
INTRODUCTION
The constitutive ubiquitin-proteasome system is mainly implicated in the controlled degradation of proteins in eukaryotic cells [1]. Since an increased degradation by proteasome of cell cycle inhibitors or proapoptotic proteins contributes to malignant transformation [2, 3], considerable efforts to develop proteasome inhibitors were made and led to three approved drugs, bortezomib for the treatment of multiple myeloma [4] and mantle lymphoma [5], carfilzomib [6] and the orally available ixazomib [7] for the treatment of multiple myeloma (Figure 1A). These covalent inhibitors inhibit mainly the β5 activity of the catalytic core of the constitutive proteasome (cCP) but also, indiscriminately, that of the catalytic core of the immunoproteasome (iCP). A large variety of other covalent inhibitors of the cCP have been reported [8, 9] whereas noncovalent ones are less frequent. Some molecules contain a N-capped dipeptide backbone (compounds A1a-b, Figure 1B) [10, 11] or mimic the natural cyclic tripeptide TMC-95 (compound A2) [12–14] (Figure 1B). Recently reported noncovalent inhibitors are now essentially organic compounds [15–21] (compounds A3-A7, Figure 1B). Since noncovalent inhibitors are in general devoid of reactive groups, they do not have the drawbacks generally associated with the presence of a warhead such as lack of specificity, instability, and excessive reactivity [9]. Moreover, the reversible binding provides enzyme-inhibitor complexes with a limited life-time and favors inhibitor widespread tissue distribution [10].
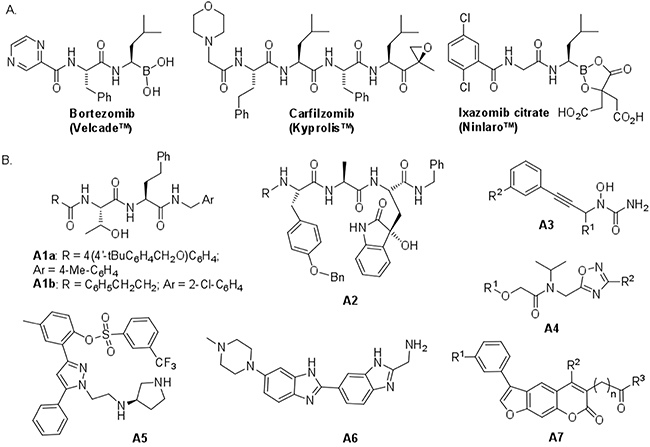
Figure 1: Chemical structures of some known proteasome inhibitors. A. Inhibitors used in cancer therapy. B. Selected noncovalent inhibitors of proteasomes. Compounds A1a, A4, A5, A6 and A7 display noticeable activity for iCP.
The immunoproteasome is found in lymphoid cells and cells exposed to cytokines like interferon-γ or tumor necrosis factor [22]. The lack of selectivity of bortezomib and carfilzomib against the cCP and iCP may in part explain some of the side effects and resistance observed during treatments with these molecules and that arise from undesired inhibition of the constitutive proteasome in normal cells [23, 24]. Expression of the immunoproteasome was found highly increased in a number of diseases [22] including cancer, autoimmune diseases and multiple myeloma [23] as well as rheumatoid arthritis [25], Huntington disease [26] and Alzheimer’s disease [27]. Taking together these observations, it would seem that targeting selectively the iCP could be critical in several disease indications [28].
The catalytic cores iCP and cCP are closely related with two outer rings of 7 α-subunits and two inner rings of β-subunits with subtle differences in the catalytic site subpockets [29]. The constitutive β-subunits β1, bearing the so-called caspase–like or post-acid activity (PA, often cleaves after acidic amino acids), β2, bearing the trypsin-like activity (T-L, often cleaves after basic amino acids) and β5 bearing the chymotrypsin-like activity (ChT-L, often cleaves after hydrophobic amino acids) are replaced in the iCP by three homologous subunits (β1i, LMP2; β2i, MECL; β5i, LMP7). Of importance, small compounds interfering with one type of catalytic activity can also block the others (cross-react) as observed for instance for the TMC-95 mimic A2 (Figure 1B) [12–14] that binds to all three proteosomal active centers. Some compounds can cross-react between iCP and cCP and also between proteasomes of different species [8, 9, 29, 30]. Furthermore, catalytic activities most often, depending on the chemistry and the nature of the ligand, require the presence of the adjacent subunit, with for instance, for the ChT-L activity, the catalytic site on the β5 subunit with additional contacts for ligand binding and orientation involving residues located on the β6 subunit. The enzymatic activity of each catalytic center is associated with the Thr1 N-terminal residue, in which the gamma hydroxyl group acts as a nucleophile in the hydrolysis of the peptide bonds.
Only a small number of iCP-selective inhibitors have been described [8, 31, 32]. They are mainly covalent inhibitors bearing an electrophilic warhead (peptide epoxyketone [33–35], peptide aldehyde [23], peptide sulfonyl fluoride [36], peptide chloroacetamide [37], oxathiazolone [38] or β-lactone [39]). Conversely, noncovalent inhibitors of the immunoproteasome are still poorly described. Some N-capped dipeptides (e.g., molecule A1a) are selective inhibitors of the ChT-L activity of iCP [10, 40] (Figure 1B). Pyrazole compound A5 inhibits the ChT-L and PA activities of both iCP and cCP [19]. The oxadiazole scaffold of compound A4 was discovered by structure-based virtual screening and led to the development of potent inhibitors of the ChT-L activity of the cCP [17] that are less active against iCP (unpublished result; IC50 = 0.14 ± 0.02 µM instead of 0.037 ± 0.003 µM for cCP, R1 = 4-BrC6H4, R2 = 3,4-di-MeOC6H3). Benzimidazole derivative A6 acted selectively on the T-L activity of iCP (subunit β2i) [20]. Psoralene based inhibitors (e.g., compound A7), selective of the ChT-L activity of iCP, were also recently reported [21].
Overall, it would be highly desirable to identify novel immunoproteasome-selective inhibitors with new scaffolds. For the reasons commented above, noncovalent iCP inhibitors may constitute a valuable alternative to covalent ones. For example, the knowledge of cCP binders can be exploited to find iCP inhibitors as observed with the β1/β2 specific sulfonamide derivative that noncovalently binds between subunits β1 and β2 [41].
