

Introduction
The worldwide incidence of skin cancer, including non melanoma skin cancer (NMSC) and cutaneous melanoma (CM), is rapidly increasing, being the most common human cancer[1]. While, both entities are influenced by the carcinogenic effect of UV exposure, their incidence and tumor aggressiveness differ considerably, CM being the least frequent and most aggressive form[2] .
Approximately, 10% of total melanoma cases originate in individuals belonging to high-risk melanoma pedigrees. To date, CDKN2A is the major gene associated with the high risk inherited in melanoma prone families [3] and in multiple primary melanoma patients [4]. CDKN2A acts as a tumor suppressor gene, negatively regulating cell cycle progression and promoting cellular senescence. Recently, a role for CDKN2A in cellular oxidative stress regulation has been suggested [5, 6]. Recurrent CDKN2A mutations have been identified in melanoma families [7-11], mutation p.G101W being the most frequent one detected in Mediterranean pedigrees [12]. Although heterozygous loss of CDKN2A is sufficient to confer a 67% lifetime risk of melanoma [13], the mechanisms responsible for tumor enhancement still have to be clarified [14]. In contrast, the role of high-penetrance CDKN2A mutations in NMSC susceptibility has not been clearly elucidated [15, 16].
Skin cancer epidemiology is complex due to the multigenic nature of the disease. In particular, the MC1R gene is a key regulator of skin pigmentation and melanocyte differentiation, playing a central role in determining the pigmentation phenotype, sun sensitivity and tanning ability[17]. Certain MC1R polymorphisms are responsible for the red hair color (RHC) phenotype (red hair, fair skin and poor tanning response)[18] which is associated with high UV-radiation sensitivity and skin cancer susceptibility (CM and NMSC)[19, 20]. Furthermore, MC1R variants are a modifying factor for melanoma risk in CDKN2A mutation carriers, [21-23] suggesting that multiple medium- and low-penetrance genes may influence the risk conferred by high penetrance melanoma genes.
To date, there is a lack of information about the mechanisms underlying the increased skin cancer risk in carriers of CDKN2A mutations in association with MC1R variants. The effect of both genes has been separately investigated using murine-derived melanocytes and/or focused on mono-cultured melanocytes [6, 24-26]. However, these mono-cultures exhibit phenotypic characteristics closely mimicking those observed in transformed melanocytes, such as elevated growth rate [27, 28] and acquired expression of melanoma-associated antigens[29, 30] which are abolished when they are co-cultured with keratinocytes[30-32]. This data indicates that melanocyte proliferation and behavior is controlled by surrounding basal keratinocytes which regulate epidermal homeostasis [33, 34]. Thus, the establishment and characterization of a melanocyte–keratinocyte co-culture system is essential for elucidating the early molecular events leading to skin cancer.
The aim of the present study was to investigate the global molecular effect of germinal CDKN2A mutations (p.G101W) and MC1R RHC variants in the transcriptome of primary skin cells from individuals belonging to skin cancer prone families (familial melanoma pedigrees).
Results
Impact of CDKN2A germinal mutation p.G101W on Global Transcript Profiles of skin primary cultures:
Whole genome expression profiling was performed on melanoctye-keratinocyte co-cultures from CDKN2A mutation carriers (samples A1 and B1 described in Table 1) and wild-type CDKN2A individuals (samples A2 and B2). After stringent microarray data filtering, 1536 of the initial 19596 transcripts on the array were found to be differentially expressed in CDKN2A mutated cultured cells. Specifically, 60.7% of transcripts were found to be up-regulated (933/1536) and 39.3% of them down-regulated (603/1536).
Table 1: Genotype and phenotype features of four individuals from two melanoma pedigrees.
Pedigree A |
||||||
Individual |
MM* |
CDKN2A |
MC1R |
Skin phototype |
Eye color |
Hair color |
A1 |
5 |
p.G101W |
p.R160W, p.R151C |
II |
Green |
Red |
A2 |
0 |
Wild-type |
p.R160W, p.R151C |
II |
Green |
Red |
Pedigree B |
||||||
Individual |
MM* |
CDKN2A |
MC1R |
Skin phototype |
Eye color |
Hair color |
B1 |
0 |
p.G101W |
Wild-type |
II |
Blue |
Black |
B2 |
0 |
Wild-type |
Wild-type |
II |
Blue |
Brown |
Two siblings from each pedigree (A and B) were included. Each individual was considered as a “genomic condition” in the study. A total of two and three melanoma cases have been found in pedigree A and B, respectively. Abbreviations: CDKN2A: cyclin-dependent kinase inhibitor 2A; MC1R: melanocortin 1 receptor; MM: melanomas. * Number of melanomas in each individual.
Class comparison generated a signature of 108 unique genes that significantly discriminated between CDKN2A mutation and non-mutation (p-value <0.0001, listed in Table S1). Functional categorization of those transcripts belonging to known genes (Figure 1a, Figure 1b) showed that the mutant CDKN2A signature included deregulation of a vast number of genes involved in cell component and metabolism (37% up-regulated and 42% down-regulated,). Notably, immunomodulation and interferon response genes represented the second most prevalent subset of up-regulated transcripts (24%) and third down-regulated subgroup (13%) which may reflect a constitutively altered cytokine and chemokine profile in mutant cells. The set of deregulated genes involved in immunomodulation and interferon response includes: CLEC2B, IFI44L, IFIT1, IFI44, IFI27, IFNK, DDX58, RNF182, IFIT2, IL15, SP110, BTN3A2, RFX2, IL17D and HLA-DPB1 (Figure 2). The molecular profile also included deregulation of genes which may confer a phenotype with the capability for malignant transformation since they are involved in cell adhesion, cell growth and proliferation. Over-expression of apoptosis related genes CASP3, XAF1 and SAMD9 was also detected. Transcription factors such as CEBPZ, LEF1, MACC1, TBX1, ZNF404 and ZNF8, epidermal differentiation and melanogenesis genes such as KRT2, TYPR1, LIMS3, EEA1 and MFI2 were also components of the mutant CDKN2A signature.
