
INTRODUCTION
Breast cancer (BC) is the most common malignancy in women, accounting for 22% of new cases of cancer each year [1]. It is estimated that for BC, as known for most malignant tumors, 5 to 10% are hereditary [2]. Germline mutations in the tumor suppressor genes, BRCA1 and BRCA2 account for approximately 20% of cases of hereditary breast cancer cases [3]. Furthermore, germline mutation in BRCA1 gene carriers have a cumulative risk of developing breast cancer ranging from 44% to 68% by 70 years of age [4]. Mutations in BRCA1 and BRCA2 genes are associated with the Hereditary Breast and Ovarian Cancer Predisposition Syndrome (HBOC). Patients who have HBOC syndrome have a personal and a strong family history of cancer mainly in the following organs: breast, ovarian, pancreas and prostate. HBOC families, like other families with hereditary cancer predisposition syndromes, are characterized by early age on diagnosis, multiple primary tumors, bilateral tumors or multiple rare tumors and two or more generations affected by cancer [5, 6].
BRCA1 and BRCA2 are involved in the maintenance of genomic integrity through various cellular processes such as DNA damage recognition, transcription and cell cycle regulations and repair of DNA damage [7]. Given this, pathological alterations in these genes may cause changes in the function of its proteins. To date more than 2,500 proved pathogenic mutations have been described throughout the coding sequence BRCA1 and BRCA2, which can be found in the database ClinVar [8] as well as in the Biobase portal - HGMD (Human Genome Mutation Database) [9]. In addition to genetic alterations, the occurrence of epigenetics alterations, such as hypermethylation of tumor suppressor genes, could be responsible for silencing this gene and consequently increase the risk of cancer development [10, 11].
The evaluation of the status of gene promoter methylation has been considered a potential biomarker for various tumor types and an useful tool for tumor detection or as a prognostic factor, as described for many different cancers such as lung cancers [12], colorectal [13], head and neck [14], and even for breast tumors [15–18].
Although the correlation between promoter hypermethylation of BRCA1 gene with transcriptional inactivation has been reported in several studies [19, 20], as well as the association of the methylation level with clinical stage [20], histologic grade [20], triple negative phenotype [20, 21], and ancestry [21], there is little evidence on the correlation between the BRCA1 methylation status and hereditary breast cancer. The methylation profile of mutation carriers was first described in 2001 by Esteller et al., who proposed that in hereditary tumors caused by alterations in tumor suppressor genes, the importance of inactivation by epigenetic events depends on which genes are involved in the tumor under study. According to the author, carriers of germline BRCA1 mutations present a higher frequency of genetic events (and not epigenetic) as the “second event”.
In this study, our objective was to characterize women at-risk for hereditary BC regarding their status of BRCA1 mutation and methylation and to correlate these results with the levels of gene expression, histopathological and clinical data, as well as with prognosis and family history of cancer.
RESULTS
Patient and tumor characteristics
The average age at diagnosis of the patients included was 39.0 years (SD = 9.4). When patients are grouped according to germline BRCA1 mutational status, the mean age at diagnosis for women of the BRCA1-pathogenic groups, BRCA1-VUS and BRCA1-WT were respectively 41.6; 37.2 and 36.5 years (SD = 7.3, 9.4 and 10.1 years, respectively).
Additionally, the age at diagnosis was categorized and the information, as well as the clinical and pathological data of the cases included in the study, are detailed in Table 1.
Table 1: Clinicopathological characterization of the patients
BRCA1-pathogenic |
BRCA1-VUS |
BRCA1-WT |
p-value |
||
|---|---|---|---|---|---|
n = 23 |
n = 22 |
n = 43 |
|||
Age at diagnosis |
≤ 30 years |
0 (0.0%) |
7 (31.8%) |
10 (23.3%) |
|
> 30 and ≤ 50 years |
21 (91.3%) |
12 (54.5%) |
27 (62.8%) |
0.020** |
|
> 50 years |
2 (8.7%) |
3 (13.6%) |
6 (14.0%) |
||
Bilateral tumor |
No |
13 (56.5%) |
15 (68.2%) |
33 (76.7%) |
0.247* |
Yes |
10 (43.5%) |
7 (31.8%) |
10 (23.3%) |
||
Histological type |
Ductal/lobular carcinoma “in situ” |
4 (17.4%) |
2 (9.1%) |
4 (9.3%) |
0.676** |
Invasive ductal/lobular carcinoma |
19 (82.6%) |
20 (90.9%) |
39 (90.7%) |
||
Histological grade |
I |
1 (4.3%) |
1 (4.5%) |
7 (16.2%) |
|
II |
4 (17.4%) |
11 (50.0%) |
15 (34.9%) |
0.072** |
|
III |
14 (61.0%) |
7 (31.9%) |
17 (39.6%) |
||
Unknown |
4 (17.3%) |
3 (13.6%) |
4 (9.3%) |
||
Clinical staging |
0 or I |
5 (26.3%) |
6 (37.5%) |
18 (48.6%) |
|
II |
9 (47.4%) |
6 (37.5%) |
15 (40.5%) |
||
III |
5 (26.3%) |
4 (25.0%) |
3 (8.1%) |
0.332** |
|
IV |
0 (0.0%) |
0 (0.0%) |
1 (2.7%) |
||
Tumor size |
T1 |
7 (30.4%) |
8 (36.4%) |
20 (46.5%) |
0.415* |
T2-T4 |
16 (69.6%) |
14 (63.6%) |
23 (53.5%) |
||
Lymph node status |
N0 |
11 (47.8%) |
13 (59.1%) |
28 (65.1%) |
0.396* |
N1-N3 |
12 (52.2%) |
9 (40.9%) |
15 (34.9%) |
||
Distant metastasis |
M0 |
23 (100%) |
22 (100%) |
40 (93.0%) |
0.431** |
M1 |
0 (0.0%) |
0 (0.0%) |
3 (7.0%) |
*Chi-square test / ** Fisher’s Exact Test.
Regarding hormone receptors, estrogen (ER) and progesterone (PR) were predominantly negative in the BRCA1-pathogenic group, unlike that observed in the BRCA1-VUS and BRCA1-WT groups (p = 0.008 and p = 0.003, respectively; Table 2). On the other side, the negativity of the human epidermal growth factor receptor-type 2 (HER2) was frequent in all three groups. Thirty patients were triple negative, with 53.3% of them carrying BRCA1 pathogenic mutations, 16.7% with a VUS on BRCA1 and 30% were BRCA1 WT (p < 0.005).
