
INTRODUCTION
5-fluorouracil (5-FU) is a uracil analogue and widely used antimetabolite agent for the treatment of various cancer types, including colorectal, breast, and head and neck cancer [1, 2]. 5-FU does not exhibit cytotoxic properties prior to intracellular conversion to one of its active metabolites, namely 5-fluorodeoxyuridine monophosphate (5-FdUMP), 5-fluorodeoxyuridine triphosphate (5-FdUTP) or 5-fluorouridine triphosphate (5-FUTP) [1]. 5-FdUMP is a suicide inhibitor of thymidylate synthase (TYMS), the only cellular de novo- source of deoxythymidine monophosphate (dTMP) [1]. Depletion of dTMP leads to deoxynucleotide pool imbalances, resulting in disruption of DNA synthesis and repair [3, 4]. 5-FUTP is misincorporated into RNA, whereby RNA processing and function are impaired [1, 4, 5]. Furthermore, 5-FdUTP incorporation into DNA inhibits DNA synthesis and induces single- and double strand breaks, leading to DNA fragmentation [6-8]. Despite its benefits as a chemotherapeutic agent in neoadjuvant and adjuvant treatment, especially for patients suffering from (metastatic) colorectal cancer, several studies indicate that 5-FU can also induce liver injury [9-17].
Drug-induced liver injury shares several features with non-alcoholic fatty liver disease (NAFLD). In the majority of patients, NAFLD is associated with risk factors reflecting the metabolic syndrome such as obesity, insulin resistance or dyslipidemia. With the continued rise of obesity in the Western countries, the prevalence of NAFLD has followed a similar trend and is today recognized as the most common liver disease worldwide [18, 19]. NAFLD is characterized by hepatic steatosis, which is also the most documented liver pathology observed in association with 5-FU administration [14, 16, 17, 20]. In general, hepatic steatosis is characterized by intravesicular accumulation of fat in the form of triglycerides within hepatocytes [18, 19]. This is due to an imbalance between lipid uptake on the one hand and their combustion or secretion on the other hand [18]. In NAFLD, “simple” steatosis can progress to non-alcoholic steatohepatitis (NASH), a more serious liver condition characterized by death of hepatocytes and liver inflammation with and without hepatic fibrosis [18, 19].
Generally, the presence of steatosis renders the liver more susceptible to hepatic injury [21]. Oxidative stress, mitochondrial dysfunction and cytokine induction have been identified as further factors that sensitize the liver to an inflammatory reaction [18, 19].
Several studies showed that 5-FU induces oxidative stress in cancer cells [22-24]. Therefore, 5-FU might also be able to promote the development of steatohepatitis in the presence of steatosis. Indeed, the so-called chemotherapy-associated steatohepatitis (CASH) is frequently observed in patients treated with combination therapies of 5-FU and irinotecan [9-13]. Although this damage is mostly ascribed to irinotecan, an (additional) pathological role of 5-FU cannot be excluded. To date, the capability of 5-FU to induce CASH has not been investigated in detail. Thus, the aim of this study was to develop in vitro and in vivo models for 5-FU-induced steatohepatitis and to identify the underlying mechanisms for the induction of steatosis and its progression to inflammation.
RESULTS
Effect of 5-FU on hepatocellular viability and lipid accumulation in vitro
Initially, we determined the dose range in which 5-FU did not affect viability of HepG2 cells. Microscopic analysis (Figure 1A) and assessment of LDH levels in the supernatant (Figure 1B) showed that concentrations up to 250 μM 5-FU were non-toxic for HepG2 cells. In response to this 5-FU dose TYMS expression was significantly increased (Supplementary Figure 1A) while expression levels of dihydropyrimidine dehydrogenase (DPD), the 5-FU metabolizing enzyme, did not significantly change (Supplementary Figure 1B). It has been described that expression of genes encoding enzymes relevant to 5-flurouracil metabolism correlates with histologically diagnosed chemotherapy-induced hepatic injury [25]. The missing increase of DPD in our experimental setting may be explained by the application of non-toxic 5-FU doses. However, in this dose range, incubation with 5-FU significantly increased intracellular free fatty acid (FFA) and triglyceride (TG) levels (Figure 1C). Compared to control cells, FFA and TG content approximately doubled in the presence of 200 μM 5-FU. This finding was supported by oil red O staining, showing microvesicular steatosis in 5-FU-treated cells (Figure 1D).
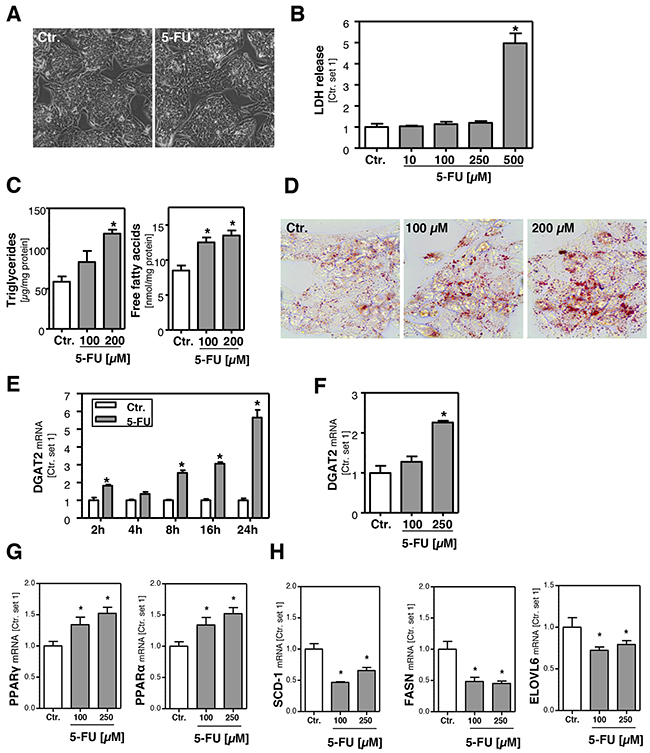
Figure 1: Effect of 5-FU on hepatocellular viability and lipid accumulation. A. Microscopic images of HepG2 cells incubated with 5-FU (250 μM) for 24 h and control cells. B. Quantification of LDH release into the supernatant of HepG2 cells treated with different 5-FU doses as indicated. C. Analysis of intracellular triglycerides and free fatty acids content normalized to total cellular protein. D. Oil red O staining of HepG2 cells incubated with 5-FU for 24 h and control cells. Furthermore, HepG2 cells were incubated with 5-FU (200 μM) for different times as indicated. Moreover, cells were incubated with two different 5-FU doses for 24 h. Analysis of mRNA levels of E, F. DGAT2, G. PPARγ and PPARα and H. SCD-1, FASN and ELOVL6 by quantitative RT-PCR (*: p<0.05 compared to control).