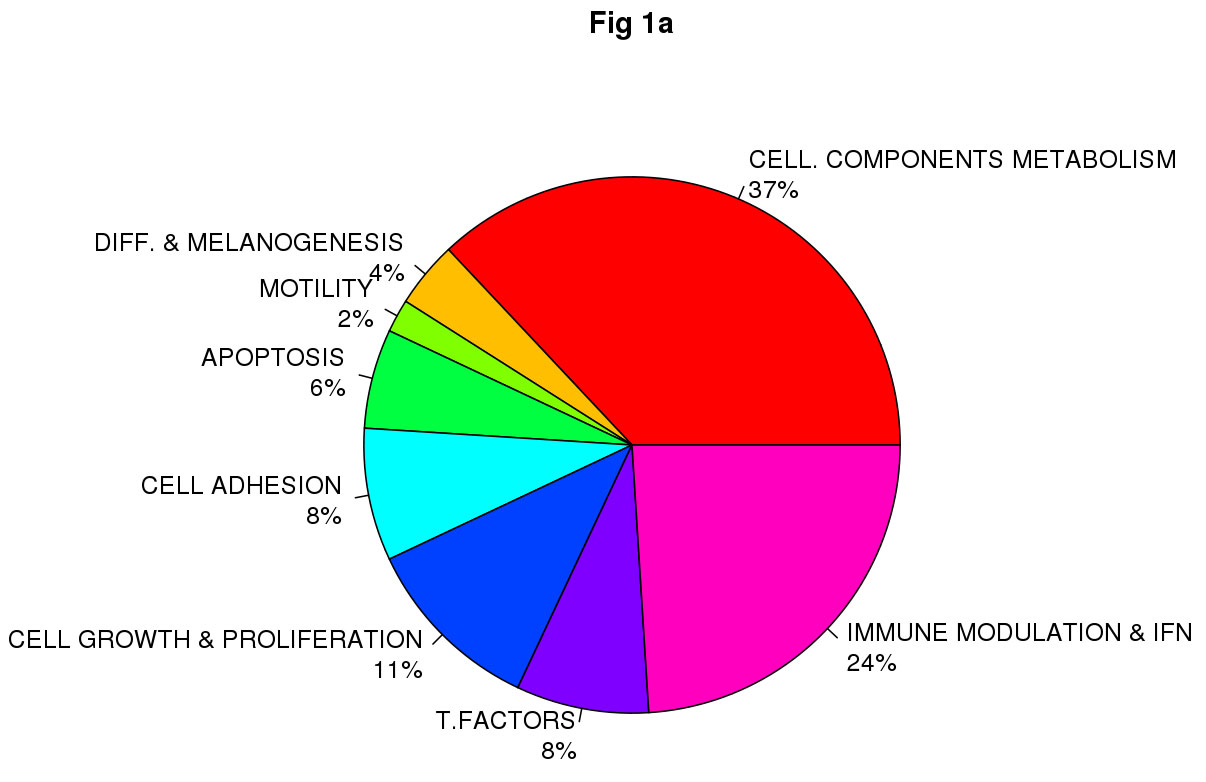
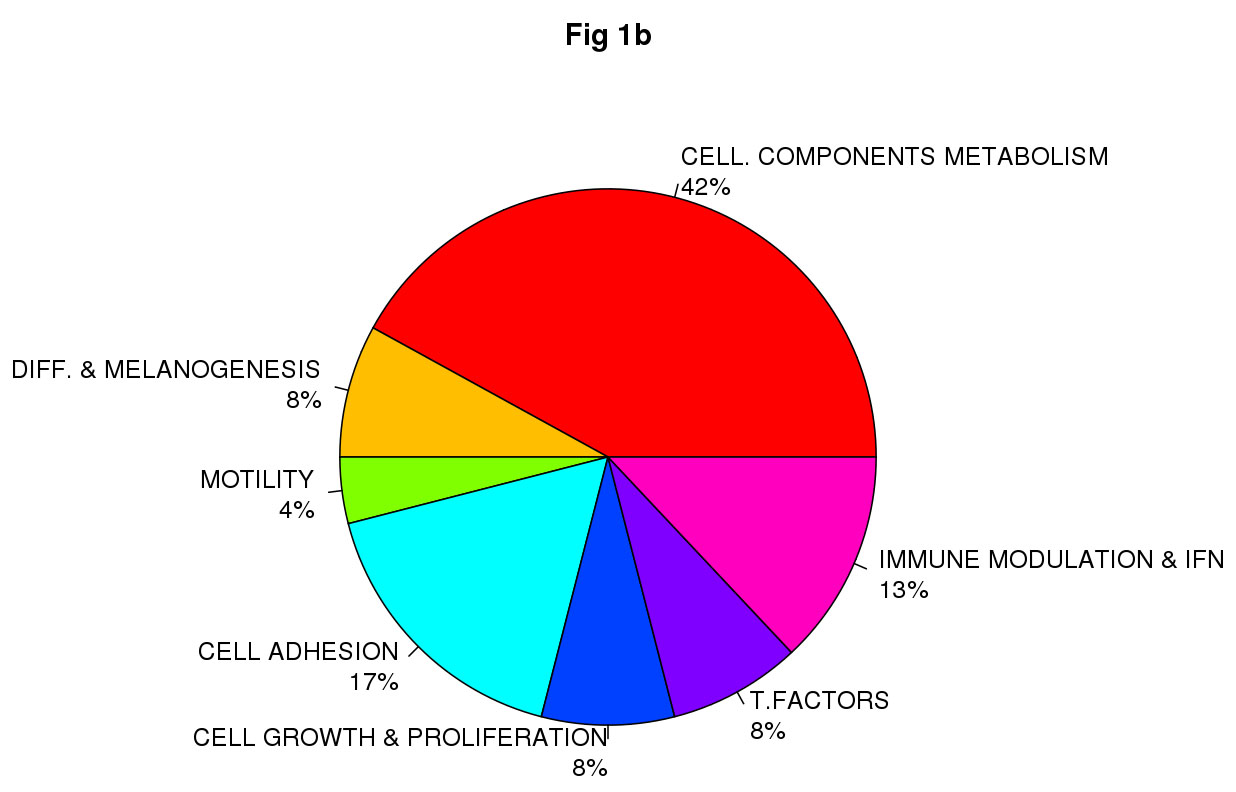
Figure 1: Impact of germinal CDKN2A mutation on global transcript profile in melanocyte-keratinocyte co-culture system. Functional classification of the set of 78 of 108 deregulated genes in CDKN2A mutants are indicated in Fig.1a Up-regulated genes and Fig.1b Down-regulated genes. T. Factors (transcription factors), Immune modulation & IFN (Immunomodulation and interferon related genes); Cell. Components and metabolism (Cellular components and metabolism), Diff & Melanogenesis (differentiation and melanogenesis)
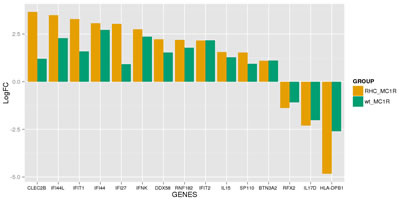
Figure 2: Set of immunomodulation and interferon response deregulated in mutant CDKN2A co-cultures. The logarithm of the Fold change (LogFC) for the set of genes is indicated for wild-type MC1R co-cultures (wt_MC1R; green) and for Red Hair Colour MC1R variants co-cultures (RHC_MC1R; yellow).
Further examination by functional enrichment analysis revealed an over-representation of down-regulated genes from the Notch signaling pathway (hsa_04330; p=0.038) in mutant CDKN2A skin cells including NCOR2, PSEN, DLLC1, DVL3, NOTCH3, RFNG and the co-activator MAML1 which controls the growth-promoting effect of Notch1.