Table 2: Expression of hormone receptors, HER2 and Ki67
BRCA1-pathogenic |
BRCA1-VUS |
BRCA1-WT |
p-value |
||
|---|---|---|---|---|---|
n = 23 |
n = 22 |
n = 43 |
|||
Estrogen receptor (ER) |
Negative |
16 (69.6%) |
7 (31.8%) |
14 (32.6%) |
0.008* |
Positive |
7 (30.4%) |
15 (68.2%) |
29 (67,4%) |
||
Progesterone receptor (PR) |
Negative |
19 (82.6%) |
8 (36.4%) |
19 (44,2%) |
0.003* |
Positive |
4 (17.4%) |
14 (63.6%) |
24 (55,8%) |
||
HER-2 |
Negative |
20 (90.9%) |
17 (85.0%) |
32 (74,4%) |
0.270** |
Positive |
2 (9.1%) |
3 (15.0%) |
11 (25,6%) |
||
Ki67 |
≤ 14% |
2 (8.7%) |
2 (9.0%) |
4 (9.3%) |
|
> 14% |
14 (60.9%) |
16 (72.8%) |
32 (74.5%) |
1.000* |
|
Unknown |
7 (30.4%) |
4 (18.2%) |
7 (16.2%) |
*Chi-square test / **Fisher’s Exact Test.
Family history
To obtain the family history of cancer, the pedigree of the 88 families included in the study were reviewed. In this analysis, information regarding the presence, frequency and age at diagnosis of primary tumors related to HBOC were considered, such as the presence of breast cancer (female and male), ovary, pancreas and prostate. The average age at breast cancer diagnosis in the family was 42.6 (SD = 9.4). When the three groups were considered separately, the average age at diagnosis of breast cancer in the family, in the BRCA1-pathogenic, BRCA1-VUS and BRCA1-WT groups were respectively 44.1; 41.2 and 42.5 years (SD = 5.7, 9.7 and 10.7 years, respectively).
Table 3 depicts the family history in relation to the BRCA1 germline mutational status. We observed a higher proportion of reports of breast cancer before the age of 50 in families with pathogenic BRCA1 mutations (p = 0.005). In addition, presence of breast cancer among mother and daughter was seen in all three groups, with a greater propensity for such phenomenon among patients in the BRCA1-pathogenic group (p = 0.055). The total number of breast cancer cases in the family was also evaluated. As expected, the majority of BRCA1 mutated patients had 3 or more cases of breast cancer in their families (73.9%), while most patients from BRCA1-VUS and BRCA1-WT groups (63.6% and 64.3%) reported up to two cases of breast cancer in the family (p = 0.007).
Table 3: Family history according to the BRCA1 mutational status
BRCA1-pathogenic |
BRCA1-VUS |
BRCA1-WT |
||
|---|---|---|---|---|
n = 23 |
n = 22 |
n =4 3 |
||
Presence of bilateral breast cancer |
No |
13 (56.5%) |
15 (68.2%) |
33 (76.7%) |
Yes |
10 (43.5%) |
7 (31.8%) |
10 (23.3%) |
|
p-value |
0.235* |
|||
Presence of pancreatic cancer |
No |
21 (91,3%) |
20 (90.9%) |
42 (97.7%) |
Yes |
2 (8,7%) |
2 (9.1%) |
1 (2.3%) |
|
p-value |
0.296** |
|||
Presence of ovarian cancer |
No |
16 (69.6%) |
18 (81.8%) |
41 (95.3%) |
Yes |
7 (30.4%) |
4 (18.2%) |
2 (4.7%) |
|
p-value |
0.015** |
|||
Presence of breast and ovarian cancer |
No |
20 (87.0%) |
21 (95.5%) |
43 (100.0%) |
Yes |
3 (13.0%) |
1 (4.5%) |
0 (0.0%) |
|
p-value |
0.039** |
|||
Presence of male breast cancer |
No |
23 (100.0%) |
22 (100.0%) |
42 (97.7%) |
Yes |
0 (0.0%) |
0 (0.0%) |
1 (2.3%) |
|
p-value |
1.000** |
|||
Presence of prostate cancer |
No |
20 (87.0%) |
15 (68.2%) |
35 (81.4%) |
Yes |
3 (13.0%) |
7 (31.8%) |
8 (18.6%) |
|
p-value |
0.300** |
|||
Presence of breast cancer among mother and daughter |
No |
12 (52.2%) |
17 (77.3%) |
34 (79.1%) |
Yes |
11 (47.8%) |
5 (22.7%) |
9 (20.9%) |
|
p-value |
0.055* |
|||
Breast cancer cases < 50 years |
0 |
0 (0.0%) |
2 (9.1%) |
3 (7.0%) |
1–2 |
12 (52.2%) |
18 (81.8%) |
35 (81.4%) |
|
≥ 3 |
11 (47.8%) |
2 (9.1%) |
5 (11.6%) |
|
p-value |
0.005** |
|||
Generations affected by breast cancer |
1–2 |
21 (91.3%) |
20 (90.9%) |
38 (88.4%) |
≥ 3 |
2 (8.7%) |
2 (9.1%) |
5 (11.6%) |
|
p-value |
1.000** |
|||
Total number of breast cancer cases in the family |
1–2 |
6 (26.1%) |
14 (63.6%) |
27 (64.3%) |
≥ 3 |
17 (73.9%) |
8 (36.4%) |
15 (35.7%) |
|
p-value |
0.007** |
|||
*Chi-square test / **Fisher’s Exact Test.
Methylation analysis
First, the Percentage of Relative Methylation (PRM) between all normal and tumor samples was compared independently using the Mann-Whitney test, in order to check if there is difference in the level of methylation profile between these two groups. Through this analysis, it was observed that there is a statistically significant difference in the profile of methylation in the promoter region of BRCA1 between normal and tumor tissues, with a higher level of methylation being observed in tumor samples when comparing to normal samples (p = 0.0001) (Figure 1).