In line with these results, expression levels of diacylglycerol acyltransferase 2 (DGAT2), which catalyzes the terminal step in the formation of triglycerides, were time- and dose-dependently increased in response to 5-FU (Figure 1E, 1F). Interestingly, 5-FU also caused a significant induction of peroxisome proliferator activated receptor alpha (PPAR-α) and PPAR-γ, both critical regulators of hepatic lipid metabolism (Figure 1G). However, expression levels of stearoyl-Coenzyme A desaturase 1 (SCD-1) and fatty acid synthase (FASN) were not increased but even lower in 5-FU-treated compared to untreated cells (Figure 1H). Also expression of fatty acid elongases 6 (ELOVL6), which has been described as critical promoter of nonalcoholic steatohepatitis [26], was reduced by 5-FU (Figure 1H). Together, these findings indicate that the 5-FU-induced FFA and TG accumulation in HepG2 cells is not mediated via de novo lipogenesis.
Effect of 5-FU on β-oxidation and oxidative stress in hepatoma cells in vitro
In search for the underlying mechanisms of 5-FU induced lipid accumulation, we analyzed expression levels of genes that are involved in combustion of fatty acids. Carnitine palmitoyltransferase-1 (CPT-1) is the key enzyme for the transport of fatty acids into mitochondria for β-oxidation [27, 28]. 5-FU caused a dose-dependent induction of CPT-1 expression in HepG2 cells (Figure 2B), whereby the maximum was reached 8 h after stimulation (Figure 2A). ATP-citrate lyase (ACLY) has been described as a regulator of the carnitine system [29], however, also this lipogenic enzyme was downregulated in response to 5-FU treatment (Supplementary Figure 2A, 2B). Interestingly, the increased CPT-1-expression was paralleled by reduced mitochondrial function as analyzed by XTT-assays (Figure 2C, 2D). Impairment of mitochondrial fatty acid combustion can lead to the activation of extra-mitochondrial pathways. Indeed, 5-FU-induced the expression of fatty acid acyl-CoA oxidase 1 (ACOX1) (Figure 2E, 2F). This enzyme catalyzes the initial step for peroxisomal β-oxidation [28]. The 5-FU-induced ACOX1 expression continued to increase beyond the observed time period of 24 h (Figure 2E) and resulted in approximately 4-fold higher expression levels after 24 h stimulation (Figure 2F). Peroxisomal β-oxidation is known to lead to the production of reactive oxygen species (ROS) [27, 30-32], and the antioxidant enzyme heme oxygenase 1 (HMOX1) has been shown to be increased in response to oxidative stress [33]. Stimulation with 5-FU caused a time- and dose-dependent induction of HMOX1 expression levels (Figure 2G, 2H) that paralleled the pattern of 5-FU effects on ACOX1 mRNA levels. Together these data suggest that in response to 5-FU-induced lipid accumulation, mitochondrial and peroxisomal β-oxidation are up-regulated and thus induce oxidative stress, a known inducer of an inflammatory response.
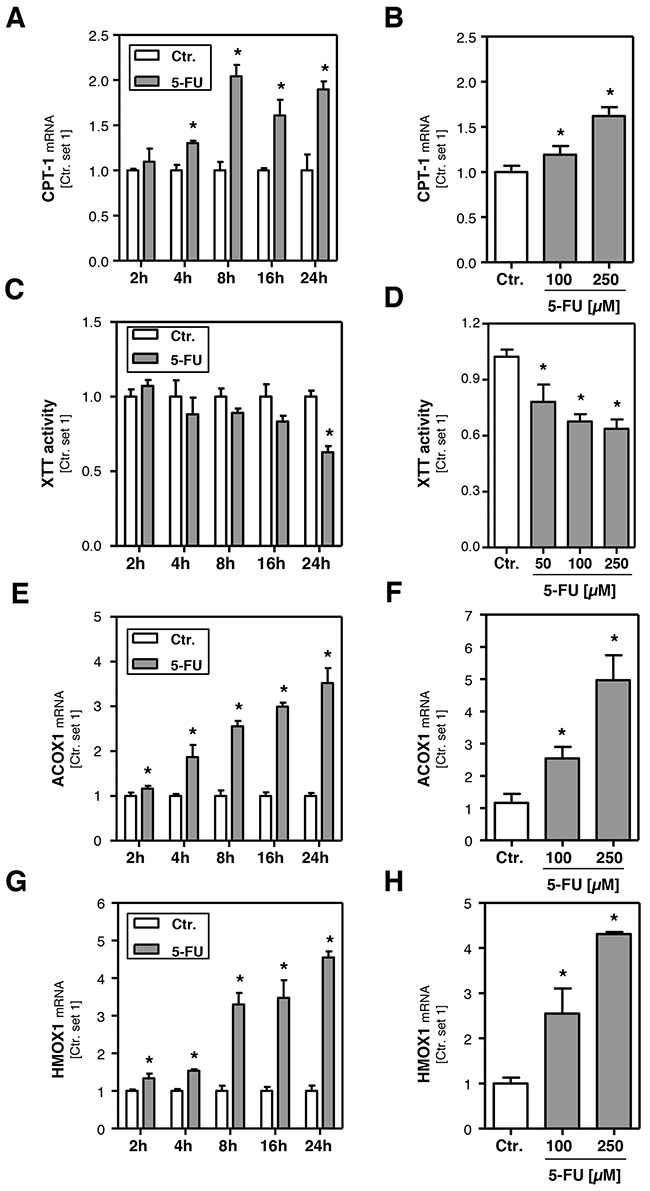
Figure 2: Effect of 5-FU on hepatocellular β-oxidation and oxidative stress. HepG2 cells were incubated with 5-FU (200 μM) for different times as indicated. Furthermore, cells were incubated with two different 5-FU doses for 24 h. Analysis of mRNA levels of A, B. CPT-1, E, F. ACOX1 and G, H. HMOX1 by quantitative RT-PCR. C, D. Analysis of XTT activity. (*: p<0.05 compared to control).
Effect of 5-FU on hepatocellular inflammatory response in vitro
To investigate the potential of 5-FU to induce an inflammatory response the expression levels of interleukin-8 (IL-8), a cytokine involved in the initiation of an inflammatory reaction [34], and intercellular adhesion molecule-1 (ICAM-1), a cell surface molecule that facilitates the adhesion of leukocytes [34], were analyzed. Both IL-8 (Figure 3A, 3B) and ICAM-1 (Figure 3C, 3D) expression levels were markedly increased in response to 5-FU. Expression levels showed a time-dependent increase as early as 2 h after 5-FU treatment and reached a plateau at 16 h (Figure 3A, 3C). Expression of IL-8 and ICAM-1 is dependent on c-Jun N-terminal kinase (JNK) activity [35, 36], and increased JNK activation has also been identified as critical pathological factor in the progression from steatosis to inflammation in NAFLD [37]. Western blot analysis revealed that also 5-FU treatment significantly induced levels of phospho-c-Jun N-terminal kinase 1/2 (phospho-JNK1/2) and phosphorylated c-Jun (Figure 3E). These data suggest that activation of the JNK pathway is mediating 5-FU-induced pro-inflammatory gene expression in hepatoma cells in vitro.