In this study, we investigate small organic inhibitors that were initially identified using hierarchical structure-based virtual ligand screening computations [42] performed in the ligand-binding subsites present on the β5/β6 subunits of the cCP [15]. Displaying a new piperazinyl sulfonamide scaffold (Table 1), some inhibitors act selectively on the β2c and β2i subunits (with a preference for the β2i) or, simultaneously on the β5 and β1 subunits of both the iCP and cCP. Their differential inhibitory efficiency on the three types of proteasomal activities was evaluated in vitro together with their cytotoxicity, anti-proliferative and anti-invasive effects on two cancer cell lines.
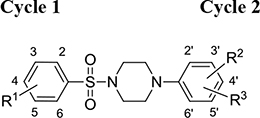
Table 1: Inhibition profile of human iCP (in bold) and cCP (plain text) at pH 8.0 and 37°C by piperazinyl sulfonamidesa
Compd |
|
IC50 (µM)b, % inhibition or x (activation factor) at 50 µM |
||||
|---|---|---|---|---|---|---|
R1 |
R2 |
R3 |
ChT-L |
PA |
T-L |
|
B1a |
4-CH2CH(CH3)2 |
4’-NO2 |
H |
ni ni |
25 % ni |
7.6 ± 0,6 10.3 ± 0.5 |
B1b |
4-C(CH3)3 |
2’-CH3 |
3’-Cl |
ni ni |
ni ni |
2.6 ± 0.4 12.0 ± 1.1 |
B1c |
4-C(CH3)3 |
2’-NO2 |
4’-Cl |
ni ni |
ni ni |
3.3 ± 0.4 8.1 ± 0.8 |
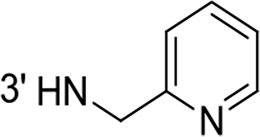
B1d |
4-Cl |
|
4’-NO2 |
ni |
46 % |
x 4.7 |
B1e |
4-CH3 |
| 4’-NO2 |
5.5 ± 0.3 2.5 ± 0.1 |
3.3 ± 0.3 4.2 ± 0.1 |
x 3 ni |
B1f |
4-Cl |
| 4’-NO2 |
5.8 ± 0.2 3.2 ± 0.1 |
3.7 ± 0.2 7.4 ± 0.3 |
x 3 ni |
B1g |
4-C(CH3)3 |
| 4’-NO2 |
24.6 ± 3.5 |
13.9 ± 1.4 |
ni |
B1h |
4-NHCOCH3 |
| 4’-NO2 |
50.5 % |
45 % |
ni |
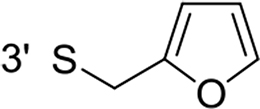
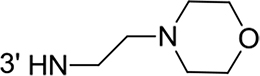
B1i |
3,4-OCH3 |
3’-NHCH2C(CH3)3 |
4’-NO2 |
ni |
ni |
ni |
|
||||||
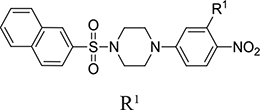
B2a |
|
32.4± 0,7 |
31.2 ± 1.0 |
ni |
||
B2b |
H |
58.9 % |
50 % |
x 4.5 |
||
B3 |
|
6.7 ± 0.2 6.5 ± 0.3 |
7.1 ± 0.2 8.1 ± 0.6 |
ni ni |
||
B4 |
|
ni |
ni |
ni |
||
aThe inhibition was evaluated after 15 min incubation of the enzyme with the compounds before adding the appropriate fluorogenic substrate to evaluate the remaining ChT-L, PA and T-L activities. bThe IC50 values were calculated by fitting the experimental data to eq 1 or eq 2. ni: inhibition < 25% at 50 µM.
RESULTS AND DISCUSSION
Compounds B1a-c target the 20S proteasome β2 subunits with a preference for the immunoproteasome whereas the other compounds target simultaneously the β1 and β5 subunits
Virtual screening was carried out on the mammalian cCP proteasome using several computational approaches and a short consensus list of molecules were selected, purchased from the ChemBridge corporation and evaluated experimentally. These hits are here further characterized using several complementary approaches. The in silico screening efforts [15] led to the discovery of a family of piperazine sulfonamide binders inhibiting at least one type of activity of cCP. Among the 65 hits that we identified, a dozen of molecules belonged to this category (Table 1). We decided to explore further their properties by testing their potential to inhibit human iCP versus cCP. They were assayed for in vitro inhibition of purified human cCP and iCP by measuring the hydrolysis of the β5 (ChT-L) substrate Suc-LLVY-AMC, β1 (PA) substrate Z-LLE-βNA, and β2 (T-L) substrate Boc-LRR-AMC in the presence of various concentrations of the tested molecules. When a noticeable inhibition was detected, the IC50 values were determined (Figure 2). Structures and inhibition efficacies of the most efficient molecules are summarized in Table 1.
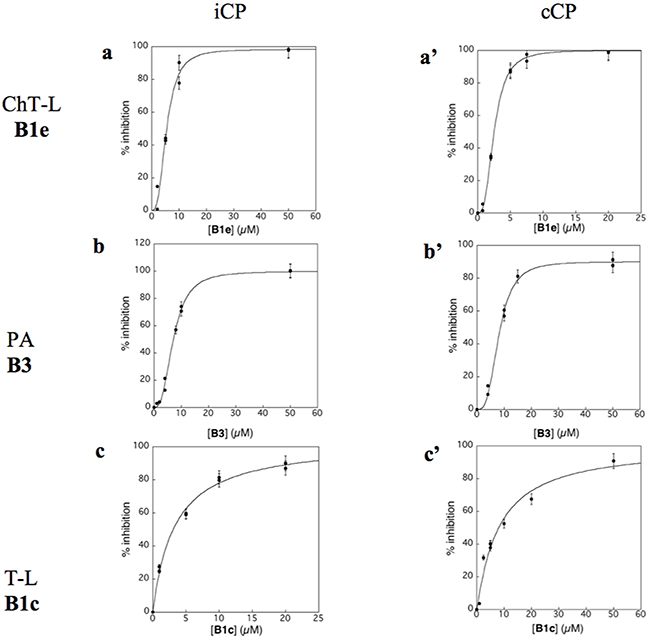
Figure 2: Inhibition profile of human iCP (a,b,c) and cCP (a’, b’, c’) by compounds B1e (a,a’), B3 (b,b’) and B1c (c,c’) at pH 8.0 and 37°C. [iCP] = [cCP] = 0.3 nM; ChT-L activity, [Suc-LLVY-AMC]0 = 20 µM; PA activity, [Z-LLE-βNA]0 = 100 µM (iCP) and 50 µM (cCP); T-L activity, [Boc-LRR-AMC]0 = 50 µM. The experimental data are adjusted to equation 1 (c) or equation 2 (a, a’, b, b’, c’).