Impact of non-functional MC1R gene on Global Transcript Profiles of skin primary cultures:
MC1R analysis was carried out following the same workflow used in the CDKN2A analysis. Whole genome expression profiling displayed 3570 transcripts differentially expressed in RHC variant (A1 and A2) vs. wild-type MC1R skin cells (B1 and B2). Specifically, 54% of transcripts (1954/3570) were up-regulated while 46% (1616/3570) were down-regulated. By class comparison, 159 genes showed highly statistically significant differences (p < 0.0000001) (listed in Table S2). RHC skin cells showed a down-regulation of a subset of genes categorized as melanocyte differentiation and pigmentation genes. The transcription of most of these genes is regulated directly via the MC1R signaling pathway, or indirectly through keratinocyte differentiation genes (TYRP1, MLANA, TYR, CRNN, PADI1, GDF15, MLPH, KIT, INHBB, DAB2, KLK6, FOXC1, and NDRG4). Furthermore, RHC variant skin cells exhibited up-regulation of DNA damage and/or DNA repair response genes (BUB1, CHEK1, POLQ, RAD51AP1, RAD54B, RAD51C, BLM, UBE2T, GINS2, EXO1, FAM33A, PTTG2, PTTG1, RAD54L, DTL), cell cycle and proliferation genes (CDKN3, CDC25, CDK1, CCNA2, CDC20, CCNB2, CCNB1, CKS, CDCA3, PLK1, PRIM, CDCA8, GMNN, CDC6, VRK1, CKS1B, RFC4, CDC45, CENPN, HAUS8, PSRC, MCM6, MELK, AURKA, NCAPG) and microtubule-based motors kinesin superfamily genes (KIF11, KIF15, KIF2C, KIF4A,KIF23).
The functional enrichment analysis showed a considerably higher number of altered pathways in non-functional MC1R cells than in CDKN2A mutant cells. The RHC skin cells displayed over-expression of genes playing a role in major DNA repair and cell homeostasis pathways such as DNA replication, cell cycle and also oxidative phosphorylation which leads to production of reactive oxygen species (Figure 3 and Table 2). The lysosome and endocytosis pathways, which are intrinsically related to melanosome transfer from melanocytes to keratinocytes, were over-represented in the set of down-regulated transcripts as a direct consequence of low pigment production in RHC skin cells. Interestingly, RHC skin cells also exhibited an up-regulation of genes involved in neurodegenerative disorders such as Parkinson´s disease (hsa05012), Huntington´s disease (hsa5016) and Alzheimer´s disease (hsa05010). Although most of the genes were involved in several of these pathways, some of them were restricted to Huntington’s disease (AP2B1, CREB3L4, POLR2H and SOD1), to Alzheimer´s disease (NAE1, FAS) or to Parkinson’s disease (PARK7).
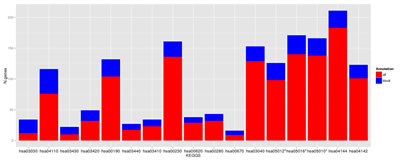
Figure 3: Deregulated pathways in RHC MC1R melanocyte-keratinocyte co-culture system. In blue are the number of total genes annotated within the pathway, in red are the number of deregulated genes in the RHC MC1R co-cultures. The KEGG term is indicated for each pathway (axis X). * Pathways related to neurodegenerative diseases.
Table 2: Deregulated pathways in RHC MC1R melanocyte-keratinocyte co-culture system.
KEGG Term |
STATUS |
Adj. P-Value |
DNA replication (hsa03030) |
UP |
3.83E-14 |
Cell cycle (hsa04110) |
UP |
7.87E-14 |
Mismatch repair (hsa 03430) |
UP |
1.56E-06 |
Nucleotide excision repair (hsa03420) |
UP |
7.01E-06 |
Oxidative phosphorylation (hsa00190) |
UP |
6.91E-05 |
Homologous recombination (hsa03440) |
UP |
6.98E-04 |
Base excision repair (hsa03410) |
UP |
1.07E-03 |
Purine metabolism (hsa00230) |
UP |
1.80E-02 |
Pyruvate metabolism (hsa00620) |
UP |
3.47E-02 |
Valine. leucine and isoleucine degradation (hsa00280) |
UP |
8.13E-03 |
One carbon pool by folate (hsa00670) |
UP |
2.69E-03 |
Spliceosome (hsa03040) |
UP |
1.80E-02 |
Parkinson's disease (hsa05012) |
UP |
3.10E-05 |
Hugtington's disease (hsa05016) |
UP |
5.25E-04 |
Alzheimer's disease (hsa 05010) |
UP |
3.03E-03 |
Endocytosis (hsa04144) |
DOWN |
3.83E-02 |
Lysosome (hsa04142) |
DOWN |
4.70E-03 |
The KEGG term is indicated for each pathway (X-axis). Status: UP: pathway overrepresented in the group of up-regulated transcripts in RHC MC1R; DOWN: pathway over-represented in the group of down-regulated transcripts in RHC MC1R.
Besides the lack of functionality caused by p.R151C and p.R160W variants, we also analyzed a possible effect on MC1R levels, by the relative quantification of MC1R expression. We observed a statistically significant reduction in MC1R expression in RHC skin cells (ddCt=0.06±0.02) compared to wild-type MC1R skin cells (ddCt=0.50±0.07; P-value<0.0001). Furthermore, the expression of FARP1, SLFN11, GFPT2, COL5A3, TYRP1, LEF1, KRT2, ST6GALNAC3, MLANA, MSMB, SILV, A2M, and ALOX5 was evaluated by RT-PCR confirming the results obtained in the array (data not shown).
We further examined the overlap between genes differentially expressed in both mutant CDKN2A (n=1536) and in non functional MC1R cells (n=3570). A total of 485 genes were altered in the same fashion in both mutant CDKN2A and non functional MC1R cells and the expression of 136 genes was found inversely correlated between both groups. Of these 136 genes (listed in Table S3), 30 were up-regulated in mutant CDKN2A and down-regulated in non functional MC1R cells and 106 genes were down-regulated in mutant CDKN2A and up-regulated in non functional MC1R cells (Figure 4). The functional analysis of these 136 genes did not reveal over-representation of any specific pathway.
Figure 4: Genes inversely expressed in mutant CDKN2A and RHC MC1R. 136 genes were found inversely expressed between mutant CDKN2A and RHC MC1R genes (listed in Table S3). A: RHC MC1R vs wt MC1R (both mutant CDKN2A); B: mutant CDKN2A vs wt CDKN2A (both RHC MC1R); C: mutant CDKN2A vs wt CDKN2A (both wt MC1R); D: RHC MC1R vs wt MC1R (both wt CDKN2A).
In silico model evaluation
We hypothesized that if the altered gene signatures observed in non-lesional skin from CDKN2A mutation or RHC MC1R variant carriers are critical for malignant transformation, these patterns will be maintained across the carcinogenic process. To support our hypothesis, the expression pattern of the most deregulated genes in mutant CDKN2A or RHC MC1R variant skin cells was compared to their expression observed in two previously published skin cancer datasets. The GSE2503 dataset contained the whole genome expression of five squamous cell carcinomas (SCCs) and six healthy skin samples[35] and the GSE12391 dataset included 23 melanomas (CMs) at different stages (radial growth phase or vertical growth phase) and 18 common nevi[36]. Due to differences in the array platforms used among the studies, only the expression of genes present in all datasets was evaluated (Figure 5). Thus, MC1R expression pattern analyses included 76.1% (121/159) of the most deregulated genes. Primary co-cultures with the non-functional MC1R gene showed a statistically significant expression pattern (p-value <0.005) similar to those detected in SCCs (Pearson correlation=0.44) and CMs (Pearson correlation=0.25) (Figure 5a).