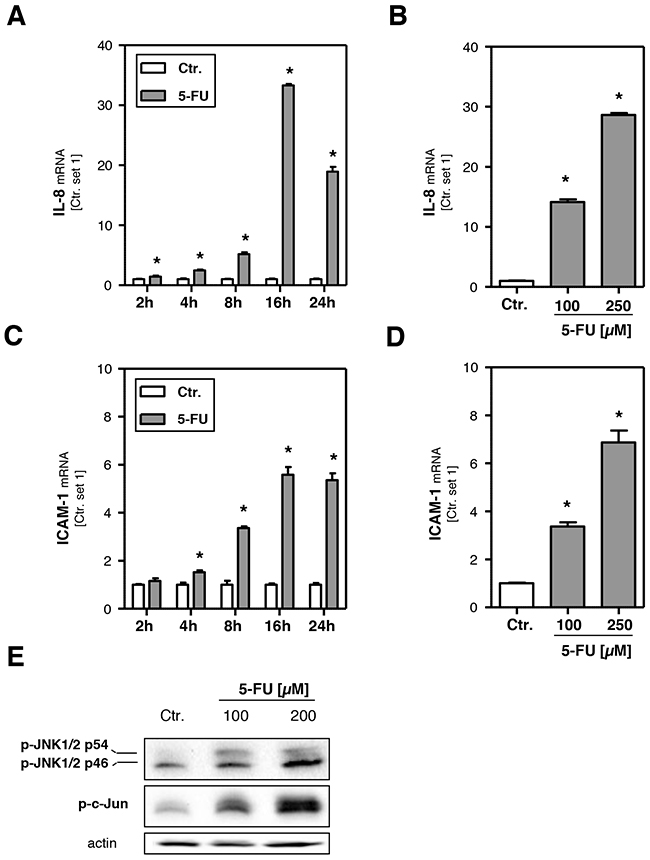
Figure 3: Effects of 5-FU on hepatocellular inflammatory response. HepG2 cells were incubated with 5-FU (200 μM) for different times as indicated and for 24 h with two different doses, respectively. Analysis of mRNA levels of A, B. IL-8 and C, D. ICAM-1 by quantitative RT-PCR (*: p<0.05 compared to control). E. Western Blot analysis of p-JNK1/2 and p-c-Jun protein levels of HepG2 cells incubated with 5-FU for 12 h. Actin protein levels were measured to ensure equal loading.
Effect of 5-FU on primary human hepatocytes in vitro
HepG2 cells are a widely used cellular in vitro model to analyze drug metabolism and toxicity. Still, we wanted to verify observed 5-FU effects in primary human hepatocytes (PHH). A 5-FU dose of 500 μM did not affect the viability of PHH (Figure 4A, 4B), and we used this concentration for subsequent in vitro analyses in PHH. 5-FU stimulation did not affect DPD expression levels in PHH (Supplementary Figure 3A), which were similar as in HepG2 cells (Supplementary Figure 3B). Moreover, also TYMS expression was not altered in primary PHH in response to 5-FU treatment (Supplementary Figure 3C). This difference to HepG2 cells may be explained by the fact that expression levels of this 5-FU target enzyme were almost absent in PHH as compared to HepG2 hepatoma cells (Supplementary Figure 3D). However, also in PHH, 5-FU caused a significant increase of the intracellular FFA and triglyceride level (Figure 4C, 4D). PPAR-α and PPAR-γ expression levels were not significantly altered by 5-FU treatment in PHH (Figure 4E) and also SCD-1, FASN, ACLY and ELOVL6 expression were similar or lower in 5-FU stimulated PHH compared to control cells (Figure 4F and Supplementary Figure 2C). These findings indicated that also in PHH 5-FU-induced steatosis is not mediated via de novo lipogenesis. However, incubation with 5-FU significantly induced ACOX1 and HMOX1 expression (Figure 4G, 4H), as well as expression of pro-inflammatory genes (Figure 4I) in PHH. In summary, the 5-FU effects on PHH reveal a high congruence with the data obtained with HepG2 cells and indicate that this human cell line is a suitable in vitro model to study 5-FU-mediated effects related to hepatocellular steatosis and inflammation.
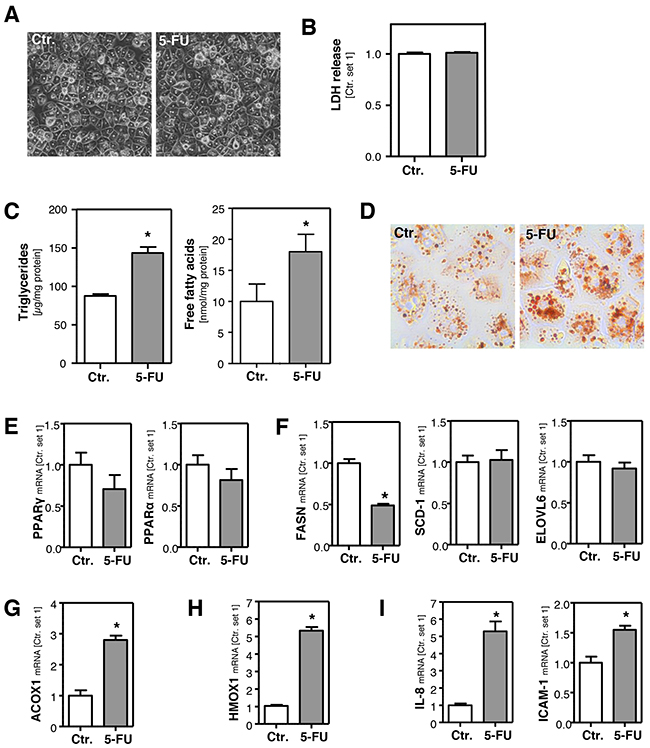
Figure 4: Effect of 5-FU in primary human hepatocytes. Primary human hepatocytes were incubated with 5-FU (500 μM) for 24 h. A. Microscopic images. B. Quantification of LDH release into the supernatant. C. Analysis of intracellular triglycerides and free fatty acids content normalized to total cellular protein. D. Oil red O staining. Analysis of mRNA levels of E. PPARγ and PPARα, F. FASN, SCD-1 and ELOVL6, G. ACOX1, H. HMOX1 and I. IL-8 and ICAM-1 by quantitative RT-PCR (*: p<0.05 compared to control).
