
Introduction
Over 60% of currently used antitumor drugs come from natural sources such as plants, fungi and microorganisms. The large scale screening programs for natural products with anticancer activities, e.g. those launched in 1950s by Italian research company or in 1960s by the National Cancer Institute (NCI), allowed for identification of bacteria-produced doxorubicin and taxol (paclitaxel), derived from the bark of the yew tree. Both of these compounds are widely used in chemotherapy regimens in different cancer types. Though their mechanism of action is different as doxorubicin intercalates into DNA and abrogates replication [1] and taxol inhibits microtubules depolymerization during mitosis [2], they both induce strong oxidative stress, though by different means [3-5]. Total cellular antioxidant capacity is a known determinant of cancer susceptibility to these drugs [6-8]. Oxidative stress induced by chemotherapeutics is crucial for their efficacy, but, on the other hand, contributes to the cumulative and irreversible cardiotoxicity observed clinically [9, 10]. These side effects highlight the lack of selectivity of chemotherapy [11]. Therefore, non-toxic natural substances that potentiate action of chemotherapeutics and allow for lowering their concentration are of a particular interest to the anticancer drug field.
ROS in cellular transformation
Majority of cellular reactive oxygen species (ROS) is produced during aerobic respiration by electrons released from the electron transport chain (ETC) in mitochondria. Incomplete oxygen reduction creates superoxide anion (O2.-), the precursor of three remaining species: hydroxyl radical (OH.), hydrogen peroxide (H2O2) and peroxynitrite (OONO-) [12] (Figure 1). Mitochondrial electron leakage increases with age pointing to the imbalance between mitochondrial biogenesis and degradation - a root cause of neurodegenerative and cardiovascular diseases, diabetes and cancer [13]. The second largest contributor to cellular ROS are NADPH oxidases (NOX) residing in cytoplasm, catalyzing the production of superoxide from O2 and NADPH [14, 15]. At low concentrations, superoxide production may be involved in cellular signal transduction, but high concentrations of radicals cause oxidative damage due to their high reactivity towards other cellular compounds [16].
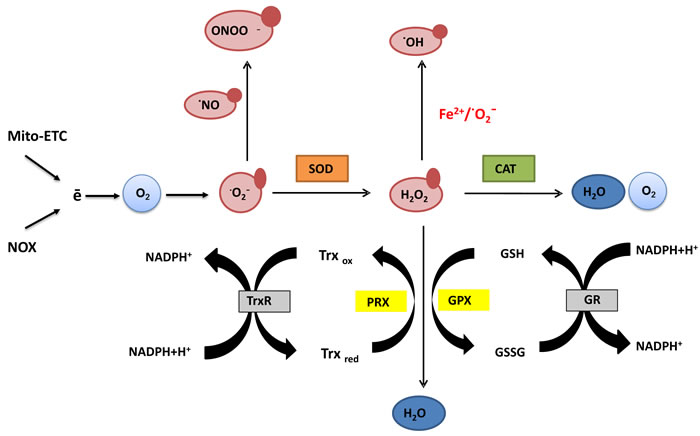
Figure 1: Generation and scavenging of reactive oxygen species (ROS). Electrons released from the mitochondrial electron transport chain (Mito-ETC) and produced by NADPH oxidases (NOX) are the major source of endogenous reactive oxygen species. Coupled to molecular oxygen they give rise to the primary free radical and the precursor of remaining species - superoxide (·O2-). In the reaction with a short-lived nitric oxide (·NO), superoxide forms a highly reactive peroxynitrate (ONOO-) able to modify structure and function of proteins. Alternatively, superoxide dismutase (SOD) converts superoxide to hydrogen peroxide (H2O2), which can be further transformed in several ways. In the presence of transition metal ions like Fe2+ (Fenton’s reaction) or in reaction with superoxide, H2O2 forms highly reactive hydroxyl radical (·OH) which damages lipids, proteins and DNA. Peroxysomal enzyme catalase (CAT) neutralizes H2O2 to water and oxygen. H2O2 might be also utilized in the reaction of oxidation of monomeric glutathione (GSH) to the glutathione disulfide (GSSG) or reduced thioredoxin (Trxred) to the oxidized thioredoxin (Trxox) catalyzed by glutathione peroxidase (GPX) or peroxidases involved in the thioredoxin turnover (PRX). Reduced glutathione pool is restored by glutathione reductase (GR) which reduces oxidized glutathione with the use of NADPH. Similarly, thioredoxin reductase (TrxR) balances the amount of reduced Trx by transferring electrons from NADPH to oxidized catalytic sites. Thanks to the thiol groups in the Cys residues both glutathione and thioredoxin participate in the reduction of oxidized proteins. Their synthesis as well as the turnover are under tight homeostatic control creating a system responsible for reduction of redox-sensitive proteins upon oxidative stress.