Because of platform disparities, the mutant CDKN2A expression pattern was evaluated analyzing 53.7% of the most deregulated transcripts (58/108). The expression pattern correlated to those observed in SCCs (Pearson correlation=0.35) and CMs (Pearson correlation=0.28) showing a statistical tendency among datasets (p-value <0.06), in which immunomodulation and interferon response genes were also up-regulated (SP110, BTN3A2, IL15, IFIT2, IFI44, IFIT1, IFI44L and CLEC2B; Figure 5b). A correlated expression pattern was found within a subset of genes between CMs and mutant CDKN2A skin cells (Pearson Correlation=0.66, P-value = 0.009; Figure 5c). Remarkably, this subset of genes presented an expression pattern that inversely correlated between SCCs and CMs (Pearson correlation= -0.88, p-value<0.001) and also between SCCs and mutant CDKN2A skin cells (Pearson correlation= -0.58, p-value=0.027). Specifically, the expression pattern of TBX1, CLDN17, RFX2, COL5A3, IFIT2, CEACAM1, WDR78, LXN, SLC39A8, ZNF83, RPS27L, LSM3, CTSK, and PMP22 was exclusively correlated to MM and mutant CDKN2A MM prone skin cells, and inversely with NMSC.
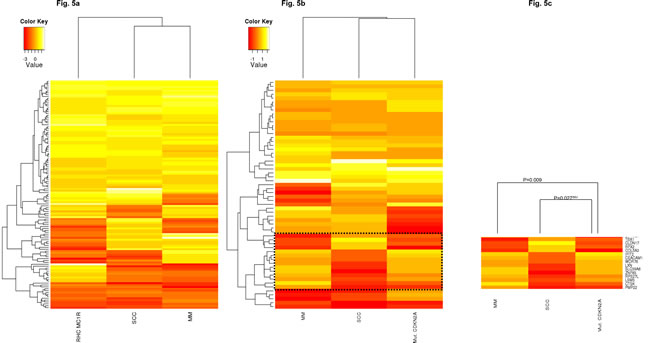
Figure 5: In-silico model evaluation. Comparison of the expression patterns detected in our study to those observed in Squamous cell Carcinomas (SSC; GSE2503 dataset) and Melanomas (MM; GSE12391 dataset).
Fig. 5A: Heatmap MC1R expression pattern showed a positive correlation among datasets (p-value <0.005).
Fig. 5B: Heatmap CDKN2A expression pattern showed a positive correlation among datasets (p-value <0.06).
Fig. 5C: Subset of genes with an inverse expression pattern between mutant CDKN2A skin cells and SSC and MM. INV: inversely correlated pattern.
Discussion
The aim of the study was to determine the constitutional effect of germline CDKN2A mutations or MC1R variants in skin cancer, in order to identify early critical molecular targets implicated in the disease. To do so we analyzed co-cultured melanocyte-keratinocyte systems derived from two siblings belonging to two melanoma prone families with a founder CDKN2A mutation and/or carrying non-functional MC1R alleles to compare global gene expression profiles.
To date, CDKN2A is the major gene responsible for increased melanoma susceptibility in high-risk pedigrees. Germline high-penetrance mutations are found in 10% of melanoma prone families and somatic CDKN2A alterations are also recurrent events in primary melanomas[37] and melanoma cell lines[38]. Alterations in gene expression in as yet phenotypically normal cells but bearing single-hit mutations in tumor suppressor genes appear to be the earliest molecular change during cancer development[39].
Altered baseline expression signatures associated with CDKN2A mutations in cultured normal skin fibroblasts from familial melanoma patients have already been reported [40]. However, the transcriptome deregulation within keratinocytes and melanocytes where cutaneous carcinomas and melanomas arise have not been previously assessed. We found that transcriptome signatures were altered by single hit CDKN2A mutations in co-cultured melanocyte-keratinocyte systems. CDKN2A mutant cells exhibit mainly baseline differences in genes related to cell components, metabolism and immune response. Melanoma cells are addicted to oncogene-driven energy production which can be mediated by somatic BRAF mutations [41], our findings indicate that non transformed skin cells already have a deregulated metabolic profiling which may be necessary for the skin cancer initiation.
Previous studies suggest that genetic variation of some cytokines or their receptors influence melanoma susceptibility [42, 43] and even modify the risk in melanoma-prone families with CDKN2A mutations [44]. Consistent with this hypothesis, our data reflects the important role of innate immunity and immune response pathways which may be deregulated in early steps of melanoma development. Skin cells also exhibited deregulation of epidermal differentiation and melanogenesis genes. Thus, we found down-regulation of the TYRP1 gene, a melanocyte differentiation marker and over expression of MFI2 gene, a cell-surface glycoprotein playing a role in melanoma cell proliferation and tumorigenesis [45] and EEA1 gene, a marker of early endosomes [46]. Interestingly, EEA1 interacts with MFI2 regulating endosome fusion and trafficking [47]. Early endosomes are direct precursors of melanosomes, which are the specialized organelles for the biosynthesis and storage of melanins[46]. These findings may indicate that constitutive disorganized melanosomal structures and changes in pigment production may favor malignant transformation which in turn, could be related to autoimmune response deregulation [48, 49].
Via functional analyses, we observed a down-regulation of the Notch signaling pathway in CDKN2A mutant cells. This pathway is essential in epidermal-melanocyte interactions [50, 51] and recent evidence suggests Notch pathway as a link between the control of epidermal differentiation and proliferation and skin homeostasis [52, 53]. Notch signaling has a dual action (as oncogene or as tumor suppressor) in skin cancer. While, up-regulation of the Notch pathway is observed in CMs or in SCCs at sun-protected sites, it is down-regulated in UV-related solar keratoses SCCs or in common melanocytic nevi (reviewed in Panelos and Massi, 2009 [54]). A high melanocytic nevi count is the strongest known risk factor for melanoma[55] and a potent predictor of mutant CDKN2A gene carrier status[56]. Thus, the baseline down-regulation of the Notch signaling pathway in CDKN2A mutation carriers may proffer a melanocyte proliferation advantage, which can trigger common nevi development in human skin.
The pigmentation related gene MC1R acts as a moderate melanoma risk gene and variants in this gene are modifying factors for melanoma risk in CDKN2A mutation carriers [21-23]. Furthermore, MC1R variants have also been clearly associated with elevated NMSC susceptibility [19, 20]. We observed that the number of genes differentially expressed in non-functional MC1R was considerably higher than in mutant CDKN2A. Both p.R151C and p.R160W alleles halted MC1R activity and also induced an altered cell surface molecule expression due to impaired export traffic [57] that could lead to a reduction in MC1R gene expression levels as observed in our study. We found that RHC MC1R skins cells differ from wild-type in the deregulation of genes involved in key physiological processes such as differentiation, cell adhesion and cell cycle progression which may be directly related to the etiology of skin cancer. We found that in non-functional MC1R cells, pathways associated with increased DNA damage without UV radiation exposure were up-regulated, indicating that RHC variants contribute to cancer risk in humans increasing the DNA damage mediated by intrinsic UV-independent mechanisms as recently observed in mice models [58]. Notably, RHC MC1R skin cells also showed up-regulated pathways related to neurodegenerative diseases. The MC1R gene is expressed in cells of the nervous system and its activation decreases oxidative stress and has anti-inflammatory and immunomodulatory effects [59]. Co-occurrence of Parkinson’s disease (PD) and CMs [60, 61] has been reported in epidemiological studies. The diagnosis of CM is associated with an approximate 50% increased risk of subsequent PD[61] development whereas individuals with PD have a two-fold increase in risk of subsequent CM[62] development. Previous data suggests that this relation is associated with pigment-related genes[63] rather than Parkinson’s-related genes[64, 65]. Our study further supports that MC1R is involved in the cross-link between both diseases. Thus, MC1R variants may increase the oxidative damage in brain cells and deregulate inflammatory processes which consequently, increase the susceptibility to neurodegenerative disorders.