Effect of 5-FU on hepatic steatosis and inflammation in mice
Next, we wanted to verify our in vitro findings in an in vivo model of 5-FU-induced steatohepatitis in mice. For that, mice were intraperitoneally injected with a single dose of 5-FU (200 mg/kg body weight) and liver tissue and blood samples were collected 24 h after injection. Plasma aspartate transaminase (AST) levels were not elevated (Figure 5A) and also H/E staining of liver tissue did not show significant histological pathologies (Figure 5B) in 5-FU-treated mice under this experimental conditions. As observed in PHH, DPD and TYMS expression were not altered in the livers of 5-FU-treated mice (Supplementary Figure 4A). Also FFA levels did not significantly differ, but triglyceride levels were significantly elevated in the livers of 5-FU-treated mice (Figure 5C). In contrast,PPAR-α and PPAR-γ expression levels were not significantly altered by 5-FU treatment (Supplementary Figure 4B), and SCD-1, FASN, ACLY and ELOVL6 expression was similar or lower in the livers of 5-FU-treated mice compared to control animals (Supplementary Figure 4C). These findings indicated that also in vivo, 5-FU-induced steatosis is not mediated via de novo lipogenesis. However, both hepatic CPT-1 (Figure 5D) and ACOX1 expression (Figure 5E) were significantly induced by 5-FU. Furthermore, hepatic HMOX1 expression was significantly higher in 5-FU-treated mice compared to controls (Figure 5F), indicative for increased oxidative stress. Moreover, 5-FU caused an induction of the JNK pathway in the liver of treated mice (Figure 5G). Furthermore, hepatic CXCl1 and ICAM-1 expression were significantly induced (Figure 5H, 5I) and also immunohistological CD3 staining indicated an inflammatory response in the liver of 5-FU-treated mice (Figure 5J). Pro-ininflammatory cytokines and hepatic inflammation, respectively, have been suggested as regulators of hepatic plasminogen activator inhibitor-1 (PAI-1) expression. Accordingly, we observed a significantly increased expression of this serine protease inhibitor in the livers of 5-FU-treated mice (Figure 5K). Results of studies performed in animal models of alcoholic and non-alcoholic fatty liver disease suggest that PAI-1 is a key modulator of hepatic lipid transport and also contributes to hepatic inflammation and fibrosis [38]. Actually, alpha-smooth muscle actin (α-sma) expression (Figure 5K) was significantly increased in the livers of 5-FU-treated mice indicating the activation of hepatic stellate cells (HSC), a key event of hepatic fibrosis [39]. Although the expression levels of transforming growth factor-β (TGF-β) and collagen I were not yet significantly elevated (data not shown), repeated and long-term application of 5-FU may also induce hepatic fibrosis in mice similarly as observed in patients receiving long-term chemotherapy [40].
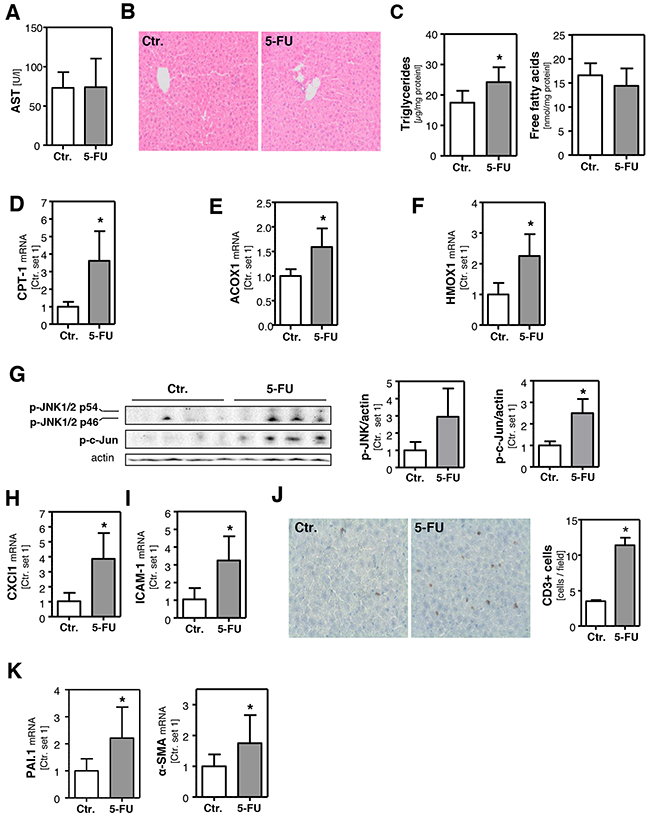
Figure 5: Effect of 5-FU on hepatic steatosis and inflammation in mice. Mice were intraperitoneally injected with a single dose 5-FU (200 mg/kg) and liver tissue samples were collected 24 h after injection. Control mice were injected with solvent isotonic saline solution. A. Plasma levels of aspartate transaminase (AST). B. H/E staining of liver tissue samples. C. Hepatic triglycerides and free fatty acids content normalized to total protein. Analysis of mRNA levels of D. CPT-1, E. ACOX1 and F. HMOX1 by quantitative RT-PCR. G. Western blot analysis of hepatic p-JNK and p-c-Jun protein levels (left panel). Densitometric analysis of p-JNK/actin ratio (middle panel) and p-c-Jun/actin ratio (right panel). Analysis of mRNA levels of H. CXCl1 and I. ICAM-1 by quantitative RT-PCR. J. CD3 staining of liver tissue samples (left panel). Quantification of CD3-positive cells (right panel). Analysis of mRNA levels of K. PAI-1 and α-SMA by quantitative RT-PCR (*: p<0.05 compared to control).
In summary, the in vivo findings were consistent with the observations made in vitro. Expression of enzymes involved in mitochondrial and peroxisomal β-oxidation was significantly induced and paralleled by an upregulation of the antioxidant enzyme HMOX1, indicative for oxidative stress in response to 5-FU treatment. Moreover, 5-FU induced JNK-activation, hepatocellular steatosis and an inflammatory reaction in both models.
DISCUSSION
The aim of this study was to investigate molecular mechanisms by which 5-FU affects hepatic steatosis and inflammation.