From a structural point of view, the compound sulfonyl group is linked to the piperazinyl moiety and to cycle 1, a diversely substituted cycle by phenyl (B1a-i, B3), naphthyl (B2a-b) or benzyl (B4) groups. The second piperazinyl nitrogen is linked to monosubstituted (B1a, B2b) or disubstituted (B1b-i, B2a, B4) phenyl cycle 2. For B3, the spacer CO-CH2-O ensures the covalent link between piperazinyl nitrogen and a naphthyl group. Compounds B1a-c inhibited selectively the T-L activity of both, the iCP and cCP. A poor influence of the nature of cycle 1 R1 substituent, [isobutyl (B1a) or tert-butyl (B1b-c)] and cycle 2 ones [R2 = 4’-NO2 (B1a), R2 = 2’-CH3 and R4 = 3’-Cl (B1b); R2 = 2’-NO2 and R4 = 4’-Cl (B1b)] was observed for cCP with IC50 values ranking from 8.1 ± 0.5 to 10.3 ± 0.8 µM. Compounds B1a, B1b and B1c were more efficient on iCP than on cCP with cCP/iCP ratios for the IC50 values of 1.4, 4.6 and 2.4, respectively. The introduction of pyridyl (B1d and B1g), furanyl (B1e and B1f) or morpholino (B2a and B1h) heterocycles in the 3’-cycle 2 substituent failed to maintain the T-L inhibitory activity whereas ChT-L and PA activities were both inhibited. This indicates that cycle 2 3’-substituents were not tolerated in the β2 subunit binding region. The PA activity of iCP was slightly more efficiently inhibited by B1e and B1g than the ChT-L one (factor of ≈ 2.2) whereas the opposite was observed for the inhibition of cCP (factors of 1.7 and 2.3 for B1e and B1f respectively). The ratio cCP/iCP was respectively of 1.3 and 2 for B1e and B1f (PA activity), and 0.55 and 0.45 (ChT-L activity). For both enzymes, the attachment of the heterocyclic substituent at the 3’ position of cycle 2 seemed more favorable using the arm S(or NH)CH2 (B1e-g) than a shorter (B1d) or a longer (B1h) one. Changing cycle 1 R1, 4-CH3 (B1e) to 4-Cl (B1f), retained the in vitro ChT-L and PA inhibitory activities. The replacement of the phenyl cycle 1 by the bicyclic naphthyl (B2a) was detrimental, mostly if the 3-substituent (NH-CH2-furane) was replaced by hydrogen. Conversely, the replacement of cycle 2 by the motif COCH2O-naphthyl (B3) led to a molecule inhibiting quasi-equally the ChT-L and PA activities of both iCP and cCP. These results highlight the importance of the nature of the substituent present on cycle 2 of the piperazine sulfonylamide central core for either the selective inhibition of the T-L activity, or for the simultaneous inhibition of the ChT-L and PA activities. Moreover, these substitutions may be used to guide preferentially the inhibition towards iCP versus cCP. A selective inhibition of the proteasomal T-L activity was also observed with a related quinoline-sulfonyl hybrid [43].
The subunit preference is supported by computational analysis of selected compounds
Crystal structures of murine iCP and cCP complexes provided valuable insights over the subtle and complex structural differences among the different active sites [29]. For instance, subunit β5i can accommodate larger amino acid side chains at position P1 than β5c. This difference in size was partly attributed to distinct conformation of Met 45. The opposite is expected for the P3 residue due to Ser 27, which is substituted by Ala in β5c. The S3 pocket is more hydrophilic in β5i as compared to β5c. Differences were also noticed for the S1 pocket of β1i, which is more hydrophobic and smaller than that of β1c, and for the subunit β1i where the S3 pocket is of smaller size and more polar than that of β1c. On the other hand, all β2 subunits harbor a relatively large S1 pocket and overall β2c and β2i are structurally very similar in the substrate-binding channel.
The exact position of the here identified inhibitors in the different catalytic centers would require X-ray crystallography studies but attempts to obtain crystals for the presented compounds have so far failed. Yet, it has been possible to crystallize some sulfonamide ligands at the β1/β2 interface [41] related to the compounds that we identified by virtual screening [15], indicating that molecules selected by computational means can help gaining knowledge over this highly complex enzymatic system. To facilitate the reading of the structural and chemical data, we decided to present the analysis of two inhibitors that are chemically related but possess different properties and to rationalize their likely mechanism of actions in the light of the recently reported human cCP crystal structure [44] and of several other structural studies [11, 29, 45, 46]. The B1b and B1e inhibitors were docked with two different methods in all three binding centers of the constitutive human proteasome and compared with the mouse immuno- and constitutive proteasomes.
The most likely position of inhibitor B1e in the ChT-L binding site of cCP is shown in Figure 3. In this orientation the sulfonyl group would make hydrogen bonds with Thr 1 and Gly 47, two important residues of the active site known from X-ray crystallography studies to interact with such chemical group in a similar fashion [41, 46, 47]. In this orientation, the 4-CH3-phenyl group of molecule B1e could nicely fit into the hydrophobic and relatively large S1-specificity pocket and have favorable interactions with Met 45, an important amino acid of this subpocket. This orientation would thus mimic the P1-leucine side chain of the covalent bortezomib inhibitor co-crystallized with the human proteasome molecule or of the C-terminal benzylic group of compound A1b [11] and A2 [12] also co-crystallized with the proteasome. The piperazine group of B1e could have additional hydrophobic interactions and form several hydrogen bonds in the S2-S3 subsites. The nitrogen atoms would indeed adopt a position relatively similar to the ones observed for bortezomib. Other hydrophobic contacts and polar contacts are seen in the large S4 and S5 subsites and the NO2 group of B1e is here expected to form hydrogen bonds with the protein backbone and possibly with the Lys 136 side chain of the β6 subunit. Such interactions of a NO2 group would be consistent with the type of interactions that such chemical function would make. In fact we analyzed over 50 X-ray structures downloaded from the Protein Data Bank that contain a NO2 group including the nitro-biaryl ether macrocycle derived from TMC-95 in complex with the proteasome [45]. Nitro groups NO2 tend to have interactions with backbone atoms, but interact very often with the Lys and Arg side chains and can also have favorable interactions with Asn and His. All these contacts could take place in the iCP as suggested by the structural alignment of mouse iCP and cCP over the structure of human cCP. Interestingly, none of the 20 generated poses allowed similar types of positioning and interactions for the inhibitor B1b that was found inactive on the ChT-L site. In fact, even if the sulfonyl group were to be oriented differently, the hydrophobic regions of the B1b molecule would tend to point toward polar or charged amino acid side chains. Thus, B1b, with its two hydrophobic heads, does not seem to fit properly in the chymotrypsin-like sites.