Previous studies showed that many of the alterations associated with transcriptome and proteome signatures of as yet phenotypically normal cells bearing inherited alterations are also present in the corresponding form of cancer [39]. Thus, in our study, expression patterns detected in MM prone cells from non-lesional areas of the skin were compared with those observed in skin cancer tumors (SCCs and CMs) [35, 36]. Although differences in the experimental designs among studies (normal skin cells vs tumoral tissue or cultured cells vs fresh-frozen tissue) may interfere in the analysis, the expression patterns detected in our study correlate to those observed in skin tumors. Interestingly, the MC1R expression pattern was more similar among studies than the CDKN2A pattern (p-value<0.005 and p-value<0.06, respectively). These differences are caused in part by a subset of genes showing a converse pattern which allows us to distinguish between SSCs versus CMs and mutant CDKN2A skin cells (MM prone cells). Accordingly, previous studies have detected the inverse expression of two genes from this subset (CEACAM1 and CTSK) between SSCs and MMs [66, 67]. Such differences may underlie the epidemiological differences observed between MC1R and CDKN2A and skin cancer susceptibility since MC1R polymorphisms are involved in both CMM and NMSC susceptibility while CDKN2A mutations are closely related to CM susceptibility rather than SCC susceptibility.
In conclusion, we have identified baseline expression signatures in skin cells carrying germline CDKN2A mutations and RHC MC1R variants which are maintained in skin tumors (melanoma and squamous cell carcinomas). The study identified a large catalogue of genes in CDKN2A mutant skin cells that are closely related to skin cancer, highlighting the role of genes involved in immune response, in melanosome biogenesis and the Notch signaling pathway Also, our data indicates that non functional MC1R variants promote DNA damage by intrinsic UV-independent mechanisms in human skin cells. Furthermore, our study revealed a role for MC1R in the susceptibility to neurodegenerative diseases which may be related to its role in oxidative stress and inflammatory processes.
Methods
Study Design
To detect the effect of CDKN2A and MC1R genes, two melanoma families (A and B) were selected (Table I). The most frequent mutation, p.G101W, resulting from a common ancestor in Mediterranean pedigrees, was chosen in relation to the CDKN2A status. Regarding MC1R status, the presence of two red hair variants in each individual (RHC: p.R151C and p.R160W) was analyzed. To reduce the effect of intra-individual variability, two siblings from each family were selected, resulting in four different extreme genotypic conditions: a carrier of variants in both genes (sample A1), an individual without variants in any gene (sample B2) and two carriers of variants in one of these genes (samples A2 and B1), respectively.
The study was approved by the IRB and signed informed consents were obtained from all individuals.
Primary keratinocyte and melanocyte co-culture
Skin biopsies from non-lesional areas were taken and human keratinocytes and melanocytes were obtained by mechanical fragmentation and enzymatic digestion [68]. Briefly, skin biopsy fragments were digested with collagenase type I solution (2mg/ml) (Sigma–Aldrich, Gillingham, UK) for 90 minutes. The collagenase solution was then completely eliminated and the remaining skin biopsy was introduced into a mixture of 0.05% trypsin /0.02% EDTA (T/E) (Sigma). Every 30 min, T/E was changed for a fresh T/E mixture. Collected T/E was inactivated with serum containing culture medium and was centrifuged at 1400 rpm for 10 minutes. Primary keratinocytes and melanocytes obtained by this method were cultured on a feeder layer of lethally irradiated (X-ray; 50 Gy) 3T3-J2 cells (a gift from Dr J. Garlick, SUNY), as previously described [69, 70]. The seeding media was a 3:1 mixture of Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO-BRL, Barcelona, Spain) and HAM’S F12 (GIBCO-BRL) containing 10% FCS, 0.1 nM choleric toxin, 2 nM T3, 5 µg/mL insulin, and 0.4 µg/mL hydrocortisone. Cells were cultured at 37ºC in a humid atmosphere containing 5% CO2. The culture medium was changed every two days. This co-culture system has been extensively proven to preserve stemness of keratinocytes[69-71] as well as to maintain the physiological melanocyte:keratinocyte ratio (1:40)[72, 73]. Moreover, by using the skin cells from this type of co-culture, the donor’s phenotypic pigmentation and UV-response features are preserved in vivo on a humanized skin mouse model[74].
RNA extraction
Total RNA isolation from primary cultures on passage 3-4 was performed using the Trizol extraction method (Invitrogen Life Technologies, Carlsbad, CA) followed by purification in commercial columns (Qiagen, Valencia, CA). Total isolated RNA was further purified using an RNeasy kit (Qiagen, Valencia, CA). RNA concentration was determined using a NanoDrop Spectrophotometer (Thermo Scientific) and integrity of the RNA was verified by Bioanalyzer 2100 (Agilent, USA). The RNA integrity number was in all cases higher than 8.
Expression array
Analysis of global expression was performed using the Whole Human Genome (4x44k) Oligo Microarray kit (G4112F, Agilent, US). The microarray contains probes from over 41,000 unique human genes and transcripts, all with public OMIM annotations (RefSeq, Goldenpath, Ensembl, Unigene Human Genome and GenBank databases). Overall, 50 ng of RNA were labeled using Low input Quickamp Labeling kit (Agilent, US). In all samples 10 commercial controls probes were added in order to standardize the results (RNA Spike-in kit, one color, Agilent, US). The arrays were scanned using the DNA Microarray Scanner G2565CA (Agilent, US). Finally, Feature Extraction Software (FES, Agilent, USA) was used both to perform the quality control process and to extract the information. Three replicates from each primary culture were analyzed.
Quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) of selected genes
To confirm the microarray results, the expression of 13 genes (FARP1, SLFN11, GFPT2, COL5A3, TYRP1, LEF1, KRT2, ST6GALNAC3, MLANA, MSMB, SILV, A2M, and ALOX5) was validated by RT-PCR. Also, MC1R expression was evaluated. The ACTB gene was used for normalization. RNA derived from donors’ healthy skin was used as a calibrator in each reaction. A total of 200 ng of RNA was retrotranscribed to cDNA using Taqman PCR Core Reagent kit (Roche Applied Science, Penzbergf, Germany). Real-time PCR was performed using Taqman Universal PCR master Mix (Roche Applied Science, Penzbergf, Germany). Reaction was performed in an ABI Step One plus RT-PCR sequence detection instrument (Applied Biosystems, CA, US). Primer design and gene-specific probe selection were carried out by the Universal Probe Library software (UPL, Roche, Mannheim, Germany).
Data was evaluated using the relative quantification method of ddCt[75]. Expression values were evaluated by T-test for equality means using the SPSS 17.0. P-values less than or equal to 0.05 were considered statistically significant.
Microarray data and statistical analyses
The Agilent Processed Signal was standardized across arrays using quantile normalization[76]. Differential gene expression analysis was carried out using the limma package from Bioconductor. Multiple testing adjustments of p-values were performed according to Benjamini and Hochberg methodology[77].