Once, we established an in vitro model applying HepG2 cells. This cell line has been most extensively employed in many studies analyzing drug metabolism and toxicity, since the cells retain a large part of cellular functions similar to those of normal hepatocytes [41, 42]. Also because of the high degree of morphological and functional differentiation, HepG2 cells are a suitable model to study intracellular trafficking and drug targeting in vitro [43]. In the present study, we found that 5-FU-mediated effects in HepG2 cells and in primary human hepatocytes were very similar. Therefore, we propose that also for future studies analyzing 5-FU-mediated effects on hepatocytes, such as screening of drugs that interfere with the observed pathological mechanisms, respectively, HepG2 can be a suitable model. 5-FU was applied in a dose range not causing cellular injury to focus on direct, cell specific effects. The applied 5-FU doses were in line with previous studies of Novak et al. [44] and Gajski et al. [45], who also found that doses up to 250 μM are non-toxic for HepG2 cells. Also for analyzing the effect of 5-FU in primary human hepatocytes, a dose was chosen in accordance with previous studies showing that it does not affect hepatocellular viability [46]
For in vivo analyses, we administered a single dose of 200 mg/kg 5-FU to mice. This dose was below LD50 determined earlier in mice (250-500 mg/kg, depending on the time of day) [47] and in line with previous studies analyzing the anti-tumorigenic effect of 5-FU in murine cancer models [48]. Also here, we chose experimental conditions not causing significant hepatocellular injury to specifically address direct 5-FU effects on liver cells. And we applied only a single 5-FU dose to focus on early 5-FU-mediated effects.
An important observation in our study was that 5-FU treatment induced a significant triglyceride accumulation in hepatocytes and HepG2 cells, an incidence known from clinical trials, where up to half of patients receiving 5-FU monotherapy were found to develop hepatic steatosis [14-17]. One factor contributing to hepatocellular lipid accumulation is endogenous de novo lipogenesis [49, 50]. However, key enzymes of hepatic lipogenesis were not significantly increased or even downregulated, respectively, in response to 5-FU treatment. Another possible trigger for lipid accumulation is impaired fatty acid degradation. Generally, fatty acid molecules are broken down via beta-oxidation in the mitochondria to generate acetyl-CoA. However, steatohepatitis-inducing drugs are often inhibitors of the mitochondrial respiratory chain [51] and also 5-FU has been shown to be related to a reduction of mitochondrial membrane potential and mitochondrial membrane collapse [52]. Fitting to this, we observed a dose- and time- dependent impairment of mitochondrial function in response to 5-FU treatment. An impairment of the respiratory chain leads to an accumulation of electrons, which directly react with oxygen to produce harmful ROS [51]. Furthermore, lack of cofactors for β-oxidation such FAD and NAD+ may additionally decrease metabolism of FFAs, which accumulate and thus induce CPT-1 mRNA expression [53]. Fitting to this, induction of CPT-1 expression was also observed in the livers of mice after acute inhibition of mitochondrial β-oxidation [54]. Thus, it appears that also increased CPT-1 expression in 5-FU-treated cells and livers was caused by impaired mitochondrial β-oxidation.
A consequence of impaired mitochondrial β-oxidation is a shift to extra-mitochondrial pathways. In line with this, we observed a significant upregulation of ACOX1, the initial enzyme for peroxisomal β-oxidation. Also in rodent models of (non-alcoholic) steatosis peroxisomal β-oxidation was shown to be increased [55]. This form of FFA combustion causes increased hydrogen peroxide generation, thus contributing to harmful ROS production in addition to an impaired respiratory chain. Also in our in vitro and in vivo models, 5-FU caused a marked upregulation of HMOX1 expression indicative for oxidative stress. This is in accordance with other studies that showed that 5-FU induces oxidative stress in cancer cells [22, 56]. Our in vitro data indicate that the 5-FU-induced shift to peroxisomal β-oxidation and consecutive induction of oxidative stress and inflammation occur already after several hours. Accordingly, we also found increasing ACOX1 expression, elevated HMOX1 and pro-inflammatory gene expression as well as CD3-infiltration (Supplementary Figure 5A-5F) in vivo in mouse livers already 12 h after 5-FU application.
Taken together, we propose the hypothesis that the observed triglyceride accumulation induced by 5-FU is most likely the result of mitochondrial β-oxidation impairment, possibly by affecting the respiratory chain. Although fatty acids are progressively oxidized in peroxisomes, they are insufficiently metabolized and accumulate. Together, impaired mitochondrial and enhanced peroxisomal fatty acid metabolism lead to oxidative stress.
ROS are known inducers of pro-inflammatory genes [30, 31], and indeed we observed increased IL-8 and ICAM-1 expression as well as induced hepatic infiltration with immune cells in response to 5-FU treatment. Moreover, 5-FU treatment led to a significant activation of JNK. This stress-activated protein kinase is an important modulator in response to liver injury [57], and we and others have shown that it plays a critical role in the development and progression of hepatic inflammation in alcoholic as well as non-alcoholic fatty liver disease (NAFLD) [37, 58]. Obesity is the major risk factor for NAFLD and it is also an important risk factor for different types of cancer [59]. Therefore, it is likely that a significant number of cancer patients receiving chemotherapy have also NAFLD. Future experimental and epidemiological studies are warranted to study whether already present hepatic steatosis is a trigger for more hepatic inflammation and injury. Furthermore, hepatic JNK-activation and inflammation may play a crucial role in the progression and development of hepatocellular cancer (HCC) [60, 61] as well as chemotherapy resistance of HCC cells [62, 63]. Moreover, hepatic inflammation and expression of IL-8 have been shown to promote hepatic metastasis of several types of cancers [64, 65]. Therefore, it is intriguing to speculate whether our findings may also have impact on progression and chemotherapy (resistance) of primary and secondary liver cancer, which needs to be addressed in future studies.
In summary, our study indicates a crucial role of 5-FU in the development of chemotherapy-associated steatohepatitis. As pointed out in the introduction, a two hit model for the development and progression of non-alcoholic fatty liver disease has been proposed [18, 66]. Critical steps are hepatic steatosis and its progression to steatohepatitis. In this study, we identify molecular mechanisms by which 5-FU can induce both hepatocellular lipid accumulation and inflammatory gene expression. Herewith, 5-FU alone as well as together with other factors, such as irinotecan, may be responsible for the critical hit tipping the balance towards hepatic steatosis or from steatosis to steatohepatitis, respectively. Our newly developed in vitro and in vivo models can be used to get further insight into the pathogenesis of 5-FU-induced steatohepatitis and to develop therapeutic strategies to inhibit the pathological effects of this chemotherapeutic drug on hepatocytes.
MATERIALS AND METHODS
Cells and cell culture model
Isolation and culture of primary human hepatocytes (PHH) were performed as described [67, 68]. Human liver tissue for cell isolation was obtained from the charitable state controlled foundation HTCR [69], with informed patient consent and approved by the local Ethics Committee. HepG2 hepatoma cells (ATCC No. HB-8065) were cultured as described [70].
For stimulation experiments with 5-FU, cells were seeded in standard 6-well-plates at a density of 5x105 cells/well and were allowed to adhere for 24 h before treatment. Cytotoxic effects were monitored by analysis of LDH release into the supernatant (cytotoxicity detection kit; Roche Applied Sciences, Indianapolis, IN).