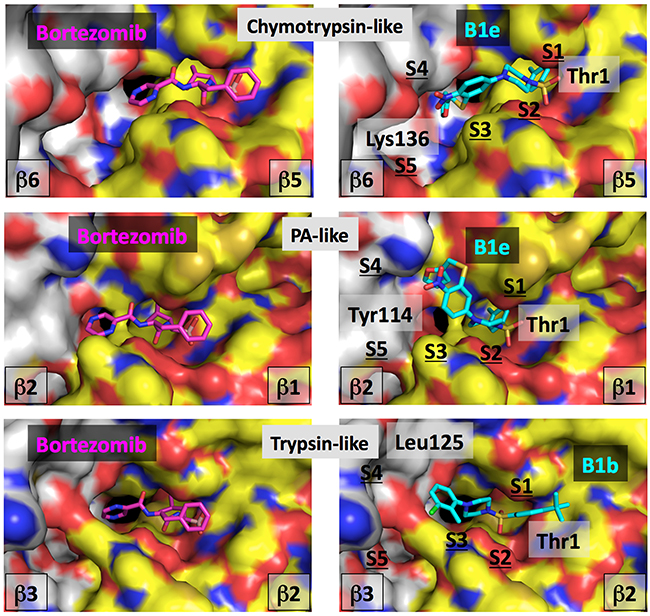
Figure 3: Computational analysis of compounds B1e and B1b. Left, bortezomib is shown co-crystallized with the human cCP in the catalytic centers. Right, compounds B1e or B1b are positioned into the equivalent centers by computational approaches. The same color-coding is applied in all the images. The molecular surface of the catalytic subunit is shown in yellow (C atoms), blue (N atoms), red (O atoms) and brown (S atoms) while for the surface of the adjacent subunit, the color is white (C atoms), blue (N atoms), red (O atoms) and brown (S atoms). The color code for the bortezomib atoms is: C atoms are in magenta, N atoms in blue, O atoms in red and the B atom is in light red. For compounds B1e and B1b, C atoms are in cyan, O are in red, N are in blue and S are in yellow. Some residues are labeled for orientation (see text).
With regard to the PA site, it can be seen in Figure 3 that compound B1e could fit in this active center in a tilted position as compared to the one observed in the ChT-L center. In the β1 subunit, the S1 specificity pocket contains for instance Arg 45 but the hydrophobic head of the B1e inhibitor can make some favorable contacts with the hydrophobic moiety of the Arg residue, a situation in part observed in the co-crystal structure of the bortezomib-human constitutive proteasome complex. The somewhat titled orientation of B1e as compared to bortezomib (Figure 3) in this catalytic center is in fact very similar to the tilt of inhibitor A1b co-crystallized with the proteasome ChT-L center [11]. The B1e inhibitor would make additional contacts with for instance Tyr 114 of the adjacent β2 subunit (an Asp in the equivalent position of the β6) and possibly hydrogen bonds can be formed between the NO2 group of the B1e compound and the nearby β2 His 116. Superimposition of the mouse cCP and iCP onto the human docked structure suggests that molecule B1e could interact equally well with both types of proteasome. Possibly, molecule B1b, that does not inhibit significantly the β1 site, is too small to make sufficient hydrophobic contacts with residues of the β2 subunit in the S4-S5 subpocket area while hydrogen bonds cannot form in the S4 region.
In the T-L center (β2), molecule B1e was found to be essentially inactive while molecule B1b was active. In Figure 3, a possible position of B1b is seen. The molecule occupies the site in a similar fashion than bortezomib except for the S1 site. The T-L S1 pocket is large and it was found that the P1-leucine residue of bortezomib does not form hydrophobic contacts there [48]. In our docking study, it is not clear if the hydrophobic and aromatic cycle 1 of inhibitor B1b points in the S2 pocket, like the phenylalanine P2 residue of bortezomib or if this cycle points into the S1 pocket. Considering the size of the S1 pocket, this could be definitively possible without changing the overall orientation of the compound although this may not be favorable from an energetic point of view as this pocket contains in the human cCP the negatively charged Asp 53 residue, also present in the mouse enzyme while it is Glu 53 in the mouse immunoproteasome. The S1 pocket is thus essentially hydrophilic and in part negatively charged and this might be the reason why no docking poses could fit the molecule such that it makes contact with the S1 subpocket and still has a relatively good predicted binding score. In this orientation, hydrogen bonds could form between the B1b molecule and sub-pockets S2 and S3.
The other hydrophobic ring of inhibitor B1b could have hydrophobic interactions in the area of the S4 subpocket, for example with residues Leu 125 and Ile 126, a situation highly related to the interaction found for a phenyl group distal to the P1 of the larger LU-102 inhibitor (azido-Phe-Leu-Leu-4-aminomethyl-Phe(P1 residue)-methyl vinyl sulfone) co-crystallized with yeast proteasome (LU-102 is interacting with the equivalent Leu and Ile residues in yeast) [46]. Nearby Leu 125, three negatively charged residues are found in the human cCP, β2 Glu 22 and β3 Asp 124 and Glu 105 that should repel the NO2 group of B1e. This situation is conserved in the mouse iCP although Glu 22 is an Asn but at position 23, an Asp is found (Gly in the human cCP). Taken together, the presence of a negative charge in the S1 pocket and in the S3-S4-S5 areas would not favor the binding of molecule B1e due to unfavorable contacts with cycle 1 and the NO2 group of cycle 2 while it could be possible to fit the more hydrophobic B1b molecule.
Inhibition of tumor cells proliferation and invasion by B1f and B3 compounds
We first analyzed the cytotoxic effects of B1f and B3 treatment on colon (CT-26) and breast cancer cells (MDA-MB-231 cells) using the MTT cell viability assay. B1f and B3 inhibited the viability of these cells in a dose-dependent manner after 48 h treatment. Higher doses of B1f and B3 lead to 100% cytotoxicity (Figure 4A). For MDA-MB-231 cells the LC50 values were 15.4 ± 4.7 and 45.8 ± 5.2 μM for B3 and B1f, respectively. For the CT26 cells the LC50 values were 26.4 ± 1.7 and 24.4 ± 8.6 μM for B3 and B1f, respectively. We next assessed the effect of B1f and B3 on the proliferation of these cells during 24h. As can be seen, in the presence of 10 μM of B1f and B3 compounds, tumor cells exhibited significant reduction in their proliferation in comparison to control cells (Figure 4B). Similarly, to evaluate whether these inhibitors can affect their invasion, tumor cells were incubated for 24 h in a microchemotaxis chamber pre-coated with collagen IV (Figure 4C) in the presence of the compounds B1f or B3. Here again, treatment of these cells with the inhibitors (10 μM) clearly decreased their ability to invade (Figure 4C).