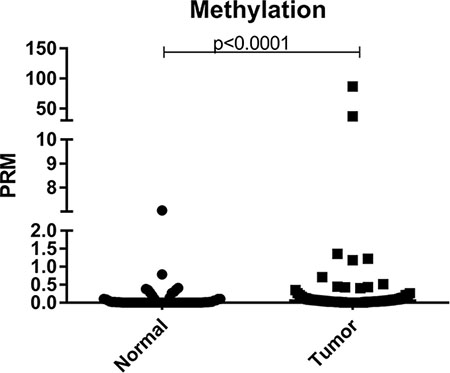
Figure 1: Scatter plot of the percentage of relative methylation (PRM) of the samples analyzed in the study. Left: normal samples and right: tumor samples. The Y-axis shows the PMR level. The p-value (Mann-Whitney) from each comparison is provided.
Secondly, we compare the level of methylation of mutated versus non-mutated patients, and no difference was observed between the groups (p = 1.000). Additionally, the level of methylation of normal and tumor samples was analyzed according to the mutational status of BRCA1 (mutated vs. non-mutated). No statistical difference was observed when comparing tumor and normal samples in the mutated group (p = 0.0097), however, this comparison was significant in the non-mutated group. The results of this analysis are shown in Figure 2.
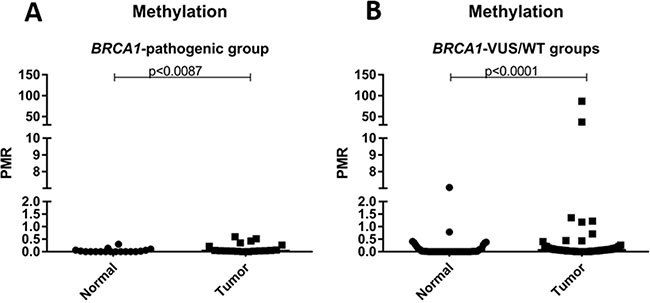
Figure 2: Scatter plot of the percentage of relative methylation (PRM) of normal and tumor samples according to mutation status. (A) PRM representation only in patients with deleterious germline mutation in BRCA1 (BRCA1-pathogenic group). (B) PRM representation only in patients without deleterious germline mutation in BRCA1 (BRCA1-VUS groups and BRCA1-WT). The Y-axis shows the PMR level. The p-value (Mann-Whitney) from each comparison is provided.
In the sequence, to classify the samples as methylated or unmethylated, a cut-off of 4% was set, and, using this stratification, BRCA1 promoter hypermethylation was found in 2 of 88 (2.3%) breast tumor samples analyzed. When the methylation status was evaluated according to the BRCA1 mutational status, patients with deleterious germline mutation showed no hypermethylation of the BRCA1 gene, whereas 2/43 patients of the BRCA1-WT group showed methylation in the promoter region of the BRCA1 gene. An interesting finding was the identification of BRCA1 hypermethylation in the normal adjacent tissue in one sample (PRM = 37% in the tumor tissue and 7% in the normal tissue). To eliminate the possibility of a contamination from tumor cells, a blood sample from that patient was investigated and the BRCA1 hypermethylation was verified (PRM = 9%), confirming the presence of a constitutive BRCA1 epimutation. The PRM of the second methylated sample was 86.7%, however for this sample only the tumor tissue was hypermethylated.
Gene expression analyses
The evaluation of BRCA1 expression was performed by RT-qPCR for 68 tumor samples and 49 matched normal samples (54 samples were excluded due to poor RNA quality). Among the tumor samples, 16/68 (23.5%) belonged to the BRCA1-pathogenic group (mutated BRCA1), 16/68 (23.5%) were from BRCA1-VUS group and 36/68 (53%) were BRCA1 WT. Regarding normal samples, 11/49 (22.5%) belonged to the BRCA1-pathogenic group, 12/49 (24.5%) to the BRCA1-VUS group and 26/49 (53.0%) were BRCA1 WT.
The Mann-Whitney test showed a statistically significant difference (p < 0.0001) between the gene expression of normal samples vs tumor samples (Figure 3). In addition, when using the ΔCT method to estimate the level of expression and the median value of all samples evaluated as the cutoff point, all normal samples analyzed showed high levels of BRCA1 expression. Unfortunately, gene expression analysis could not be performed on the sample with constitutive BRCA1 epimutation due to low RNA quality. However, if only one endogenous gene is used as a reference for the assessment of gene expression, we note that those tumor samples showed low BRCA1 mRNA expression.
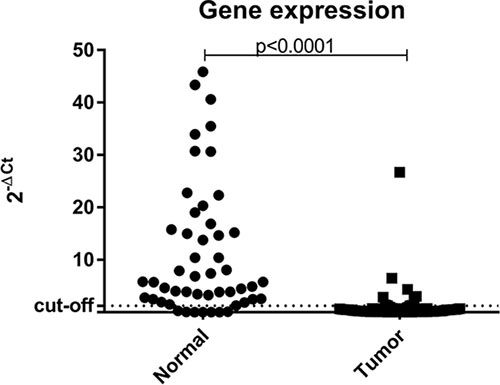
Figure 3: Scatter plot of the expression profile of BRCA1 in normal and tumor samples suitable for analysis. The Y-axis shows the fold-change of the relative expression (2-ΔCt). The p-value (Mann-Whitney) from each comparison is provided. The dotted line indicates the median value used as cutoff. Left: normal samples. Right: tumor samples.
The great majority of the tumor samples analyzed, (57/68; 83.8%) had low expression levels of BRCA1. When comparing gene expression results among the three groups separately, low levels of BRCA1could be observed in all patients (16/16; 100%) with deleterious germline mutation in BRCA1, in 11/16 (68.8%) of BRCA1-VUS patients, and 30/36 (83.3%) of those WT for BRCA1. When patients were categorized into only two groups (non-mutated vs. mutated) it was observed that 16/16 (100%) patients with deleterious germline mutation in BRCA1 have low gene expression levels in tumor samples, as well as 41/52 (78.8%) of those patients without deleterious germline mutation (BRCA1-VUS and BRCA1-WT groups) (Figure 4).