Murine models of 5-FU induced steatohepatitis
Female C57BL/6N mice (n=5/group), aged 7-9 weeks, were intraperitoneally injected with a single dose 5-FU (200 mg/kg) or isotonic saline solution (control group). Liver tissue and blood samples were collected 12 h or 24 h after injection and immediately frozen and stored at -80°C until subsequent analyses.
Analysis of cellular lipid content
Cellular lipid droplets were visualized by Oil Red O staining as described [71]. Furthermore, total free fatty acids (FFA) and cellular triglycerides were extracted and quantified with the free fatty acids kit (half micro test) (Roche diagnostics, Mannheim, Germany) and the triglyceride determination kit (GPO) (Sigma, Deisenhofen, Germany) [71].
Mitochondrial activity assay
For quantification of hepatocellular mitochondrial activity, the colorimetric XTT assay (Roche Diagnostics, Mannheim, Germany) was used according to the manufacturer’s instructions.
Quantitative real-time-PCR analysis
RNA isolation from cells and tissues and subsequent reverse transcription were performed as described [72]. Quantitative real-time-PCR was performed applying LightCycler technology (Roche) [72]. The following sets of primers were used: human ACLY (forward: 5’-CGGACTTCGGCAGAGGTAGA-3’, reverse 5’-GGG GAG GGA ACT CGA TGT CA-3’), human DPD (forward: 5’-AGG ACG CAA GG AGG GTT TG-3’, reverse: 5’-GTC CGC CGA GTC CTT ACT GA-3’), human ELOVL6 (forward: 5’-CCA GTC AAC TCC TCG CAC TTT-3’, reverse: 5’-TGA CCG TGT CCG GTA TTT CC-3’), human HMOX1 (forward: 5’-GAG TGT AAG GAC CCA TCG GA-3’, reverse 5’-GCC AGC AAC AAA GTG CAA G-3’), human ICAM-1 (forward: 5’-CTG TCA CTC GAG ATC TTG AGG-3’, reverse: 5’-CCT GCA GTG CCC ATT ATG A-3’), human IL-8 (forward: 5’-TCT GCA GCT CTG TGT GAA GGT GCA GTT-3’, reverse: 5’-AAC CCT CTG CAC CCA GTT TTC CT-3’), human TYMS (forward: 5’-CCT CTG CTG ACA ACC AAA CG, reverse: 5’-GAA GAC AGC TCT TTA GCA TTT G-3’); murine ACLY (forward: 5’-GCA CCC AGA AGG CAA GAT CC-3’, reverse: 5’-CTT GGG ACT GAA TCT TGG GGC-3’), murine DPD (forward: 5’-TCT CAG CCT ACA ATG CCC CT-3’, reverse: 5’-GTT GTC CCC CGG ATG ATT CTT-3’), murine ELOVL6 (forward: 5’-AGA ACA CGT AGC GAC TCC GA-3’, reverse: 5’-AGC GTA CAG CGC AGA AAA CA-3’) and murine TYMS (forward: 5’-GAC TGC TCC GTT ATG CTG GTG-3’, reverse: 5’-CGT TTG GTT GTG AGC AGA GGA-3’). Other human and murine mRNA expression analyses were performed using QuantiTect Primer Assays according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Amplification of cDNA derived from human and murine 18S rRNA (forward: 5’-AAA CGG CTA CCA CAT CCA AG-3’, reverse: 5’-CCT CCA ATG GAT CCT CGT TA-3’) was used for normalization.
Protein analysis
Protein extraction and Western blotting were performed as described [73] applying the following primary antibodies: rabbit polyclonal anti-phospho-JNK (#9251) and rabbit monoclonal anti-phospho-c-Jun (#3270) from Cell Signaling Technology (Danvers, MA, USA; all diluted 1:1,000), and mouse monoclonal anti-actin (Merck Millipore, Billerica, MA, USA; MAB1501, 1:10,000). Goat anti-rabbit (Santa Cruz Biotechnology, Heidelberg, Germany; sc-2030, 1:2,000) and goat anti-mouse (Santa Cruz Biotechnology, sc-2005, 1:3,000) were used as secondary antibody.
Histological and immunohistological analyses
For hematoxylin and eosin (HE) staining and immunohistochemical analysis, standard 5 μm sections of formalin-fixed and paraffin-embedded tissue blocks were used. Immunohistochemical CD3 staining was performed using a rabbit anti-human T cell CD3 peptide antibody (Sigma, Saint Louis, Missouri, USA) as described [37].
Statistical analysis
Values are presented as mean ± SEM or as mean ± SD in case of in vivo experiments. Comparison between groups was made using the Student’s unpaired t-test. A p value <0.05 was considered statistically significant. All calculations were performed using the statistical computer package GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, USA).
Microscopical images were taken using an Olympus™ CKX41 microscope with the ALTRA 20 Soft Imaging System™ and CellA software version 2.6 (Olympus Soft Imaging Solutions GmbH, Münster, Germany). Images were processed using IrfanView™ software version 4.36 (Irfan Skiljan, Jajce, Bosnia).
ACKNOWLEDGMENTS
We want to thank Rudolf Jung for excellent technical assistance. We acknowledge the Human Tissue and Cell Research (HTCR) Foundation for making human tissue available for research and hepacult GmbH (Regensburg, Germany) for providing primary human hepatocytes for in vitro studies.
CONFLICTS OF INTEREST
Nothing to declare
FUNDING
This work was supported by grants from the German Research Association (DFG) to A.K.B. and C.H. (FOR 2127) and C.H. (KFO262), and from the German Cancer Aid and BioSysNet to A.K.B.
REFERENCES
1. Longley DB, Harkin DP and Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003; 3:330-338.
2. Pinedo HM and Peters GF. Fluorouracil: biochemistry and pharmacology. J Clin Oncol. 1988; 6:1653-1664.
3. Miura K, Kinouchi M, Ishida K, Fujibuchi W, Naitoh T, Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, Shibata C and Sasaki I. 5-fu metabolism in cancer and orally-administrable 5-fu drugs. Cancers (Basel). 2010; 2:1717-1730.
4. Zhang N, Yin Y, Xu SJ and Chen WS. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules. 2008; 13:1551-1569.
5. Parker WB and Cheng YC. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol Ther. 1990; 48:381-395.
6. Abdel-Hamid NM and Morsy MA. Novel biochemical pathways for 5-Fluorouracil in managing experimental hepatocellular carcinoma in rats. J Membr Biol. 2010; 234:29-34.
7. Schilsky RL. Biochemical and clinical pharmacology of 5-fluorouracil. Oncology (Williston Park). 1998; 12:13-18.
8. Ghoshal K and Jacob ST. An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug. Biochem Pharmacol. 1997; 53:1569-1575.