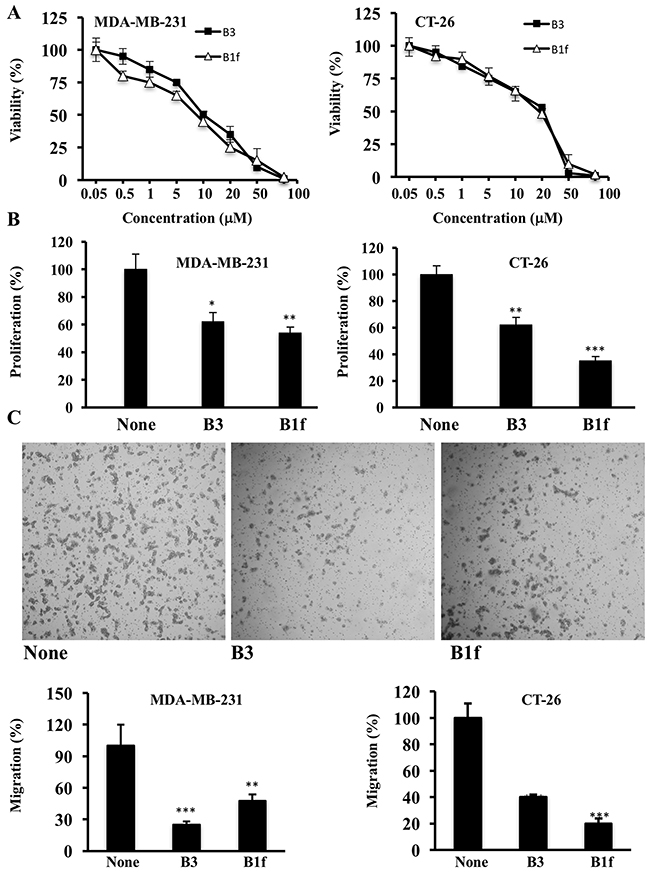
Figure 4: Effect of compounds B1f and B3 on tumor cell proliferation and invasion. A. CT-26 and MDA-MB 231 cells were treated with various concentrations of indicated compounds for 48h and cells viability was measured by MTT assay. B. Tumor cells were serum deprived overnight and then treated for 24 h with control media (None) or media containing compound B3 or B1f. Cell proliferation was assessed using Cell Titer96 non-radioactive cell proliferation assay. Results are shown as means ± S.E. of three experiments performed in triplicate. *P < 0.05. **P < 0.001. *** P < 0.0001. C. Indicated tumor cells were incubated in a microchemotaxis chamber pre-coated with collagen IV for cell invasion assay as described in the Materials and Methods section. The results are represented as the percentage of invading cells. Data are representative of 3 experiments and shown as mean ± S.E. (n=3 per group). **P < 0.001. *** P < 0.0001.
The discovery of piperazine sulfonamides broadens the range of potential scaffolds able to inhibit the immunoproteasome. Moreover, compounds that inhibit the trypsin activities of iCP and cCP as summarized in Figure 5 are still relatively rare. Several here described compounds constitute interesting starting points for the development of noncovalent immunoproteasome-selective inhibitors susceptible to target selectively the malignant phenotype of cancer cells.
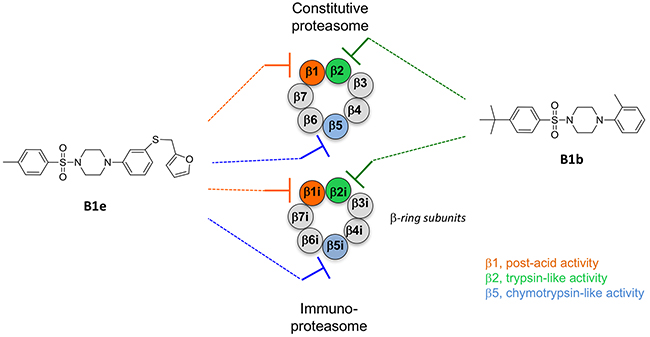
Figure 5: Schematic representation of the β-ring subunits of the cCP and iCP and their inhibition by representative sulfonyl piperazines.
MATERIALS AND METHODS
The compounds selected by virtual ligand screening were purchased from ChemBridge corporation (www.chembridge.com). Purified human constitutive 20S proteasome cCP and human 20S immunoproteasome iCP from erythrocyte were obtained from Boston Biochem (Cambridge, USA). All fluorogenic substrates (Suc-LLVY-AMC, Boc-LRR-AMC, and Z-LLE-βNA) were obtained from Bachem (Weil am Rhein, Germany). Other reagents and solvents were purchased from commercial sources. Fluorescence was measured using a BMG Fluostar microplate reader (black 96-well microplates).