Gene set analysis was carried out for the Kyoto Encyclopedia of Genes and Genomes pathways (KEGG) using FatiScan[78] in Babelomics[79]. This tool detects significantly up- or down-regulated blocks of functionally related genes in lists of genes ordered by different criteria such as differential expression, KEGG pathways and others. The core of the method is based on an algorithm to test whether a set of genes, labeled with terms (biological information), contain significant enrichments of one or several of these terms with respect to another set of reference genes. FatiScan uses a Fisher’s exact test for 2×2 contingency tables for comparing two groups of genes and extracting a list of KEGG terms whose distribution among the groups is significantly different. Given that many KEGG terms are simultaneously tested, the results of the test are corrected for multiple testing to obtain an adjusted p-value. FatiScan returns adjusted p-values based on the False Discovery Rate (FDR) method[77]. KEGG Pathways for the genes in the microarray where taken from the KEGG web.
In order to gain insight into the co-regulation patterns on transcript expression profiles, data from our microarray experiments were clustered together with expression data obtained from two previous experiments focused on NMSC (GSE2503 dataset[35]) and MM (GSE12391 dataset[36]). Raw data were obtained from the Gene Expression Omnibus. For each dataset we analyzed the differential expression and determined the fold-changes. We selected two subsets: genes differentially expressed in our study for the MC1R comparison (RHC alleles vs. wild-type MC1R) and a second group of genes differentially expressed in our study for the CDKN2A comparison (p.G101W vs. wild-type CDKN2A). In each scenario, fold-changes were normalized using the quantile method[76] to compare differential expression results in all experiments. We performed a hierarchical clustering analysis of differentially expressed transcripts and the graphical representation showed the relationship between experiments. The correlation analysis quantified the linear relationship between the different studies m
Supplemental Data Description
Supplemental Data includes two tables with the list of the most deregulated genes associated with CDKN2A mutations or MC1R variants; and one table containing the list of genes inversely deregulated between CDKN2A and MC1R.
Acknowledgments
The present project was founded by a grant from CIBER de Enfermedades Raras of the Instituto de Salud Carlos III, Spain (INTRA/07/726.2). The research at the Melanoma Unit in Barcelona is partially funded by Grants from Fondo de Investigaciones Sanitarias (P.I. 09/01393 and P.I. 12/00840, co-funded by “Fondo Europeo de Desarrollo Regional (FEDER). Unión Europea. Una manera de hacer Europa”); by the AGAUR 2009 SGR 1337 of the Catalan Government, Spain; by the European Commission under the 6th Framework Programme, Contract nr: LSHC-CT-2006-018702 (GenoMEL) and by the National Cancer Institute (NCI) of the US National Institute of Health (NIH) (CA83115). This work is also partly supported by grants BIO2008-04212 and SAF2010-16976 from the Spanish Ministry of Science and Innovation (MICINN), PROMETEO/2010/001 from the GVA-FEDER and S2010/BMD-2420 (CELLCAM) from Comunidad de Madrid.
Web Resources:
Bioconductor (http://www.bioconductor.org)
Babelomics (http://babelomics.bioinfo.cipf.es/ )
Kyoto Encyclopedia of Genes and Genomes pathways (KEGG) (http://www.genome.jp/kegg/)
Ensembl 56 release (http://www.ensembl.org)
Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/)
Accession Numbers The GEO accession number for the expression microarray data reported in this paper is GSE44805.
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ptorruosgomwqds&acc=GSE44805
Conflict of Interest:
The authors state no conflict of interest.
References
1. Siegel R, Naishadham D and Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62(1):10-29.
2. Thompson JF, Scolyer RA and Kefford RF. Cutaneous melanoma in the era of molecular profiling. Lancet. 2009; 374(9687):362-365.
3. Goldstein AM, Chan M, Harland M, Hayward NK, Demenais F, Bishop DT, Azizi E, Bergman W, Bianchi-Scarra G, Bruno W, Calista D, Albright LA, Chaudru V, Chompret A, Cuellar F, Elder DE, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007; 44(2):99-106.
4. Puig S, Malvehy J, Badenas C, Ruiz A, Jimenez D, Cuellar F, Azon A, Gonzalez U, Castel T, Campoy A, Herrero J, Marti R, Brunet-Vidal J and Mila M. Role of the CDKN2A locus in patients with multiple primary melanomas. J Clin Oncol. 2005; 23(13):3043-3051.
5. Liontos M, Pateras IS, Evangelou K and Gorgoulis VG. The Tumor Suppressor Gene ARF as a Sensor of Oxidative Stress. Curr Mol Med. 2012.
6. Jenkins NC, Liu T, Cassidy P, Leachman SA, Boucher KM, Goodson AG, Samadashwily G and Grossman D. The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene. 2011; 30(3):265-274.
7. Lang J, Boxer M and MacKie RM. CDKN2A mutations in Scottish families with cutaneous melanoma: results from 32 newly identified families. Br J Dermatol. 2005; 153(6):1121-1125.
8. Larre Borges A, Cuellar F, Puig-Butille JA, Scarone M, Delgado L, Badenas C, Mila M, Malvehy J, Barquet V, Nunez J, Laporte M, Fernandez G, Levrero P, Martinez-Asuaga M and Puig S. CDKN2A mutations in melanoma families from Uruguay. Br J Dermatol. 2009; 161(3):536-541.
9. Yakobson E, Eisenberg S, Isacson R, Halle D, Levy-Lahad E, Catane R, Safro M, Sobolev V, Huot T, Peters G, Ruiz A, Malvehy J, Puig S, Chompret A, Avril MF, Shafir R, et al. A single Mediterranean, possibly Jewish, origin for the Val59Gly CDKN2A mutation in four melanoma-prone families. Eur J Hum Genet. 2003; 11(4):288-296.
10. Majore S, De Simone P, Crisi A, Eibenschutz L, Binni F, Antigoni I, De Bernardo C, Catricala C and Grammatico P. CDKN2A/CDK4 molecular study on 155 Italian subjects with familial and/or primary multiple melanoma. Pigment Cell Melanoma Res. 2008; 21(2):209-211.
11. Eliason MJ, Larson AA, Florell SR, Zone JJ, Cannon-Albright LA, Samlowski WE and Leachman SA. Population-based prevalence of CDKN2A mutations in Utah melanoma families. J Invest Dermatol. 2006; 126(3):660-666.
12. Ciotti P, Struewing JP, Mantelli M, Chompret A, Avril MF, Santi PL, Tucker MA, Bianchi-Scarra G, Bressac-de Paillerets B and Goldstein AM. A single genetic origin for the G101W CDKN2A mutation in 20 melanoma-prone families. Am J Hum Genet. 2000; 67(2):311-319.
13. Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA, Mann GJ, Mantelli M, Nancarrow D, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002; 94(12):894-903.
14. Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D and Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005; 11(20):7234-7242.
15. Jefferies S, Edwards SM, Hamoudi RA, A’Hern R, Foulkes W, Goldgar D and Eeles R. No germline mutations in CDKN2A (p16) in patients with squamous cell cancer of the head and neck and second primary tumours. Br J Cancer. 2001; 85(9):1383-1386.