Higher steady-state levels of ROS in cancer cells relative to normal cells have been known for around 35 years [17]. Increased ROS are crucial in the initiation of carcinogenesis when acquiring new mutations and clonal expansion of initiated cells are needed to establish a tumor. This renders them both the cause and the result of cellular transformation: ROS-induced oxidative damage favors production of more radicals and establishes a feed-in loop, increasing mutations rate, activating oncogenes, enhancing metabolic reprogramming and progression of tumors. The enhanced ROS generation is induced by oncogenic signaling with main drivers: V-Ras, K-Ras, mtp53 and c-Myc [18, 19] and involves both mitochondrial and cytoplasmic ROS. K-Ras-induced cellular transformation was shown to require NOX1 activation through p38/PDPK1/PKCδ/p47phox cascade [20], while expression of Myr-Akt, H-RasG12V and K-RasG12D in murine embryonic fibroblasts (MEFs) conferred increased mitochondrial ROS-dependent soft agar colony formation [21]. Mutations in tumor suppressors genes are often associated with the induction of strong oxidative stress and promote the survival of cells with high ROS levels. Mutant BRCA1 and p53 were shown to attenuate antioxidant signaling driven by the nuclear factor (erythroid-derived 2)-like 2 (Nrf2), contributing to cancer initiation [22, 23]
One of the consequences of the excessive damage caused by ROS are changes in mitochondrial membrane permeability that result in cytochrome C release and apoptotic death [24-29]. In defense, cancer cells boost their antiapoptotic mechanisms like nuclear factor kappa-light-chain-enhancer of activated B cells (NFĸB) pathway to escape cell death [30, 31]. Decreased mitochondrial activity triggers the glycolytic switch and upregulates glycolytic pathway in order to produce more energy and biomass (ribose, amino acids, fatty acids) for rapidly proliferating cancer cells [32]. Moreover, exposure to oxidative stress induces mutations in mitochondrial DNA as well as in VEGF (Vascular Endothelial Growth Factor) and HIF-1α (Hypoxia Inducible Factor-1α) genes, promoting angiogenesis and further enhancing metabolic reprogramming of cells [33]. Oxidative stress also changes the tumor microenvironment to support growth and cell spread. Hydrogen peroxide produced by tumor tissue can initiate destruction of non-tumor surrounding tissue to obtain nutrients and promote growth [34]. This explains why tumors are said to be “addicted to ROS signaling”.
ROS adaptations in tumors
Distinct redox homeostasis and higher intracellular ROS levels in cancer cells drive their growth and metastasis but might also pose a threat of oxidative damage and death. Moderate expression of NADPH oxidase NOX5-L induced cancer cells proliferation accompanied by AKT and ERK phosphorylation, whereas an increase in NOX5-L above a certain threshold promoted apoptosis [35]. Tumors need to adapt to the oxidative stress conditions and they do that by enhancing their antioxidative defense to lower ROS levels and by inducing autophagy to reduce the oxidative damage to biomolecules and organelles [36-39]. These two mechanisms constitute finely orchestrated and interconnected repair system in oxidatively stressed cells seeking homeostasis [36]. Interestingly, the same oncogene signals that boost ROS signaling, promote antioxidant adaptive mechanisms to stand this constant stress and minimize oxidative damage. Activation of endogenous K-Ras(G12D), B-Raf(V619E) and Myc(ERT2) led to lowering of intracellular ROS due to the increased transcription of Nrf2 and elevation of the basal Nrf2 antioxidant program [40]. Furthermore, genetic targeting of the Nrf2 pathway impaired K-Ras(G12D)-induced proliferation and tumorigenesis in vivo pointing that the Nrf2 pathway represents a previously unappreciated mediator of oncogenesis [40]. Accordingly, it was reported that genetic mutations that occur in cancer cells led to constant Nrf2 activity and enhanced antioxidant capacity [41]. Harris et al. (2015) showed that synthesis of the antioxidant glutathione (GSH) was required for cancer initiation in vivo [42]. Genetic loss of the enzyme driving GSH synthesis, glutamate-cysteine ligase modifier subunit (GCLM), prevented a tumor’s ability to drive malignant transformation. Interestingly, at later stages of tumor progression GSH became dispensable potentially due to the compensation from an alternative antioxidant pathway - thioredoxin pathway, demonstrating the importance of GSH and thioredoxin to tumor progression and indicating them as potential targets for therapeutic intervention.