9. Pilgrim CH, Thomson BN, Banting S, Phillips WA and Michael M. The developing clinical problem of chemotherapy-induced hepatic injury. ANZ J Surg. 2012; 82:23-29.
10. Lehmann K, Rickenbacher A, Weber A, Pestalozzi BC and Clavien PA. Chemotherapy before liver resection of colorectal metastases: friend or foe? Ann Surg. 2012; 255:237-247.
11. Fernandez FG, Ritter J, Goodwin JW, Linehan DC, Hawkins WG and Strasberg SM. Effect of steatohepatitis associated with irinotecan or oxaliplatin pretreatment on resectability of hepatic colorectal metastases. J Am Coll Surg. 2005; 200:845-853.
12. Vauthey JN, Pawlik TM, Ribero D, Wu TT, Zorzi D, Hoff PM, Xiong HQ, Eng C, Lauwers GY, Mino-Kenudson M, Risio M, Muratore A, Capussotti L, Curley SA and Abdalla EK. Chemotherapy regimen predicts steatohepatitis and an increase in 90-day mortality after surgery for hepatic colorectal metastases. J Clin Oncol. 2006; 24:2065-2072.
13. Gomez-Ramirez J, Martin-Perez E, Amat CG, Sanz IG, Bermejo E, Rodriguez A and Larranaga E. [Influence of pre-surgical chemotherapy on liver parenchyma and post-surgical outcome of patients subjected to hepatectomy due to colorectal carcinoma metastases]. Cir Esp. 2010; 88:404-412.
14. Zorzi D, Laurent A, Pawlik TM, Lauwers GY, Vauthey JN and Abdalla EK. Chemotherapy-associated hepatotoxicity and surgery for colorectal liver metastases. Br J Surg. 2007; 94:274-286.
15. Miyake K, Hayakawa K, Nishino M, Morimoto T and Mukaihara S. Effects of oral 5-fluorouracil drugs on hepatic fat content in patients with colon cancer. Acad Radiol. 2005; 12:722-727.
16. Peppercorn PD, Reznek RH, Wilson P, Slevin ML and Gupta RK. Demonstration of hepatic steatosis by computerized tomography in patients receiving 5-fluorouracil-based therapy for advanced colorectal cancer. Br J Cancer. 1998; 77:2008-2011.
17. Pawlik TM, Olino K, Gleisner AL, Torbenson M, Schulick R and Choti MA. Preoperative chemotherapy for colorectal liver metastases: impact on hepatic histology and postoperative outcome. J Gastrointest Surg. 2007; 11:860-868.
18. Dietrich P and Hellerbrand C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2014; 28:637-653.
19. Hardy T, Oakley F, Anstee QM and Day CP. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu Rev Pathol. 2016.
20. Jimenez R, Hijona E, Emparanza J, Alustiza JM, Hijona L, Macarulla MT, Portillo MP, Herreros-Villanueva M, Beguiristain A, Arenas J and Bujanda L. Effect of neoadjuvant chemotherapy in hepatic steatosis. Chemotherapy. 2012; 58:89-94.
21. Lionarons DA, Heger M, van Golen RF, Alles LK, van der Mark VA, Kloek JJ, de Waart DR, Marsman HA, Rusch H, Verheij J, Beuers U, Paulusma CC and van Gulik TM. Simple steatosis sensitizes cholestatic rats to liver injury and dysregulates bile salt synthesis and transport. Sci Rep. 2016; 6:31829.
22. Alexandre J, Nicco C, Chereau C, Laurent A, Weill B, Goldwasser F and Batteux F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006; 98:236-244.
23. Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW and Vogelstein B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001; 7:1111-1117.
24. Eichhorst ST, Muller M, Li-Weber M, Schulze-Bergkamen H, Angel P and Krammer PH. A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol Cell Biol. 2000; 20:7826-7837.
25. Pilgrim CH, Brettingham-Moore K, Pham A, Murray W, Link E, Smith M, Usatoff V, Evans PM, Banting S, Thomson BN, Michael M and Phillips WA. mRNA gene expression correlates with histologically diagnosed chemotherapy-induced hepatic injury. HPB (Oxford). 2011; 13:811-816.
26. Matsuzaka T, Atsumi A, Matsumori R, Nie T, Shinozaki H, Suzuki-Kemuriyama N, Kuba M, Nakagawa Y, Ishii K, Shimada M, Kobayashi K, Yatoh S, Takahashi A, et al. Elovl6 promotes nonalcoholic steatohepatitis. Hepatology. 2012; 56:2199-2208.
27. Fromenty B and Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995; 67:101-154.
28. Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002; 16:663-678.
29. Migita T, Okabe S, Ikeda K, Igarashi S, Sugawara S, Tomida A, Soga T, Taguchi R and Seimiya H. Inhibition of ATP citrate lyase induces triglyceride accumulation with altered fatty acid composition in cancer cells. Int J Cancer. 2014; 135:37-47.
30. Reddy JK and Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006; 290:G852-858.
31. Gusdon AM, Song KX and Qu S. Nonalcoholic Fatty liver disease: pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid Med Cell Longev. 2014; 2014:637027.
32. Mannaerts GP, Debeer LJ, Thomas J and De Schepper PJ. Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate-treated rats. J Biol Chem. 1979; 254:4585-4595.
33. Kim CS, Kwon Y, Choe SY, Hong SM, Yoo H, Goto T, Kawada T, Choi HS, Joe Y, Chung HT and Yu R. Quercetin reduces obesity-induced hepatosteatosis by enhancing mitochondrial oxidative metabolism via heme oxygenase-1. Nutr Metab (Lond). 2015; 12:33.
34. Marra F and Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014; 147:577-594 e571.
35. Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P and McClain CJ. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007; 46:823-830.
36. Lehnert M, Relja B, Sun-Young Lee V, Schwestka B, Henrich D, Czerny C, Froh M, Borsello T and Marzi I. A peptide inhibitor of C-jun N-terminal kinase modulates hepatic damage and the inflammatory response after hemorrhagic shock and resuscitation. Shock. 2008; 30:159-165.
37. Dorn C, Engelmann JC, Saugspier M, Koch A, Hartmann A, Muller M, Spang R, Bosserhoff A and Hellerbrand C. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab Invest. 2014; 94:394-408.
38. Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Konigsrainer A, Maier KP, Bischoff SC and Bergheim I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008; 138:1452-1455.
39. Hellerbrand C. Hepatic stellate cells--the pericytes in the liver. Pflugers Arch. 2013; 465:775-778.
40. Reddy SK, Reilly C, Zhan M, Mindikoglu AL, Jiang Y, Lane BF, Alexander HR, Culpepper WJ and El-Kamary SS. Long-term influence of chemotherapy on steatosis-associated advanced hepatic fibrosis. Med Oncol. 2014; 31:971.