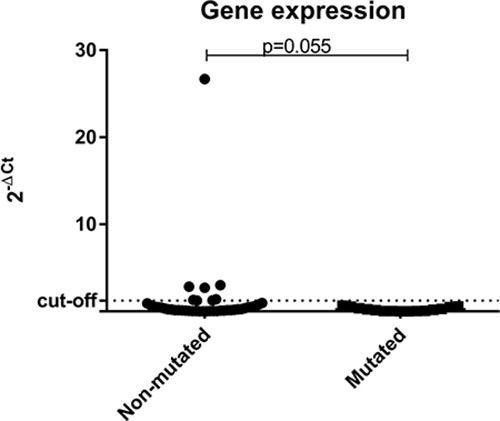
Figure 4: Scatter plot of the expression profile of BRCA1 in tumor samples according to the status of BRCA1 germline mutation. The Y-axis shows the fold-change of the relative expression (2-ΔCt). The p-value (Mann-Whitney) from each comparison is provided. The dotted line indicates the median value used as cutoff. Left: patients without deleterious BRCA1 germline mutation (BRCA1-VUS and BRCA1-WT groups). Right: patients with deleterious germline mutation in BRCA1 (BRCA1-pathogenic group).
Association between clinicopathologic, molecular and family history features
The expression levels of BRCA1 in patients with BRCA1 hypermethylation could not be evaluated due to poor RNA quality.
The description of the clinical and molecular data is shown in Figure 5. The presence of an association between the level of methylation and clinical and histopathologic variables was analyzed and the results of this analysis are described in Table 4. We did not find any statistically significant association among the variables, possibly due to the small number of methylated samples. Both cases with BRCA1 hypermethylation had invasive ductal tumors and were negative for the estrogen receptor. Moreover, the breast cancer developed by the patient carrying the epimutation was bilateral and diagnosed at 37 years of age.
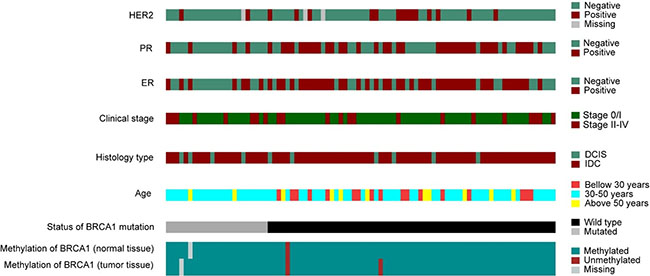
Figure 5: Molecular and pathological characteristics (rows) of the patients included in the study. Patients are organized in columns and arranged to emphasize mutual exclusivity among characteristics.
Table 4: Clinical and histopathological characteristics compared to the methylation profile
Methylated |
Non-Methylated |
|||
|---|---|---|---|---|
n = 2 |
n = 85 |
p-value |
||
Germline deleterious mutation |
No |
2 (100%) |
63 (74.1%) |
1.000** |
Yes |
0 (0.0%) |
22 (25.9%) |
||
Histological type |
Invasive ductal/lobular carcinoma |
2 (100%) |
76 (89.4%) |
1.000** |
Ductal/lobular carcinoma in situ |
0 (0.0%) |
9 (10.6%) |
||
Estrogen receptor |
Negative |
2 (100%) |
35 (41.2%) |
0.178** |
Positive |
0 (0.0%) |
50 (58.8%) |
||
Progesterone receptor |
Negative |
1 (50.0%) |
44 (51.8%) |
1.000** |
Positive |
1 (50.0%) |
41 (48.2%) |
||
HER2 |
Negative |
2 (100%) |
67 (78.9%) |
|
Positive |
0 (0.0%) |
15 (17.6%) |
1.000** |
|
Unknown |
0 (0.0%) |
3 (3.5%) |
||
Triple Negative |
No |
1 (50.0%) |
56 (65.9%) |
1.000** |
Yes |
1 (50.0%) |
29 (34.1%) |
**Fisher’s Exact Test.
Although no significant correlation between the methylation profile and clinical and histopathological features was found, we can highlight that none patient with deleterious germline mutation in BRCA1 showed hypermethylation. In addition, patients with methylation in the BRCA1 gene, showed negativity for the estrogen receptor (Figure 5).
By correlating the expression levels of BRCA1 with clinical and tumor characteristics we observed that a significant proportion of cases with high levels of BRCA1 expression (35.0%) had tumors at early stages (T1, N0 and M0). Among those less advanced and with low gene expression tumors (T1), the majority (76.2%, 16/21) had luminal subtype, followed by 19.0% (4/21) triple negatives and 4.8% (1/21) were HER2 subtype. In addition to the histopathological data, we evaluated the association between BRCA1 transcriptional levels with the status of hormone receptors (ER and PR), HER2 and Ki67. Although the relationship between transcriptional level and hormone receptors status was not significant, it can be observed that most of the triple negative patients (20/22) presented low BRCA1 expression, whereas patients with high expression were mostly Luminal (Table 5).
Table 5: Association between gene expression and molecular subtype
Low Expression |
High Expression |
p-value |
||
|---|---|---|---|---|
n = 57 |
n = 11 |
|||
Molecular Subtype |
Luminal (Luminal A, Luminal B) |
33 (57.9%) |
7 (63.6%) |
|
HER2 (ER−, PR−, HER2+) |
1 (1.7%) |
2 (18.2%) |
0.049** |
|
Triple-Negative (ER−, PR−, HER2−) |
20 (35.2%) |
2 (18.2%) |
||
Unknown |
3 (5.2%) |
0 (0.0%) |
**Fisher’s Exact Test.
Values in bold indicate statistical significance (p < 0.05).
Regarding the family history, the average age at diagnosis of breast cancer in the family in the three groups was 42.6 years (SD = 9.4). When the three groups are considered separately, the average age at diagnosis of breast cancer in the family, in the BRCA1-pathogenic, BRCA1-VUS and BRCA1-WT groups were respectively 44.1; 41.2 and 42.5 years (SD = 5.7, 9.7 and 10.7 years, respectively).
We observed a higher proportion of breast cancer cases younger than 50 years in families with pathogenic mutations in BRCA1, while most patients the BRCA1-VUS and BRCA1-WT groups reported only one or two cases diagnosed before the age of 50 (p = 0.005). Besides, 73.9% of the families in the BRCA1-pathogenic group had 3 or more cases of breast cancer in their families, while 63.6% and 64.3% of the families with VUS at BRCA1 gene and BRCA1-WT respectively, reported up to two cases of breast cancer in the family (p = 0.007). Furthermore, when we look at the family history of the patient who presented the epimutation, we can highlight the presence of two prostate tumors and only one generation affected by breast cancer.