Computational analysis
The virtual screening procedure initially used to identify the inhibitors was previously described [15]. Briefly, the in silico screening procedure was performed on the cCP ChT-L active site and using the 3D structure of the bovine proteasome [49] (the human and bovine subunits that carry the catalytic activity are 94-100% identical). The 3D structure used (PDB code 1IRU) was prepared for virtual screening computations (e.g., addition of hydrogen atoms at pH 7, short energy minimization, etc). Several docking software packages were used including Fred (OpenEye Scientific Software, Santa Fe, NM), Surflex [50] and LigandFit [51] to screen the ChemBridge compound collection (about 400,000 compounds that were filtered with our tool FAF-Drugs to remove compound with unwanted physicochemical properties [52]). The filtering process led to a collection containing about 300,000 molecules (about 180,000 clusters with families containing a few members up to about 20-30 members) that were then docked into the catalytic site. A consensus list of molecules possessing the best predicted binding scores with Surflex and LigandFit was generated and after visual inspection, the selected molecules were purchased from ChemBridge (www.chembridge.com) and tested experimentally. Prior to ordering the molecules, physicochemical properties, the search for structural alerts and for the presence of substructures potentially interfering with assays (e.g., PAINS) were investigated again with our online server FAF-Drugs3 [53]. With regard to physicochemical properties, the filtering was soft and, for instance, molecules with MW around 500 were tolerated as our goal was to probe these enzymes and find novel starting points. Similarly, the filtering on structural alerts was soft and we decided to keep aromatic NO2 as over 15 approved drugs contain such a group and because such group can be substituted during the optimization stages if needed. With regard to group potentially interfering with assays, we have noticed over the years that many such groups can be flagged after analysis of their structures but turn out to be inactive in various assays, to display structure-activity relationship (SAR), to co-crystallize with targets, to be present in approved drugs and indeed to be noninterfering and as such valuable chemical probes and starting points. We thus annotated our molecules but were permissive with this filtering step. For instance, compound B1a was flagged in silico as potential assay interfering compound but highly similar compounds are co-crystallized with targets and display SAR (e.g, PDB code 3CZR, target 11beta-HSD). The behavior of B1a (and other compounds) in our experiments support this selection process as if the molecules were to be interfering with the assays, they will be active/inactive on the different catalytic activities and small structural changes in the compounds would not impact affinity or selectivity. Selected ligands after experimental testing were prepared with our compound preparation package [53] and re-docked with the last version of Surflex and our online server MTiOpenScreen [54] into the three catalytic centers of the recently reported human cCP [44] taking into account several other structural studies [11, 29, 45, 46]. The twenty best-scored poses for each inhibitor were initially selected. The interactive structural analysis and figures were done with molecular graphics package PyMOL (Schrodinger LLC, USA). A good agreement for the lowest energy poses between the two docking packages was noticed and interestingly, even the docking performed over the entire surface of a pair of subunits via our server MTiOpenScreen led to the positioning of the compounds in the catalytic site with an orientation similar to the one observed by the two docking methods and with the lowest predicted binding scores. The selection of the final most likely pose for each molecule and for the molecules shown in this study was performed after analysis of the score, comparisons with experimental structures co-crystallized with ligands containing similar or related substructures/fragments and also after computations of various non-bonded interactions (e.g., salt-bridges, hydrogen bonds, pi-stacking…).
Proteasome inhibition assays
Proteasome activities were determined by monitoring for 45 min at 37°C the hydrolysis of the appropriate fluorogenic substrate Suc-LLVY-AMC (ChT-L activity) and Boc-LRR-AMC (T-L activity) using λexc = 360 nm, λem = 460 nm, and Z-LLE-βNA (PA activity) using λexc = 340, λem = 405 nm, in the presence of untreated (control) or proteasome (iCP or cCP). Substrates and compounds were previously dissolved in DMSO. The buffers were (pH 8.0): 20 mM Tris, 10% (v/v) glycerol, 0.01% (w/v) SDS, and 2% (v/v) DMSO (ChT-L and PA activities); 20 mM Tris, 10% (v/v) glycerol, 0.01% (w/v). The final iPR and cPR concentrations were 0.3 nM using 20 µM Suc-LLVY-AMC (ChT-L), 50 µM Boc-LRR-AMC (T-L) and 100 µM Z-LLE-βNA (PA). Using the appropriate substrate, the compounds (0.1-100 µM) were tested in duplicate for each inhibitor concentration to detect their potential to inhibit the ChT-L, T-L, and PA activities. The enzyme and the inhibitors were incubated for 15 min before the measurement of the enzyme activity. Initial rates determined in control experiments (V0) were considered to be 100 % of the peptidase activity; initial rates (Vi) that were above 100 % in the presence of a tested compound were considered to be activations (expressed as activation factor), while initial rates below 100 % were considered to be inhibitions. The inhibitory activity of compounds was expressed as IC50 (inhibitor concentrations giving 50 % inhibition). The values of IC50 were calculated by fitting the experimental data to the equation 1 or equation 2, where nH is the Hill number.
Viability and proliferation assays
For the viability assay, CT-26 (colon cancer) and MDA-MB231 (breast cancer) cells were plated in triplicate on 96 wells plate (5.103/well). After treatment with indicated drugs, cells were incubated for 48 hours at 37°C and incubated with solution of 5 mg/ml of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) tetrazolium substrate (Sigma) for 30 minutes at 37°C prior analysis on a microplate reader at 570. The LC50 value was defined as the drug concentration causing 50% cell death (LD = lethal dose) compared to growth of the untreated control cells. For cell proliferation, cells were plated in triplicate on 96 wells plate (5.103/well) under serum free conditions for 24 h. The starved cells were then incubated for 24h with fresh media containing the two selected compounds B1f and B3 that inhibit both ChT-L and PA activities of iCP and cCP. These two compounds were chosen based on reported studies describing that the simultaneous inhibition of ChT-L and PA activities by two selective inhibitors of these activities seemed to enhance the anticancer efficiency [55]. Proliferation level in cells was evaluated, as reported previously [56] using the Cell Titer96 non-radioactive cell proliferation assay kit (Promega) according to manufacturer’s protocol
Cell invasion assay
Cell invasion were determined using 24-well microchemotaxis chambers precoated with 7.5 μg collagen type IV (Becton Dickinson Labware), as previously described [57]. Tumor cells were resuspended in serum-free media and loaded into upper chamber of each well. Cells were incubated at 37°C for 24 h in the presence or the absence of B1f and B3 compounds, after which, the filters were fixed and stained with Diff-Quik (Medion Diagnostic). Cell invasion were quantified by determination of the number of cells that migrated directly through the membrane toward the medium containing 10% serum that was used as a chemoattractant. Cells detected in each well were counted and the results were represented as (number of migrated cells/number of total cells) × 100%.
Statistical analysis
All data are presented as ± standard error of mean (SE) unless specifically mentioned. Student’s t test was applied for statistical analysis, as appropriate. P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank Pr. Dr. Michael Groll (TUM, Garching, Germany) for crystallographic assays and useful comments as well as his collaborators Dr. Philipp Beck and Dr. Camille Le Chapelain. We also thank Claude Lalou for his technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by La Ligue Contre le Cancer to AMK, Association Française contre les Myopathies (AFM) to JV and MRR (grant number 14999). Supports from the Centre National de la Recherche Scientifique (JV and MRR), INSERM (BOV, MAM, AMK), the Universities Pierre et Marie Curie (UPMC-Sorbonne Universiteés) (MRR), Paris Diderot (BOV, MAM), and Rennes I (JV) are greatly appreciated. XM received financial support from the Ministère de l’Enseignement Supeérieur et de la Recherche (France).
REFERENCES
1. Hershko A and Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998; 67:425-479.
2. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004; 5:417-421.
