratefully acknowledge the support of the Department of Perinatal Imaging and Health and financial support from Wellcome Trust (WT094823), the Swedish Medical Research Council (VR 2015-02493), Governmental grants to University Hospitals (ALFGBG-426401), Action Medical Research, ERA-net (EU;VR 529-2014-7551), Hjärnfonden (Brain Foundation 2015-0004), the Leducq Foundation (DSRRP34404) and the Wilhelm and Martina Lundgren Foundation (HH, AB), to enable this study to be completed. In addition, the authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre Award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust.
REFERENCES
1. Evans K, Rigby AS, Hamilton P, Titchiner N, Hall DM. The relationships between neonatal encephalopathy and cerebral palsy: a cohort study. J Obstet Gynaecol. 2001; 21: 114-20.
2. Ellis M, Manandhar N, Manandhar DS, Costello AM. Risk factors for neonatal encephalopathy in Kathmandu, Nepal, a developing country: unmatched case-control study. BMJ. 2000; 320: 1229-36.
3. Pappas A, Shankaran S, McDonald SA, Vohr BR, Hintz SR, Ehrenkranz RA, Tyson JE, Yolton K, Das A, Bara R, Hammond J, Higgins RD, Hypothermia Extended Follow-up Subcommittee of the Eunice Kennedy Shriver NNRN. Cognitive outcomes after neonatal encephalopathy. Pediatrics. 2015; 135: e624-34.
4. Azzopardi D, Strohm B, Marlow N, Brocklehurst P, Deierl A, Eddama O, Goodwin J, Halliday HL, Juszczak E, Kapellou O, Levene M, Linsell L, Omar O, et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med. 2014; 371: 140-9.
5. Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 2014; 13: 217-32.
6. Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011; 10: 372-82.
7. Northington FJ, Zelaya ME, O’Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM, Martin LJ. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience. 2007; 149: 822-33.
8. Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997; 378: 70-87.
9. Thornton C, Rousset CI, Kichev A, Miyakuni Y, Vontell R, Baburamani AA, Fleiss B, Gressens P, Hagberg H. Molecular mechanisms of neonatal brain injury. Neurol Res Int. 2012; 2012: 506320.
10. Ginet V, Spiehlmann A, Rummel C, Rudinskiy N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC, Puyal J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy. 2014; 10: 846-60.
11. van den Tweel ER, Nijboer C, Kavelaars A, Heijnen CJ, Groenendaal F, van Bel F. Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia-ischemia in the neonatal rat brain. J Neuroimmunol. 2005; 167: 64-71.
12. Puka-Sundvall M, Gajkowska B, Cholewinski M, Blomgren K, Lazarewicz JW, Hagberg H. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res Dev Brain Res. 2000; 125: 31-41.
13. Gilland E, Puka-Sundvall M, Hillered L, Hagberg H. Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature rat brain: involvement of NMDA-receptors. J Cereb Blood Flow Metab. 1998; 18: 297-304.
14. Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, Forte MA, Hagberg H. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci. 2009; 29: 2588-96.
15. Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, Nilsson M, Eriksson PS, Hagberg H, Culmsee C, Plesnila N, Kroemer G, Blomgren K. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007; 14: 775-84.
16. Gibson ME, Han BH, Choi J, Knudson CM, Korsmeyer SJ, Parsadanian M, Holtzman DM. BAX contributes to apoptotic-like death following neonatal hypoxia-ischemia: evidence for distinct apoptosis pathways. Mol Med. 2001; 7: 644-55.
17. Wang X, Han W, Du X, Zhu C, Carlsson Y, Mallard C, Jacotot E, Hagberg H. Neuroprotective effect of Bax-inhibiting peptide on neonatal brain injury. Stroke. 2010; 41: 2050-5.
18. Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009; 10: 481-94.
19. Pirianov G, Brywe KG, Mallard C, Edwards AD, Flavell RA, Hagberg H, Mehmet H. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. J Cereb Blood Flow Metab. 2007; 27: 1022-32.
20. Carlsson Y, Schwendimann L, Vontell R, Rousset CI, Wang X, Lebon S, Charriaut-Marlangue C, Supramaniam V, Hagberg H, Gressens P, Jacotot E. Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann Neurol. 2011; 70: 781-9.
21. Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol. 2005; 17: 631-6.
22. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008; 9: 402-12.
23. Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000; 275: 16202-12.
24. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009; 458: 1127-30.
25. Marchenko ND, Moll UM. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle. 2007; 6: 1718-23.
26. Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010; 20: 14-24.
27. Hedtjarn M, Mallard C, Eklind S, Gustafson-Brywe K, Hagberg H. Global gene expression in the immature brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2004; 24: 1317-32.
28. Bolouri H, Savman K, Wang W, Thomas A, Maurer N, Dullaghan E, Fjell CD, Ek CJ, Hagberg H, Hancock RE, Brown KL, Mallard C. Innate defense regulator peptide 1018 protects against perinatal brain injury. Ann Neurol. 2014; 75: 395-410.
29. Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. Strong neuroprotection by inhibition of NF-kappaB after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke. 2008; 39: 2129-37.
30. Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke. 2008; 39: 2578-86.
31. Nijboer CH, Heijnen CJ, van der Kooij MA, Zijlstra J, van Velthoven CT, Culmsee C, van Bel F, Hagberg H, Kavelaars A. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann Neurol. 2011; 70: 255-64.
32. Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R, Komarova EA, Gudkov AV. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006; 2: 474-9.
33. Fleiss B, Chhor V, Rajudin N, Lebon S, Hagberg H, Gressens P, Thornton C. The Anti-Inflammatory Effects of the Small Molecule Pifithrin-micro on BV2 Microglia. Dev Neurosci. 2015; 37: 363-75.
34. Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009; 36: 15-27.
35. Nijboer CH, Heijnen CJ, Willemen HL, Groenendaal F, Dorn GW, 2nd, van Bel F, Kavelaars A. Cell-specific roles of GRK2 in onset and severity of hypoxic-ischemic brain damage in neonatal mice. Brain Behav Immun. 2010; 24: 420-6.
36. Hedtjarn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002; 22: 5910-9.
37. Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996; 87: 1317-26.
38. Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell death and Differ. 2005; 12: 162-76.
39. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012; 149: 1536-48.
40. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005; 102: 12005-10.
41. Matsumoto S, Friberg H, Ferrand-Drake M, Wieloch T. Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1999; 19: 736-41.
42. Puka-Sundvall M, Gilland E, Hagberg H. Cerebral hypoxia-ischemia in immature rats: involvement of mitochondrial permeability transition? Dev Neurosci. 2001; 23: 192-7.
43. Chavez-Valdez R, Martin LJ, Northington FJ. Programmed Necrosis: A Prominent Mechanism of Cell Death following Neonatal Brain Injury. Neurol Res Int. 2012; 2012: 257563.
44. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994; 4: 1-7.
45. Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994; 14: 887-91.
46. Allen Institute for Brain Science. (2015). Allen Developing Mouse Brain Atlas. Available from: http://developingmouse.brain-map.org.
47. Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000; 26: 133-5.
48. Zhang P, Lei X, Sun Y, Zhang H, Chang L, Li C, Liu D, Bhatta N, Zhang Z, Jiang C. Regenerative repair of Pifithrin-alpha in cerebral ischemia via VEGF dependent manner. Sci Rep. 2016; 6: 26295.
49. Jebelli JD, Hooper C, Garden GA, Pocock JM. Emerging roles of p53 in glial cell function in health and disease. Glia. 2012; 60: 515-25.
50. Chung YH, Shin CM, Kim MJ, Lee EY, Kim G, Cha CI. Enhanced expression of p53 in reactive astrocytes following transient focal ischemia. Neurol Res. 2002; 24: 324-8.
51. Yung HW, Bal-Price AK, Brown GC, Tolkovsky AM. Nitric oxide-induced cell death of cerebrocortical murine astrocytes is mediated through p53- and Bax-dependent pathways. J Neurochem. 2004; 89: 812-21.
52. Ladiwala U, Li H, Antel JP, Nalbantoglu J. p53 induction by tumor necrosis factor-alpha and involvement of p53 in cell death of human oligodendrocytes. J Neurochem. 1999; 73: 605-11.
53. Wosik K, Antel J, Kuhlmann T, Bruck W, Massie B, Nalbantoglu J. Oligodendrocyte injury in multiple sclerosis: a role for p53. J Neurochem. 2003; 85: 635-44.
54. Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, Raff M. Roles for p53 and p73 during oligodendrocyte development. Development. 2004; 131: 1211-20.
55. Li J, Ghiani CA, Kim JY, Liu A, Sandoval J, DeVellis J, Casaccia-Bonnefil P. Inhibition of p53 transcriptional activity: a potential target for future development of therapeutic strategies for primary demyelination. J Neurosci. 2008; 28: 6118-27.
56. Jebelli J, Hooper C, Pocock JM. Microglial p53 activation is detrimental to neuronal synapses during activation-induced inflammation: Implications for neurodegeneration. Neurosci Lett. 2014; 583: 92-7.
57. Jayadev S, Nesser NK, Hopkins S, Myers SJ, Case A, Lee RJ, Seaburg LA, Uo T, Murphy SP, Morrison RS, Garden GA. Transcription factor p53 influences microglial activation phenotype. Glia. 2011; 59: 1402-13.
58. Heffner CS, Herbert Pratt C, Babiuk RP, Sharma Y, Rockwood SF, Donahue LR, Eppig JT, Murray SA. Supporting conditional mouse mutagenesis with a comprehensive cre characterization resource. Nat Commun. 2012; 3: 1218.
59. Zhu X, Yu QS, Cutler RG, Culmsee CW, Holloway HW, Lahiri DK, Mattson MP, Greig NH. Novel p53 inactivators with neuroprotective action: syntheses and pharmacological evaluation of 2-imino-2,3,4,5,6,7-hexahydrobenzothiazole and 2-imino-2,3,4,5,6,7-hexahydrobenzoxazole derivatives. J Med Chem. 2002; 45: 5090-7.
60. Nayak SK, Panesar PS, Kumar H. p53-Induced apoptosis and inhibitors of p53. Curr Med Chem. 2009; 16: 2627-40.
61. Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001; 77: 220-8.
62. Leker RR, Aharonowiz M, Greig NH, Ovadia H. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp Neurol. 2004; 187: 478-86.
63. Luo Y, Kuo CC, Shen H, Chou J, Greig NH, Hoffer BJ, Wang Y. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann Neurol. 2009; 65: 520-30.
64. Vontell R, Supramaniam V, Wyatt-Ashmead J, Gressens P, Rutherford M, Hagberg H, Thornton C. Cellular mechanisms of toll-like receptor-3 activation in the thalamus are associated with white matter injury in the developing brain. J Neuropathol Exp Neurol. 2015; 74: 273-85.
65. Rice JE, 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Annals of neurology. 1981; 9: 131-41.
66. Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009; 183: 7471-7.
67. Svedin P, Hagberg H, Savman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. The Journal of neuroscience. 2007; 27: 1511-8.
68. Bona E, Hagberg H, Loberg EM, Bagenholm R, Thoresen M. Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: short- and long-term outcome. Pediatr Res. 1998; 43: 738-45.