DISCUSSION
The identification of individuals and families with hereditary cancer is important because affected individuals have a vital cumulative risk much higher than the population for the development of various cancers. These tumors usually occur at younger ages than it would be expected for the pathology in question. This study was conducted in order to characterize women with personal and family history of breast cancer (with and without germline mutation in BRCA1) regarding their clinical and molecular characteristics (mutation and methylation in BRCA1). The transcriptional level of BRCA1 was also tested for possible associations with pathological and morphological characteristics of breast cancer, as well as clinical characteristics, prognosis and family history.
The inclusion was based on the mutational status of BRCA1 (mutated, with a VUS or WT) and no patient harboring a BRCA2 mutation was included. The majority of patients (69.6%) with a pathogenic mutation at BRCA1 were triple negative (as expected). Conversely patients with a VUS on BRCA1 and those harboring WT BRCA1 had a similar proportion of triple negative tumors (31.8% and 32.6% respectively), a percentage slightly higher than that observed in sporadic cases of breast cancer [22].
Several studies have reported the association of triple negative breast cancer with the presence of pathogenic mutations in BRCA1. A study published by the CIMBA group reported a frequency of 69% of TN tumors in 3,797 patients with BRCA1 mutations [23]. Similarly, a study published by Carraro et al. showed a frequency of 71.4% of TN tumors among a small cohort of patients with germline BRCA1 mutation and early diagnosis of breast cancer [24].
The higher prevalence of TN cases among BRCA1 mutated patients can be one of the factors responsible for the poor prognosis observed in these patients. Studies point to the fact that tumors associated with the presence of BRCA1 mutations often have higher histologic grade, and elevated mitotic counts, are poorly differentiated, with high frequency of necrotic areas and pleomorphism. These characteristics are commonly associated with a worse prognosis [23, 25, 26]. In our study, we observed that most patients with mutation in BRCA1 showed histological grade III (61.0%), triple negativity and higher cell proliferation index.
Besides having a tumor profile characteristic of tumors with a poor prognosis, the family history associated with those carrying a pathogenic BRCA1 mutation was more severe than the WT or with a VUS, as it would be expected. The mean age at cancer diagnosis in the BRCA1-pathogenic group was 41.6 years, in the VUS-BRCA1 group this value decreased to 37.2 years, and for the WT-BRCA1 group the mean age was 36.5 years. This finding points to the fact that maybe most of those families were referred by genetic testing due to an earlier age at cancer diagnosis and not due to a strong family history of cancer. Although the presence of mutations in other breast and ovarian predisposition genes can be the explanation for the cancer cases identified in the WT and VUS groups, this was not investigated in the present study. The subsequent search for a better understanding of the characteristics of families at-risk for breast cancer, as well as to more accurately determine the risk of cancer is critical. Knowing which gene is altered and the mechanisms associated with this change makes possible to expand the range of preventive strategies to be offered.
Other than germline mutation status, we also investigated the methylation profile of BRCA1 in breast tumor and normal counterpart. The presence of methylation in the promoter region of BRCA1 in sporadic breast tumors has been reported in several studies. A study conducted in 2000 by Rice et al., detected the presence of hypermethylation in 3/21 breast tumors with concomitant lower levels of mRNA. The authors suggested that epigenetic silencing of BRCA1 can be a transcriptional inactivation mechanism responsible for the development of mammary tumorigenesis in sporadic cases [27]. A more recent study, conducted by Hsu et al., analyzed the methylation profile of 139 patients diagnosed with breast cancer at an early stage. The authors reported the presence of hypermethylation in 56% of the analyzed tumors and this alteration was correlated with the occurrence of triple negative tumors [21]. In 2014, work by Sharma and colleagues reported the presence of BRCA1 methylation in 30% of women with sporadic breast cancer [28]. However, studies involving methylation analysis in tumor tissue of patients carriers of a BRCA1 germline pathogenic mutation are scarce [29–31].
Although well known for hereditary colorectal tumors [17, 32], the presence of epigenetic mutations in hereditary breast cancer is not well understood. Results published by Dworkin et al., in 2009 reaffirmed the proposed by Esteller in 2001, that the hypermethylation of the BRCA1 promoter region is not the most frequent “second-hit” in patients with germline mutation in this gene [29]. Recently, Lips et al., found that none of the tumors analyzed in their study presented both events: germline mutation and methylation in the BRCA1 gene. These data suggest that BRCA1 is only inactivated by genetic or epigenetic events [30].
In the present study we identified two patients with hypermethylation of the BRCA1 gene, one of those with a constitutive epimutation (confirmed in DNA obtained from peripheric blood). That patient was BRCA1 and BRCA2 WT and had a personal history of bilateral breast cancer (diagnosed at 37 and 41 years old), both tumors were invasive ductal carcinomas with estrogen receptor negative. Moreover, in line with the cited studies, none of the analyzed patients had both events: germline mutation in BRCA1 and hypermethylation. In 2014, Al-Moghrabi et al. investigated the presence of constitutive BRCA1 hypermethylation in 155 breast cancer patients and 143 cancer-free females and found that constitutive epimutation can constitute a mechanism for breast cancer predisposition, being present in 14.2% of breast cancer patients and 9.1% of cancer-free females (with a positive family history of cancer) [33].
The other patient with BRCA1 hipermethylated identified in our study also presented negativity for the estrogen receptor and was BRCA1 WT. Study conducted by Birgisdottir et al. showed that the hypermethylation of BRCA1 in sporadic tumors was associated with the loss of estrogen receptor expression. Moreover, it was present in a higher frequency in women diagnosed under the age of 50, suggesting that the tumors with aberrant methylation in BRCA1 are similar to tumors with pathogenic mutation in BRCA1, leading to the same BRCA-ness phenomenon [34].
In our study, due to the poor quality of a proportion of RNA samples extracted from FFPE for the analysis of gene expression, it was not possible to verify the expression levels of BRCA1 of patients with hypermethylation. Furthermore, we demonstrate that the profile of BRCA1 gene expression was significantly different between normal vs. tumor tissue.
An interesting fact observed was that a significant number of cases, which did not have germline mutations nor methylation in BRCA1 had low levels of BRCA1 gene expression, leading us to speculate which could be the mechanisms related to this silencing. Among the mechanisms that may be involved, one could emphasize the presence of somatic BRCA1 mutation, LOH, mRNA decay, miRNA regulation, among others. Moreover, we cannot exclude the possibility of methylation in the “body of the gene” or in other regions of the promoter not covered in this study. Thus, considering the findings of this study and the possible mechanisms involved, further studies involving LOH analysis, miRNA, methylation in other regions of the promoter or the gene body, as well as protein analysis and functional studies should be conducted to analyze in detail the reason why the expression levels of BRCA1 are low.