16. Cabanillas R, Astudillo A, Valle M, de la Rosa J, Alvarez R, Duran NS and Cadinanos J. Novel germline CDKN2A mutation associated with head and neck squamous cell carcinomas and melanomas. Head Neck. 2011.
17. Rees JL. The genetics of sun sensitivity in humans. Am J Hum Genet. 2004; 75(5):739-751.
18. Valverde P, Healy E, Jackson I, Rees JL and Thody AJ. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet. 1995; 11(3):328-330.
19. Bastiaens MT, ter Huurne JA, Kielich C, Gruis NA, Westendorp RG, Vermeer BJ and Bavinck JN. Melanocortin-1 receptor gene variants determine the risk of nonmelanoma skin cancer independently of fair skin and red hair. Am J Hum Genet. 2001; 68(4):884-894.
20. Box NF, Duffy DL, Irving RE, Russell A, Chen W, Griffyths LR, Parsons PG, Green AC and Sturm RA. Melanocortin-1 receptor genotype is a risk factor for basal and squamous cell carcinoma. J Invest Dermatol. 2001; 116(2):224-229.
21. Box NF, Duffy DL, Chen W, Stark M, Martin NG, Sturm RA and Hayward NK. MC1R genotype modifies risk of melanoma in families segregating CDKN2A mutations. Am J Hum Genet. 2001; 69(4):765-773.
22. Goldstein AM, Chaudru V, Ghiorzo P, Badenas C, Malvehy J, Pastorino L, Laud K, Hulley B, Avril MF, Puig-Butille JA, Miniere A, Marti R, Chompret A, Cuellar F, Kolm I, Mila M, et al. Cutaneous phenotype and MC1R variants as modifying factors for the development of melanoma in CDKN2A G101W mutation carriers from 4 countries. Int J Cancer. 2007; 121(4):825-831.
23. Demenais F, Mohamdi H, Chaudru V, Goldstein AM, Newton Bishop JA, Bishop DT, Kanetsky PA, Hayward NK, Gillanders E, Elder DE, Avril MF, Azizi E, van Belle P, Bergman W, Bianchi-Scarra G, Bressac-de Paillerets B, et al. Association of MC1R variants and host phenotypes with melanoma risk in CDKN2A mutation carriers: a GenoMEL study. J Natl Cancer Inst. 2010; 102(20):1568-1583.
24. Herraiz C, Jimenez-Cervantes C, Zanna P and Garcia-Borron JC. Melanocortin 1 receptor mutations impact differentially on signalling to the cAMP and the ERK mitogen-activated protein kinase pathways. FEBS Lett. 2009; 583(19):3269-3274.
25. Le Pape E, Passeron T, Giubellino A, Valencia JC, Wolber R and Hearing VJ. Microarray analysis sheds light on the dedifferentiating role of agouti signal protein in murine melanocytes via the Mc1r. Proc Natl Acad Sci U S A. 2009; 106(6):1802-1807.
26. Sviderskaya EV, Hill SP, Evans-Whipp TJ, Chin L, Orlow SJ, Easty DJ, Cheong SC, Beach D, DePinho RA and Bennett DC. p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst. 2002; 94(6):446-454.
27. Eisinger M and Marko O. Selective proliferation of normal human melanocytes in vitro in the presence of phorbol ester and cholera toxin. Proc Natl Acad Sci U S A. 1982; 79(6):2018-2022.
28. Herlyn M, Rodeck U, Mancianti M, Cardillo FM, Lang A, Ross AH, Jambrosic J and Koprowski H. Expression of melanoma-associated antigens in rapidly dividing human melanocytes in culture. Cancer Res. 1987; 47(12):3057-3061.
29. Elder DE, Rodeck U, Thurin J, Cardillo F, Clark WH, Stewart R and Herlyn M. Antigenic profile of tumor progression stages in human melanocytic nevi and melanomas. Cancer Res. 1989; 49(18):5091-5096.
30. Valyi-Nagy IT, Hirka G, Jensen PJ, Shih IM, Juhasz I and Herlyn M. Undifferentiated keratinocytes control growth, morphology, and antigen expression of normal melanocytes through cell-cell contact. Lab Invest. 1993; 69(2):152-159.
31. Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE and Herlyn M. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000; 156(5):1515-1525.
32. Phillips J, Gawkrodger DJ, Caddy CM, Hedley S, Dawson RA, Smith-Thomas L, Freedlander E and Mac Neil S. Keratinocytes suppress TRP-1 expression and reduce cell number of co-cultured melanocytes - implications for grafting of patients with vitiligo. Pigment Cell Res. 2001; 14(2):116-125.
33. Haass NK, Smalley KS, Li L and Herlyn M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005; 18(3):150-159.
34. Hirobe T. Role of keratinocyte-derived factors involved in regulating the proliferation and differentiation of mammalian epidermal melanocytes. Pigment Cell Res. 2005; 18(1):2-12.
35. Nindl I, Dang C, Forschner T, Kuban RJ, Meyer T, Sterry W and Stockfleth E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer. 2006; 5:30.
36. Scatolini M, Grand MM, Grosso E, Venesio T, Pisacane A, Balsamo A, Sirovich R, Risio M and Chiorino G. Altered molecular pathways in melanocytic lesions. Int J Cancer. 2010; 126(8):1869-1881.
37. de Snoo FA and Hayward NK. Cutaneous melanoma susceptibility and progression genes. Cancer Lett. 2005; 230(2):153-186.
38. Ogbah Z, Puig-Butille JA, Simonetta F, Badenas C, Cervera R, Mila J, Benitez D, Malvehy J, Vilella R and Puig S. Molecular characterization of human cutaneous melanoma-derived cell lines. Anticancer Res. 2012; 32(4):1245-1251.
39. Herbert BS, Chanoux RA, Liu Y, Baenziger PH, Goswami CP, McClintick JN, Edenberg HJ, Pennington RE, Lipkin SM and Kopelovich L. A molecular signature of normal breast epithelial and stromal cells from Li-Fraumeni syndrome mutation carriers. Oncotarget. 2012; 1(6):405-422.
40. Fan M, Pfeffer SR, Lynch HT, Cassidy P, Leachman S, Pfeffer LM and Kopelovich L. Altered transcriptome signature of phenotypically normal skin fibroblasts heterozygous for CDKN2A in familial melanoma: relevance to early intervention. Oncotarget. 2013; 4(1):128-141.
41. Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P and Christensen C. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget. 2013; 4(4):584-599.
42. Gu F, Qureshi AA, Niu T, Kraft P, Guo Q, Hunter DJ and Han J. Interleukin and interleukin receptor gene polymorphisms and susceptibility to melanoma. Melanoma Res. 2008; 18(5):330-335.
43. Howell WM, Turner SJ, Bateman AC and Theaker JM. IL-10 promoter polymorphisms influence tumour development in cutaneous malignant melanoma. Genes Immun. 2001; 2(1):25-31.
44. Yang XR, Pfeiffer RM, Wheeler W, Yeager M, Chanock S, Tucker MA and Goldstein AM. Identification of modifier genes for cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. Int J Cancer. 2009; 125(12):2912-2917.
45. Guo J, Wen DR, Huang RR, Paul E, Wunsch P, Itakura E and Cochran AJ. Detection of multiple melanoma-associated markers in melanoma cell lines by RT in situ PCR. Exp Mol Pathol. 2003; 74(2):140-147.