41. Dehn PF, White CM, Conners DE, Shipkey G and Cumbo TA. Characterization of the human hepatocellular carcinoma (hepg2) cell line as an in vitro model for cadmium toxicity studies. In Vitro Cell Dev Biol Anim. 2004; 40:172-182.
42. Roe AL, Snawder JE, Benson RW, Roberts DW and Casciano DA. HepG2 cells: an in vitro model for P450-dependent metabolism of acetaminophen. Biochem Biophys Res Commun. 1993; 190:15-19.
43. Xia JF, Gao JJ, Inagaki Y, Kokudo N, Nakata M and Tang W. Flavonoids as potential anti-hepatocellular carcinoma agents: recent approaches using HepG2 cell line. Drug Discov Ther. 2013; 7:1-8.
44. Novak M, Zegura B, Baebler S, Stern A, Rotter A, Stare K and Filipic M. Influence of selected anti-cancer drugs on the induction of DNA double-strand breaks and changes in gene expression in human hepatoma HepG2 cells. Environ Sci Pollut Res Int. 2015.
45. Gajski G, Geric M, Zegura B, Novak M, Nunic J, Bajrektarevic D, Garaj-Vrhovac V and Filipic M. Genotoxic potential of selected cytostatic drugs in human and zebrafish cells. Environ Sci Pollut Res Int. 2015.
46. Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Buchler P, Haas TL, Schader MB, Untergasser A, Stremmel W and Walczak H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin Cancer Res. 2006; 12:2640-2646.
47. Burns ER and Beland SS. Effect of biological time on the determination of the LD50 of 5-fluorouracil in mice. Pharmacology. 1984; 28:296-300.
48. Chao TH, Chang GR, Chen WY, Chen PL and Mao FC. The synergistic effect of rapamycin combined with 5-fluorouracil in BALB/cByJNarl mice bearing CT-26 tumor cells. Anticancer Res. 2014; 34:3329-3335.
49. Nassir F and Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014; 15:8713-8742.
50. Rabinowich L and Shibolet O. Drug Induced Steatohepatitis: An Uncommon Culprit of a Common Disease. Biomed Res Int. 2015; 2015:168905.
51. Labbe G, Pessayre D and Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008; 22:335-353.
52. Chun YS, Laurent A, Maru D and Vauthey JN. Management of chemotherapy-associated hepatotoxicity in colorectal liver metastases. Lancet Oncol. 2009; 10:278-286.
53. Foster DW. The role of the carnitine system in human metabolism. Ann N Y Acad Sci. 2004; 1033:1-16.
54. van der Leij FR, Bloks VW, Grefhorst A, Hoekstra J, Gerding A, Kooi K, Gerbens F, te Meerman G and Kuipers F. Gene expression profiling in livers of mice after acute inhibition of beta-oxidation. Genomics. 2007; 90:680-689.
55. Natarajan SK, Eapen CE, Pullimood AB and Balasubramanian KA. Oxidative stress in experimental liver microvesicular steatosis: role of mitochondria and peroxisomes. J Gastroenterol Hepatol. 2006; 21:1240-1249.
56. Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A, Levy E, Goldwasser F, Panis Y, Soubrane O, Weill B and Batteux F. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005; 65:948-956.
57. Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE and Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006; 43:163-172.
58. Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA and Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009; 49:87-96.
59. Calle EE, Rodriguez C, Walker-Thurmond K and Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003; 348:1625-1638.
60. Min L, He B and Hui L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin Cancer Biol. 2011; 21:10-20.
61. Seki E, Brenner DA and Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012; 143:307-320.
62. Liu XY, Liu SP, Jiang J, Zhang X and Zhang T. Inhibition of the JNK signaling pathway increases sensitivity of hepatocellular carcinoma cells to cisplatin by down-regulating expression of P-glycoprotein. Eur Rev Med Pharmacol Sci. 2016; 20:1098-1108.
63. Lin YT and Chao CC. Identification of the beta-catenin/JNK/prothymosin-alpha axis as a novel target of sorafenib in hepatocellular carcinoma cells. Oncotarget. 2015; 6:38999-39017. doi: 10.18632/oncotarget.5738.
64. Nastase A, Paslaru L, Herlea V, Ionescu M, Tomescu D, Bacalbasa N, Dima S and Popescu I. Expression of interleukine-8 as an independent prognostic factor for sporadic colon cancer dissemination. J Med Life. 2014; 7:215-219.
65. Bai Z, Tai Y, Li W, Zhen C, Gu W, Jian Z, Wang Q, Lin JE, Zhao Q, Gong W, Liang B, Wang C and Zhou T. Gankyrin activates IL-8 to promote hepatic metastasis of colorectal cancer. Cancer Res. 2013; 73:4548-4558.
66. Day CP and James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998; 114:842-845.
67. Lee SML, Schelcher C, Laubender RP, Frose N, Thasler RMK, Schiergens TS, Mansmann U and Thasler WE. An Algorithm that Predicts the Viability and the Yield of Human Hepatocytes Isolated from Remnant Liver Pieces Obtained from Liver Resections. Plos One. 2014; 9.
68. Lee SM, Schelcher C, Demmel M, Hauner M and Thasler WE. Isolation of human hepatocytes by a two-step collagenase perfusion procedure. J Vis Exp. 2013.
69. Thasler WE, Weiss TS, Schillhorn K, Stoll PT, Irrgang B and Jauch KW. Charitable State-Controlled Foundation Human Tissue and Cell Research: Ethic and Legal Aspects in the Supply of Surgically Removed Human Tissue For Research in the Academic and Commercial Sector in Germany. Cell Tissue Bank. 2003; 4:49-56.
70. Hellerbrand C, Amann T, Schlegel J, Wild P, Bataille F, Spruss T, Hartmann A and Bosserhoff AK. The novel gene MIA2 acts as a tumour suppressor in hepatocellular carcinoma. Gut. 2008; 57:243-251.
71. Wobser H, Dorn C, Weiss TS, Amann T, Bollheimer C, Buttner R, Scholmerich J and Hellerbrand C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009; 19:996-1005.
72. Hellerbrand C, Muhlbauer M, Wallner S, Schuierer M, Behrmann I, Bataille F, Weiss T, Scholmerich J and Bosserhoff AK. Promoter-hypermethylation is causing functional relevant downregulation of methylthioadenosine phosphorylase (MTAP) expression in hepatocellular carcinoma. Carcinogenesis. 2006; 27:64-72.
73. Gabele E, Dostert K, Dorn C, Patsenker E, Stickel F and Hellerbrand C. A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol Clin Exp Res. 2011; 35:1361-1367.