A better understanding of the mechanisms involved in reducing gene expression levels of the BRCA1 gene or in its silencing is of fundamental importance, since the expression levels may influence the response to various agents, and also serve as therapeutic targets such as the new generation of target specific agents, namely PARP (poly ADP ribose polymerase) inhibitors.
In conclusion, it is worth noting that carriers of germline mutations in the BRCA1 gene have vital cumulative risk much higher than the population for the development of cancer. Because of that, the knowledge of which gene is altered and the mechanisms involved enables a significant improvement in the identification of individuals at-risk, as well as in decisions about the management of risk and preventive strategies, risk reduction (prophylactic surgery and chemoprevention) and therapeutics to be offered. Through the literature reports, along with our data, we can infer that a significant portion of patients at risk for hereditary breast cancer (mutated or not in BRCA1) show a reduction in gene expression levels of BRCA1, requiring more studies to elucidate these findings. Still, we can infer that germline mutation and presence of BRCA1 hypermethylation are mutually exclusive events and both contribute to the BRCA-ness. Furthermore, the presence of a constitutive epimutation should be taken into consideration in a family at high risk for hereditary breast cancer without any germline mutation identified, since the presence of this epigenetic alteration can constitute the explanation for the family history identified.
MATERIALS AND METHODS
Subjects
The study analyzed a convenience group of 85 women at-risk for hereditary breast cancer from the Oncogenetics Department of Barretos Cancer Hospital (BCH). In addition, 3 patients with TN tumors and presence of a germline BRCA1 mutation from the International Research Center AC Camargo were included. Those women were referred for BRCA1/2 genetic testing due to the presence of clinical criteria for HBOC (Hereditary Breast and Ovarian Cancer Predisposition Syndrome). For the purpose of the present study, these patients were grouped according to the genetic test results: 1) BRCA1-pathogenic group: 23 women (families) with personal and family history of breast cancer carrying a deleterious mutation in the BRCA1 gene; 2) BRCA1-VUS group: 22 women (families) with personal and family history of breast cancer with unknown clinical significance variants (VUS) identified in the BRCA1 gene and, 3) BRCA1-WT group: 43 women (families) with personal and family history of breast cancer without deleterious mutation and VUS identified in the BRCA1 gene.
For VUS classification (and inclusion in the BRCA1-VUS group) the following databases were considered: Clinvar, BIC (Breast Cancer Information Core), HGMD (Human Genome Mutation Database) and LOVD IARC (Leiden Open Variation Database).
This study was approved by the Ethics Committees of the two participating Institutions, Barretos Cancer Hospital and AC Camargo Cancer Center (Process numbers: 801/2014 and 1746/13, respectively).
Laboratory analyses
Analyses of mutations in BRCA1 and BRCA2
The analyses of the presence of germline mutations in BRCA1 and BRCA2 was conducted at the Center of Molecular Diagnosis of BCH as part of routine care through NGS sequencing (using the BRCA1/2 Ampliseq panel in the Ion Torrent PGM platform) as described elsewhere by Palmero and collaborators [35]. Basically, libraries containing the PCR product of 14 multiplex PCRs were pooled, purified and fragmented according to the supplier protocol. In the sequence the libraries with the adaptors were amplified by emulsion PCR, enriched and sequenced using the Ion 316 chips, which allow the simultaneous analysis of 12 patients per chip. Data analysis was performed using DNAstar Lasergene 10 software, with the following parameters: (1) Q call > = 40; (2) depth of coverage > = 100 and, (3) SNP % > = 23. All the identified variants were confirmed in a new PCR reaction followed by conventional bi-directional sequencing (Sanger).
For the screening of large rearrangements over the BRCA1 and BRCA2 genes, Multiplex Ligation-dependent Probe Amplification Kits (MLPA) were used.
Biological samples
For methylation and gene expression analysis a representative section of each FFPE normal and tumor tissue were stained by hematoxylin and eosin and evaluated by a pathologist to verify normal and tumor content (> 60% tumor) and macrodissected. For DNA extraction the DNeasy Blood and Tissue kit (Qiagen) was used following the manufacturer instructions. For RNA extraction, Recover All Total Nucleic Isolation Optimized for FFPE Samples (Ambion by Thermo Fisher Scientific) kit was used.
Analyses of the quality and integrity of the extracted DNA
To verify the quality and integrity of the extracted DNA a multiplex PCR reaction with four pairs of primers for the GAPDH gene was performed as described by Van Beers et al. [36].
Basically, the PCR was performed with a final volume of 30 μL, containing 1.5 mM MgCl2; 0.2 mM dNTP (Invitrogen™); 0,133 μM of each primer; 1 U Taq DNA polymerase (Invitrogen™) and 60 ng normal/tumor DNA. The reactions were performed in a Veriti® thermocycler (Applied Biosystems) using the following amplification parameters: 94°C for 1 minute, 35 cycles of 94°C for 1 minute, 56°C for 1 minute, and 72°C for 3 minutes. Finally, a final extension at 72°C for 7 minutes, finishing at 15°C. The successful amplification of DNA was checked by agarose gel electrophoresis on 1.5% GelRed ™ stained, visualized under UV light and documented.
Treatment with sodium bisulfite
For methylation analysis, the DNA obtained was subjected to treatment with sodium bisulfite using EpiTect bisulfite Kit (Qiagen) and following the manufacturer’s specifications. Briefly, 1 μg of DNA was mixed with 85 μl of bisulfite mix (provided in the kit), 35 μl protect DNA buffer and water to a final volume of 140 μl and placed in a thermocycler for bisulfite conversion. Next, the converted DNA was purified by several washing steps, eluted in 20 μl of ultra-pure water (Milli-Q) and stored at −80°C until use.
qMSP analysis
Bisulfite-modified DNA was used as a template in fluorogenic qMSP assays carried out in a final volume of 20 μL in a 7500 Real Time PCR System (Life Technologies). PCR was done in separate wells for each primer/probe set and each sample was run in triplicate. The final reaction mixture contained 3 μL of bisulfite-modified DNA, 1.2 μM of forward and reverse primers, 200 nM of probe, 0.6 U of platinum Taq polymerase (Life Technologies), 200 μM of dNTPs, 16.6 mM of ammonium sulfate, 67 mM of Tris-HCl pH 8.0, 6.7 mM of magnesium chloride, 10 mM of mercaptoethanol, 0.1% DMSO and 1× ROX dye (Life Technologies). PCR was conducted with the following conditions: 95°C for 2 minutes, followed by 45 cycles at 95°C for 15 seconds and 60°C for 1 minute.