3. Mani A and Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. Journal of clinical oncology. 2005; 23:4776-4789.
4. Adams J and Kauffman M. Development of the proteasome inhihitor Veleade((TM)) (Bortezomib). Cancer Investigation. 2004; 22:304-311.
5. Kane RC, Dagher R, Farrell A, Ko C-W, Sridhara R, Justice R and Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clinical Cancer Research. 2007; 13:5291-5294.
6. Shah JJ, Stadtmauer EA, Abonour R, Cohen AD, Bensinger WI, Gasparetto C, Kaufman JL, Lentzsch S, Vogl DT, Gomes CL, Pascucci N, Smith DD, Orlowski RZ and Durie BG. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood. 2015; 126:2284-2290.
7. Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, Sandhu I, Ganly P, Baker BW, Jackson SR, Stoppa AM, Simpson DR, Gimsing P, Palumbo A, Garderet L, Cavo M, et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. The New England journal of medicine. 2016; 374:1621-1634.
8. Huber EM and Groll M. Inhibitors for the Immuno- and Constitutive Proteasome: Current and Future Trends in Drug Development. Angew Chem Int Ed. 2012; 51:8708-8720.
9. Genin E, Reboud-Ravaux M and Vidal J. Proteasome inhibitors: recent advances and new perspective in medicinal chemistry. Curr Top Med Chem. 2010; 10:232-256.
10. Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS, Bump N, Olhava EJ, Fleming P, Dick LR, Tsu C, Sintchak MD, et al. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta 5-subunit. Biochem J. 2010; 430:461-476.
11. Blackburn C, Barrett C, Blank JL, Bruzzese FJ, Bump N, Dick LR, Fleming P, Garcia K, Hales P, Hu Z, Jones M, Liu JX, Sappal DS, Sintchak MD, Tsu C and Gigstad KM. Optimization of a series of dipeptides with a P3 threonine residue as non-covalent inhibitors of the chymotrypsin-like activity of the human 20S proteasome. Bioorganic & medicinal chemistry letters. 2010; 20:6581-6586.
12. Groll M, Gallastegui N, Marechal X, Le Ravalec V, Basse N, Richy N, Genin E, Huber R, Moroder L, Vidal J and Reboud-Ravaux M. 20S Proteasome inhibition: designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem. 2010; 5:1701-1705.
13. Desvergne A, Cheng Y, Grosay-Gaudrel S, Maréchal X, Reboud-Ravaux M, Genin E and Vidal J. Noncovalent Fluorescent Probes of Human Immuno- and Constitutive Proteasomes. J Med Chem. 2014; 57:9211-9217.
14. Desvergne A, Genin E, Maréchal X, Gallastegui N, Dufau L, Richy N, Groll M, Vidal J and Reboud-Ravaux M. Dimerized Linear Mimics of a Natural Cyclopeptide (TMC-95A) Are Potent Noncovalent Inhibitors of the Eukaryotic 20S Proteasome. J Med Chem. 2013; 56:3367-3378.
15. Basse N, Montes M, Marechal X, Qin LX, Bouvier-Durand M, Genin E, Vidal J, Villoutreix BO and Reboud-Ravaux M. Novel organic proteasome inhibitors identified by virtual and in vitro screening. J Med Chem. 2010; 53:509-513.
16. Gallastegui N, Beck P, Arciniega M, Huber R, Hillebrand S and Groll M. Hydroxyureas as noncovalent proteasome inhibitors. Angew Chem Int Ed. 2012; 51:247-249.
17. Maréchal X, Genin E, Qin L, Sperandio O, Montes M, Basse N, Richy N, Miteva MA, Reboud-Ravaux M, Vidal J and Villoutreix BO. 1,2,4-oxadiazoles identified by virtual screening and their non-covalent inhibition of the human 20S proteasome. Curr Med Chem. 2013; 20:2351-2362.
18. Kikuchi J, Shibayama N, Yamada S, Wada T, Nobuyoshi M, Izumi T, Akutsu M, Kano Y, Sugiyama K, Ohki M, Park SY and Furukawa Y. Homopiperazine derivatives as a novel class of proteasome inhibitors with a unique mode of proteasome binding. PloS one. 2013; 8:e60649.
19. Miller Z, Kim K-S, Lee D-M, Kasam V, Baek SE, Lee KH, Zhang Y-Y, Ao L, Carmony K, Lee N-R, Zhou S, Zhao Q, Jang Y, Jeong H-Y, Zhan C-G, Lee W, et al. Proteasome Inhibitors with Pyrazole Scaffolds from Structure-Based Virtual Screening. J Med Chem. 2015; 58:2036-2041.
20. Koroleva ON, The Hien P, Bouvier D, Dufau L, Qin L, Reboud-Ravaux M, Ivanov AA, Zhuze AL, Gromova ES and Bouvier-Durand M. Bisbenzimidazole derivatives as potent inhibitors of the trypsin-like sites of the immunoproteasome core particle. Biochimie. 2015; 108C:94-100.
21. Sosič I, Gobec M, Brus B, Knez D, Živec M, Konc J, Lešnik S, Ogrizek M, Obreza A, Žigon D, Janežič D, Mlinarič-Raščan I and Gobec S. Nonpeptidic Selective Inhibitors of the Chymotrypsin-Like (β5 i) Subunit of the Immunoproteasome. Angew Chem Int Ed. 2016; 55:5745-5748.
22. Lee W and Kim KB. The immunoproteasome: an emerging therapeutic target. Curr Top Med Chem. 2011; 11:2923-2930.
23. Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M and Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood. 2009; 113:4667-4676.
24. Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ and Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug resistance updates. 2015; 18:18-35.
25. Egerer T, Martinez-Gamboa L, Dankof A, Stuhlmuller B, Dorner T, Krenn V, Egerer K, Rudolph PE, Burmester GR and Feist E. Tissue-specific up-regulation of the proteasome subunit beta5i (LMP7) in Sjogren’s syndrome. Arthritis and rheumatism. 2006; 54:1501-1508.
26. Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, Ferrer I, Avila J and Lucas JJ. Neuronal induction of the immunoproteasome in Huntington’s disease. The Journal of neuroscience. 2003; 23:11653-11661.
27. Mishto M, Bellavista E, Santoro A, Stolzing A, Ligorio C, Nacmias B, Spazzafumo L, Chiappelli M, Licastro F, Sorbi S, Pession A, Ohm T, Grune T and Franceschi C. Immunoproteasome and LMP2 polymorphism in aged and Alzheimer’s disease brains. Neurobiology of aging. 2006; 27:54-66.