46. Raposo G and Marks MS. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002; 3(4):237-248.
47. Ramanathan HN and Ye Y. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res. 2011; 22(2):346-359.
48. Sakai C, Kawakami Y, Law LW, Furumura M and Hearing VJ, Jr. Melanosomal proteins as melanoma-specific immune targets. Melanoma Res. 1997; 7(2):83-95.
49. Hearing VJ. Biochemical control of melanogenesis and melanosomal organization. J Investig Dermatol Symp Proc. 1999; 4(1):24-28.
50. Schouwey K, Delmas V, Larue L, Zimber-Strobl U, Strobl LJ, Radtke F and Beermann F. Notch1 and Notch2 receptors influence progressive hair graying in a dose-dependent manner. Dev Dyn. 2007; 236(1):282-289.
51. Okuyama R, Tagami H and Aiba S. Notch signaling: its role in epidermal homeostasis and in the pathogenesis of skin diseases. J Dermatol Sci. 2008; 49(3):187-194.
52. Xin Y, Lu Q and Li Q. IKK1 control of epidermal differentiation is modulated by notch signaling. Am J Pathol. 2011; 178(4):1568-1577.
53. Lin HY, Kao CH, Lin KM, Kaartinen V and Yang LT. Notch signaling regulates late-stage epidermal differentiation and maintains postnatal hair cycle homeostasis. PLoS One. 2011; 6(1):e15842.
54. Panelos J and Massi D. Emerging role of Notch signaling in epidermal differentiation and skin cancer. Cancer Biol Ther. 2009; 8(21):1986-1993.
55. Chang YM, Newton-Bishop JA, Bishop DT, Armstrong BK, Bataille V, Bergman W, Berwick M, Bracci PM, Elwood JM, Ernstoff MS, Green AC, Gruis NA, Holly EA, Ingvar C, Kanetsky PA, Karagas MR, et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int J Cancer. 2009; 124(2):420-428.
56. Bishop JA, Wachsmuth RC, Harland M, Bataille V, Pinney E, Mac KP, Baglietto L, Cuzick J and Bishop DT. Genotype/phenotype and penetrance studies in melanoma families with germline CDKN2A mutations. J Invest Dermatol. 2000; 114(1):28-33.
57. Sanchez-Laorden BL, Herraiz C, Valencia JC, Hearing VJ, Jimenez-Cervantes C and Garcia-Borron JC. Aberrant trafficking of human melanocortin 1 receptor variants associated with red hair and skin cancer: Steady-state retention of mutant forms in the proximal golgi. J Cell Physiol. 2009; 220(3):640-654.
58. Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, Guerrero CR, Lennerz JK, Mihm MC, Wargo JA, Robinson KC, Devi SP, Vanover JC, D’Orazio JA, McMahon M, Bosenberg MW, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. 2012; 491(7424):449-453.
59. Catania A. Neuroprotective actions of melanocortins: a therapeutic opportunity. Trends Neurosci. 2008; 31(7):353-360.
60. Liu R, Gao X, Lu Y and Chen H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology. 2011; 76(23):2002-2009.
61. Olsen JH, Friis S and Frederiksen K. Malignant melanoma and other types of cancer preceding Parkinson disease. Epidemiology. 2006; 17(5):582-587.
62. Olsen JH, Friis S, Frederiksen K, McLaughlin JK, Mellemkjaer L and Moller H. Atypical cancer pattern in patients with Parkinson’s disease. Br J Cancer. 2005; 92(1):201-205.
63. Gao X, Simon KC, Han J, Schwarzschild MA and Ascherio A. Genetic determinants of hair color and Parkinson’s disease risk. Ann Neurol. 2009; 65(1):76-82.
64. Meng S, Song F, Chen H, Gao X, Amos CI, Lee JE, Wei Q, Qureshi AA and Han J. No association between Parkinson disease alleles and the risk of melanoma. Cancer Epidemiol Biomarkers Prev. 2011; 21(1):243-245.
65. Hassin-Baer S, Laitman Y, Azizi E, Molchadski I, Galore-Haskel G, Barak F, Cohen OS and Friedman E. The leucine rich repeat kinase 2 (LRRK2) G2019S substitution mutation. Association with Parkinson disease, malignant melanoma and prevalence in ethnic groups in Israel. J Neurol. 2009; 256(3):483-487.
66. Shinozuka K, Uzawa K, Fushimi K, Yamano Y, Shiiba M, Bukawa H, Yokoe H and Tanzawa H. Downregulation of carcinoembryonic antigen-related cell adhesion molecule 1 in oral squamous cell carcinoma: correlation with tumor progression and poor prognosis. Oncology. 2009; 76(6):387-397.
67. Quintanilla-Dieck MJ, Codriansky K, Keady M, Bhawan J and Runger TM. Cathepsin K in melanoma invasion. J Invest Dermatol. 2008; 128(9):2281-2288.
68. Rheinwald JG and Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975; 6(3):331-343.
69. Llames SG, Del Rio M, Larcher F, Garcia E, Garcia M, Escamez MJ, Jorcano JL, Holguin P and Meana A. Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation. 2004; 77(3):350-355.
70. Del Rio M, Larcher F, Serrano F, Meana A, Munoz M, Garcia M, Munoz E, Martin C, Bernad A and Jorcano JL. A preclinical model for the analysis of genetically modified human skin in vivo. Hum Gene Ther. 2002; 13(8):959-968.
71. Larcher F, Dellambra E, Rico L, Bondanza S, Murillas R, Cattoglio C, Mavilio F, Jorcano JL, Zambruno G and Del Rio M. Long-term engraftment of single genetically modified human epidermal holoclones enables safety pre-assessment of cutaneous gene therapy. Mol Ther. 2007; 15(9):1670-1676.
72. De Luca M, Franzi AT, D’Anna F, Zicca A, Albanese E, Bondanza S and Cancedda R. Coculture of human keratinocytes and melanocytes: differentiated melanocytes are physiologically organized in the basal layer of the cultured epithelium. Eur J Cell Biol. 1988; 46(1):176-180.
73. Guerra L, Capurro S, Melchi F, Primavera G, Bondanza S, Cancedda R, Luci A, De Luca M and Pellegrini G. Treatment of “stable” vitiligo by Timedsurgery and transplantation of cultured epidermal autografts. Arch Dermatol. 2000; 136(11):1380-1389.
74. Garcia M, Llames S, Garcia E, Meana A, Cuadrado N, Recasens M, Puig S, Nagore E, Illera N, Jorcano JL, Del Rio M and Larcher F. In vivo assessment of acute UVB responses in normal and Xeroderma Pigmentosum (XP-C) skin-humanized mouse models. Am J Pathol. 2010; 177(2):865-872.
75. Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25(4):402-408.
76. Bolstad BM, Irizarry RA, Astrand M and Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003; 19(2):185-193.
77. Benjamini Y, Drai D, Elmer G, Kafkafi N and Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001; 125(1-2):279-284.
78. Al-Shahrour F, Arbiza L, Dopazo H, Huerta-Cepas J, Minguez P, Montaner D and Dopazo J. From genes to functional classes in the study of biological systems. BMC Bioinformatics. 2007; 8:114.
79. Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tarraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, Garcia F, Marba M, Montaner D and Dopazo J. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010; 38(Web Server issue):W210-213.