Each plate included DNA samples, multiple water blanks and serial dilutions (90–0.009 ng) of a positive control for constructing the calibration curves. Leukocyte DNA from a healthy individual was methylated in vitro with SssI methyltransferase (New England Biolabs Inc) to generate completely methylated DNA at all CpG and used as a positive control. Additionally, the level of methylation of normal breast tissue was measured in two cell lines derived from normal tissue (HB4a and MCF10A).
Primers and probes that specifically amplify the promoter regions of the BRCA1 gene and the internal reference gene, ACTB were selected from the literature [37]. Samples were considered methylated when detecting amplification of at least two of the triplicates. The absence of amplification or amplification of only one of the triplicates indicates that the sample is non-methylated. The percentage of methylation on each sample was obtained by the equation: mean number of methylated copies of target gene/average number of copies of ACTB X 100. Cases were scored as positive if a percentage value of ≥ 4.0% was obtained, according to studies published in the literature [38, 39].
Analyses of the assessment and quality of total RNA
Quantification of total RNA samples was first performed with the spectrophotometer NanoDrop 2000 Spectrophotometer (Thermo Scientific) by analyzing the absorbance at 260 nm. Then, to ensure a more accurate quantification, a fluorescent method with Qubit equipment (Invitrogen) was used. The analyses of the quality/integrity of total RNA was performed by electrophoresis equipment microfluidics Bioanalyzer 2100 (Agilent Technologies) using RNA Pico Chip (Agilent Technologies) according to the manufacturer’s recommendations.
cDNA synthesis and real-time PCR
To obtain the cDNA, RT-PCR was performed using the High-Capacity kit (Applied Biosystems) according to the conditions provided by the manufacturer. Basically, 1 μg of total RNA was added to 2,0 μL RT buffer (10X), 0,8 μL dNTPs (100 mM), 2 μL RT Random Primers (10X), 1 μL-MultiScribe Reverse Transcriptase and RNase-free water to a volume end of 10 μL. The reactions were maintained at 25°C for 10 minutes, 37°C for 120 minutes and ended by an incubation of 5 minutes at 85°C. The reactions were performed in a Proflex Thermal Cycler (Thermo Fisher Scientific). Upon completion of synthesis, all cDNAs were kept at −20°C until use.
The real time PCR reactions were carried out in the StepOne Real-Time PCR Systems (Applied Biosystems) using the TaqMan system (Applied Biosystems). Inventoried trials “PrimeTime qPCR assay tests” (Integrated DNA Technologies) and TaqMan (Applied Biosystems) were used. Probes were used for two endogenous housekeeping genes: ACTB (Assay ID: Hs.PT.39a.22214847) and GAPDH (Assay ID: Hs.PT.39a.22214836) and for the testing gene BRCA1 (Assay ID: Hs01556193_m1). For reactions we used 2,5 μL water, 5 μL of TaqMan Fast Advanced 2X Master Mix (Applied Biosystems), 0,5 μL TaqMan Gene Expression Assay 20X (Applied Biosystems) containing primers and probe specific for each of the genes and 2 μL of each cDNA sample in a final volume of 10 uL reaction. The PCR was conducted in 40 cycles, according to the following protocol: 2 minutes at 50°C, 20 seconds at 95°C, 1 second at 95°C, 20 seconds at 60°C. Small experimental variations in pipetting and reaction efficiency were diluted by conducting experiments in triplicate. To achieve quantification, we used the average of the CT values of triplicates. The standard expression of the gene of interest wascalculated by the mathematical model 2-ΔCT [40]. The value of the median 2- ΔCT obtained for all samples was used as the cutoff point for classification of samples in low and high expression. The expression levels of the BRCA1 gene (ΔCT) were represented by the mean value of triplicates normalized by the average expression of endogenous (GAPDH and ACTB). Lower ΔCT values indicates greater expression. The value of the median ΔCT (0.55636) was used as the cutoff between high and low expression, e.g, the samples were compared independently.
Statistical analyses
Statistical analysis was performed using SPSS v.19.0 software for Windows (Chicago, IL). Categorical variables were described by absolute frequencies and percentages relative frequencies. Quantitative variables were described by mean and standard deviation when they had symmetrical distribution and the median, minimum and maximum when they ha asymmetric distribution. The significance level for all tests was 5%.
To compare the clinical and molecular characteristics we used Chi-square test (or Fisher’s exact test). Comparisons between methylated vs. mutated groups, as well as among presence vs. absence of expression of BRCA1 in relation to clinical and molecular characteristics were also performed using the Chi-square test.
ACKNOWLEDGMENTS
The authors thank the Center of Molecular Diagnosis and the Pathology Department of Barretos Cancer Hospital. PSF received a Master Fellowship from PROSUP/CAPES. MEM, LMRBA and ACC were recipient of FAPESP fellowship.
CONFLICTS OF INTEREST
All the authors declare no conflicts of interest.
GRANT SUPPORT
This study was partially supported by FAPESP (2013/24633–2 and 2013/23277–8).
REFERENCES
1. WHO. The world health report 2006: working together for health. 2016.
2. Lynch HT, Snyder C and Lynch J. Hereditary breast cancer: practical pursuit for clinical translation. Ann Surg Oncol. 2012; 19:1723–1731.
3. Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014; 343:1466–1470.
4. Marroni F, Aretini P, D’Andrea E, Caligo MA, Cortesi L, Viel A, Ricevuto E, Montagna M, Cipollini G, Federico M, Santarosa M, Marchetti P, Bailey-Wilson JE, et al. Penetrances of breast and ovarian cancer in a large series of families tested for BRCA1/2 mutations. Eur J Hum Genet. 2004; 12:899–906.
5. Gronwald J, Byrski T, Huzarski T, Oszurek O, Janicka A, Szymanska-Pasternak J, Gorski B, Menkiszak J, Rzepka-Gorska I, Lubinski J. Hereditary breast and ovarian cancer. Hereditary cancer in clinical practice. 2008; 6:88–98.