28. Singh AV, Bandi M, Aujay MA, Kirk CJ, Hark DE, Raje N, Chauhan D and Anderson KC. PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. British journal of haematology. 2011; 152:155-163.
29. Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M and Groll M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell. 2012; 148:727-738.
30. Guedes RA, Serra P, Salvador JA and Guedes RC. Computational Approaches for the Discovery of Human Proteasome Inhibitors: An Overview. Molecules. 2016; 21:927.
31. Miller Z, Ao L, Kim KB and Lee W. Inhibitors of the Immunoproteasome: Current Status and Future Directions. Curr Pharm Des. 2013; 19:4140-4151.
32. Kisselev AF and Groettrup M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr Opin Chem Biol. 2014; 23:16-22.
33. Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ and Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. 2009; 15:781-787.
34. de Bruin G, Huber EM, Xin B-T, van Rooden EJ, Al-Ayed K, Kim K-B, Kisselev AF, Driessen C, van der Stelt M, van der Marel GA, Groll M and Overkleeft HS. Structure-Based Design of β1i or β5i Specific Inhibitors of Human Immunoproteasomes. J Med Chem. 2014; 57:6197-6209.
35. Lei B, Abdul Hameed MDM, Hamza A, Wehenkel M, Muzyka JL, Yao X-J, Kim K-B and Zhan C-G. Molecular Basis of the Selectivity of the Immunoproteasome Catalytic Subunit LMP2-Specific Inhibitor Revealed by Molecular Modeling and Dynamics Simulations. J Phys Chem B. 2010; 114:12333-12339.
36. Dubiella C, Cui H, Gersch M, Brouwer AJ, Sieber SA, Krueger A, Liskamp RMJ and Groll M. Selective Inhibition of the Immunoproteasome by Ligand-Induced Crosslinking of the Active Site. Angew Chem Int Ed. 2014; 53:11969-11973.
37. Dubiella C, Baur R, Cui H, Huber EM and Groll M. Selective Inhibition of the Immunoproteasome by Structure-Based Targeting of a Non-catalytic Cysteine. Angew Chem Int Ed. 2015; 54:15888-15891.
38. Fan H, Angelo NG, Warren JD, Nathan CF and Lin G. Oxathiazolones Selectively Inhibit the Human Immunoproteasome over the Constitutive Proteasome. ACS Med Chem Lett. 2014; 5:405-410.
39. Groll M, Korotkov VS, Huber EM, de Meijere A and Ludwig A. A Minimal β-Lactone Fragment for Selective β5c or β5i Proteasome Inhibitors. Angew Chem Int Ed. 2015; 54:7810-7814.
40. Singh PK, Fan H, Jiang X, Shi L, Nathan CF and Lin G. Immunoproteasome beta5i-Selective Dipeptidomimetic Inhibitors. ChemMedChem. 2016; 11:2127-2131.
41. Beck P, Reboud-Ravaux M and Groll M. Identification of a β1/β2-Specific Sulfonamide Proteasome Ligand by Crystallographic Screening. Angew Chem Int Ed. 2015; 54:11275-11278.
42. Miteva MA, Lee WH, Montes MO and Villoutreix BO. Fast structure-based virtual ligand screening combining FRED, DOCK, and Surflex. J Med Chem. 2005; 48:6012-6022.
43. Pundir S, Vu HY, Solomon VR, McClure R and Lee H. VR23: A Quinoline-Sulfonyl Hybrid Proteasome Inhibitor That Selectively Kills Cancer via Cyclin E-Mediated Centrosome Amplification. Cancer research. 2015; 75:4164-4175.
44. Schrader J, Henneberg F, Mata RA, Tittmann K, Schneider TR, Stark H, Bourenkov G and Chari A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science (New York, NY). 2016; 353:594-598.
45. Groll M, Goetz M, Kaiser M, Weyher E and Moroder L. TMC-95-Based Inhibitor Design Provides Evidence for the Catalytic Versatility of the Proteasome. Chem Biol 2006; 13:607-614.
46. Geurink PP, van der Linden WA, Mirabella AC, Gallastegui N, de Bruin G, Blom AEM, Voges MJ, Mock ED, Florea BI, van der Marel GA, Driessen C, van der Stelt M, Groll M, et al. Incorporation of Non-natural Amino Acids Improves Cell Permeability and Potency of Specific Inhibitors of Proteasome Trypsin-like Sites. J Med Chem. 2013; 56:1262-1275.
47. Groll M, Nazif T, Huber R and Bogyo M. Probing structural determinants distal to the site of hydrolysis that control substrate specificity of the 20S proteasome. Chemistry & Biology. 2002; 9:655-662.
48. Groll M, Berkers CR, Ploegh HL and Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006; 14:451-456.
49. Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N and Tsukihara T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure. 2002; 10:609-618.
50. Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003; 46:499-511.
51. Venkatachalam CM, Jiang X, Oldfield T and Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. Journal of molecular graphics & modelling. 2003; 21:289-307.
52. Miteva MA, Violas S, Montes M, Gomez D, Tuffery P and Villoutreix BO. FAF-Drugs: free ADME/tox filtering of compound collections. Nucleic acids research. 2006; 34:W738-744.
53. Lagorce D, Sperandio O, Baell JB, Miteva MA and Villoutreix BO. FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic acids research. 2015; 43:W200-207.
54. Labbe CM, Rey J, Lagorce D, Vavrusa M, Becot J, Sperandio O, Villoutreix BO, Tuffery P, Miteva MA. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic acids research. 2015; 43:W448-54.
55. Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, Tokhunts RA, Amir O, Goddard AL, Pelphrey PM, Wright DL, Overkleeft HS and Kisselev AF. Selective Inhibitor of Proteasome’s Caspase-like Sites Sensitizes Cells to Specific Inhibition of Chymotrypsin-like Sites. Chemistry & Biology. 2009; 16:1278-1289.
56. Khatib AM, Lahlil R, Scamuffa N, Akimenko MA, Ernest S, Lomri A, Lalou C, Seidah NG, Villoutreix BO, Calvo F and Siegfried G. Zebrafish ProVEGF-C expression, proteolytic processing and inhibitory effect of unprocessed ProVEGF-C during fin regeneration. PloS one. 2010; 5:e11438.
57. Sfaxi F, Scamuffa N, Lalou C, Ma J, Metrakos P, Siegfried G, Ragg H, Bikfalvi A, Calvo F and Khatib AM. Repression of liver colorectal metastasis by the serpin Spn4A a naturally occurring inhibitor of the constitutive secretory proprotein convertases. Oncotarget. 2014; 5:4195-4210. doi: 10.18632/oncotarget.1966.