6. Petrucelli N, Daly MB, Feldman GL. BRCA1 and BRCA2 Hereditary Breast and Ovarian Cancer. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, et al. eds. GeneReviews(R). (Seattle (WA). 1993.
7. Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed. 2005; 7:60.
8. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44:D862–8.
9. Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genet. 2014; 133:1–9.
10. Nowsheen S, Aziz K, Tran PT, Gorgoulis VG, Yang ES, Georgakilas AG. Epigenetic inactivation of DNA repair in breast cancer. Cancer Lett. 2014; 342:213–222.
11. Karsli-Ceppioglu S, Dagdemir A, Judes G, Ngollo M, Penault-Llorca F, Pajon A, Bignon YJ, Bernard-Gallon D. Epigenetic mechanisms of breast cancer: an update of the current knowledge. Epigenomics. 2014; 6:651–664.
12. Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, Laird-Offringa IA. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012; 22:1197–1211.
13. Samowitz WS. The CpG island methylator phenotype in colorectal cancer. J Mol Diagn. 2007; 9:281–283.
14. Demokan S, Dalay N. Role of DNA methylation in head and neck cancer. Clin Epigenetics. 2011; 2:123–150.
15. Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jonsson G, Olsson H, Borg A, Ringner M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010; 12:R36.
16. Costa FF, Verbisck NV, Salim AC, Ierardi DF, Pires LC, Sasahara RM, Sogayar MC, Zanata SM, Mackay A, O’Hare M, Soares F, Simpson AJ, Camargo AA. Epigenetic silencing of the adhesion molecule ADAM23 is highly frequent in breast tumors. Oncogene. 2004; 23:1481–1488.
17. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001; 61:3225–3229.
18. Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, Baylin SB, Ahuja N, Viale A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011; 3:75ra25.
19. Matros E, Wang ZC, Lodeiro G, Miron A, Iglehart JD, Richardson AL. BRCA1 promoter methylation in sporadic breast tumors: relationship to gene expression profiles. Breast Cancer Res Treat. 2005; 91:179–186.
20. Saelee P, Chaiwerawattana A, Ogawa K, Cho YM, Tiwawech D, Suktangman V. Clinicopathological significance of BRCA1 promoter hypermethylation in Thai breast cancer patients. Asian Pac J Cancer Prev. 2014; 15:10585–10589.
21. Hsu NC, Huang YF, Yokoyama KK, Chu PY, Chen FM, Hou MF. Methylation of BRCA1 promoter region is associated with unfavorable prognosis in women with early-stage breast cancer. PLoS One. 2013; 8:e56256.
22. Tung N, Gaughan E, Hacker MR, Lee LJ, Alexander B, Poles E, Schnitt SJ, Garber JE. Outcome of triple negative breast cancer: comparison of sporadic and BRCA1-associated cancers. Breast Cancer Res Treat. 2014; 146:175–182.
23. Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. 2010; 4:174–191.
24. Carraro DM, Koike Folgueira MA, Garcia Lisboa BC, Ribeiro Olivieri EH, Vitorino Krepischi AC, de Carvalho AF, de Carvalho Mota LD, Puga RD, do Socorro Maciel M, Michelli RA, de Lyra EC, Grosso SH, Soares FA, et al. Comprehensive analysis of BRCA1, BRCA2 and TP53 germline mutation and tumor characterization: a portrait of early-onset breast cancer in Brazil. PLoS One. 2013; 8:e57581.
25. van der Groep P, van der Wall E, van Diest PJ. Pathology of hereditary breast cancer. Cell Oncol (Dordr). 2011; 34:71–88.
26. Cortesi L, Turchetti D, Bertoni C, Bellei R, Mangone L, Vinceti M, Federico M, Silingardi V, Ferrari S. Comparison between genotype and phenotype identifies a high-risk population carrying BRCA1 mutations. Genes Chromosomes Cancer. 2000; 27:130–135.
27. Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis. 2000; 21:1761–1765.
28. Sharma P, Stecklein SR, Kimler BF, Sethi G, Petroff BK, Phillips TA, Tawfik OW, Godwin AK, Jensen RA. The prognostic value of promoter methylation in early stage triple negative breast cancer. J Cancer Ther Res. 2014; 3:1–11.
29. Dworkin AM, Spearman AD, Tseng SY, Sweet K, Toland AE. Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam Cancer. 2009; 8:339–346.
30. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, Wesseling J, Rodenhuis S, Nederlof PM. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013; 108:2172–2177.
31. Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000; 92:564–569.
32. Gausachs M, Mur P, Corral J, Pineda M, Gonzalez S, Benito L, Menendez M, Espinas JA, Brunet J, Iniesta MD, Gruber SB, Lazaro C, Blanco I, et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur J Hum Genet. 2012; 20:762–768.
33. Al-Moghrabi N, Nofel A, Al-Yousef N, Madkhali S, Bin Amer SM, Alaiya A, Shinwari Z, Al-Tweigeri T, Karakas B, Tulbah A, Aboussekhra A. The molecular significance of methylated BRCA1 promoter in white blood cells of cancer-free females. BMC cancer. 2014; 14:830.
34. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006; 8:R38.
35. Palmero EI, Galvao HC, Fernandes GC, Paula AE, Oliveira JC, Souza CP, Andrade CE, Romagnolo LG, Volc S, C MN, Sabato C, Grasel R, Mauad E, et al. Oncogenetics service and the Brazilian public health system: the experience of a reference Cancer Hospital. Genet Mol Biol. 2016; 39:168–177.
36. van Beers EH, Joosse SA, Ligtenberg MJ, Fles R, Hogervorst FB, Verhoef S, Nederlof PM. A multiplex PCR predictor for aCGH success of FFPE samples. Br J Cancer. 2006; 94:333–337.
37. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics. 2006; 38:787–793.
38. Press JZ, De Luca A, Boyd N, Young S, Troussard A, Ridge Y, Kaurah P, Kalloger SE, Blood KA, Smith M, Spellman PT, Wang Y, Miller DM, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer. 2008; 8:17.
39. Eads CA, Lord RV, Wickramasinghe K, Long TI, Kurumboor SK, Bernstein L, Peters JH, DeMeester SR, DeMeester TR, Skinner KA, Laird PW. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001; 61:3410–3418.
40. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408.