Mitochondrial ROS are the major inducers of autophagy, however, upon chronic impairment of mitochondrial function, high extent of radicals shifts signaling into self-removal of mitochondria through a selective process called mitophagy [43, 44]. This fine mechanism allows autophagy to eliminate the source of oxidative stress and protect the cell from oxidative damage. Recently, autophagy was shown to prevent the initiation of hepatocarcinogenesis and metastasis of gastric cancer by maintaining healthy mitochondria and reducing oxidative stress and DNA damage [45-47]. On the other hand, once the cellular transformation was initiated, autophagy was required to promote cancer progression by limiting tumor suppressors [45].
Targeting ROS adaptations in cancer
Because of this sharp reliance on ROS production, cancer cells are more vulnerable to further disturbance of their red-ox status than normal cells. This difference establishes a therapeutic window allowing for an emergence of the selective anticancer strategy based on modulation of cancer cells redox potential. Due to the enhanced antioxidant capacity of tumors, just inducing ROS generation is not sufficient for a successful eradication of cancer. The drug should also inhibit the antioxidant defense system [48]. Many compounds of natural origin block Nrf2 pathway or directly inhibit endogenous antioxidants leading to the elevated ROS production. Moreover, Nrf2 inhibition results in a decrease of drug efflux transporters and a consequent increase in retention of anticancer drugs in cells. Therefore Nrf2 or cellular antioxidant inhibitors synergize with classic chemotherapeutics and decrease their toxicity. Surprisingly, among them there are polyphenols like resveratrol, quercetin, EGCG, apigenin, luteolin or chrysin which were initially reported to have ROS scavenging properties and are generally recognized as antioxidants. Therefore a considerable caution should be exercised when applying natural products as adjuvants since their effects strongly depend on concentration, cell type, exposure time and environmental conditions [49-55].
The Nrf2 pathway
Disruption of redox balance in cells results in activation of redox sensitive transcription factors like Nrf2, NFĸB and activator protein 1 (AP-1) [56]. The major driver of antioxidants expression that confers protection against endogenous and exogenous hazards, DNA damage and consequent cancer initiation is Nrf2 transcription factor [41, 57]. Activation of Nrf2 pathway allows for cell adaptation and survival by regulating expression of antioxidans, anti-inflammatory and phase II detoxification enzymes (Figure 2). Major regulator of Nrf2 activity in cells is the cytosolic inhibitor Keap1, responsible for its ubiquitination and proteasomal degradation [58, 59]. Apart from Keap 1, oncogenes like K-Ras(G12D), B-Raf(V619E) and Myc(ERT2) have been shown to stabilize Nrf2 and antioxidant proteins leading to drug resistance in tumors [40]. Nrf2 is overexpressed in several types of human cancer, including cancer of the lung, breast, oesophagus, ovary, prostate, pancreatic, colorectal, head and neck squamous cell carcinoma, gallbladder and skin which indicates that the cytoprotective properties of the Nrf2 pathway can be exploited by tumor cells to promote their survival [60, 61]. Constitutive Nrf2 activation has been reported to mediate chemoresistance in many tumor types [62, 63]. Suppression of Nrf2 activity inhibited tumor growth and enhanced the efficacy of chemotherapeutic agents. Disruption of the Nrf2 pathway in a mouse model of K-RasG12D-induced lung cancer enhanced the antitumor efficacy of cisplatin [64]. Temporal blockage of Nrf2-dependent cytoprotection using Nrf2 inhibitors is important to enhance a patient’s response to chemo- and radiotherapy but on the other hand, activation of Nrf2 pathway supports treatment of neurodegenerative diseases, multiple sclerosis and prevents cancer initiation by counteracting oxidative and electrophilic stress [60]. It means that in case of cancer, the Nrf2 pathway is a double edge sword: activating this pathway is crucial for chemoprevention but once the control is lost, it provides growth advantage to cancer cells allowing for rapid proliferation, escape from apoptosis or senescence and resistance to chemo- and radiotherapy. Thus, both activation and inhibition of Nrf2 activity could be beneficial, although in different patient cohorts (Figure 3).
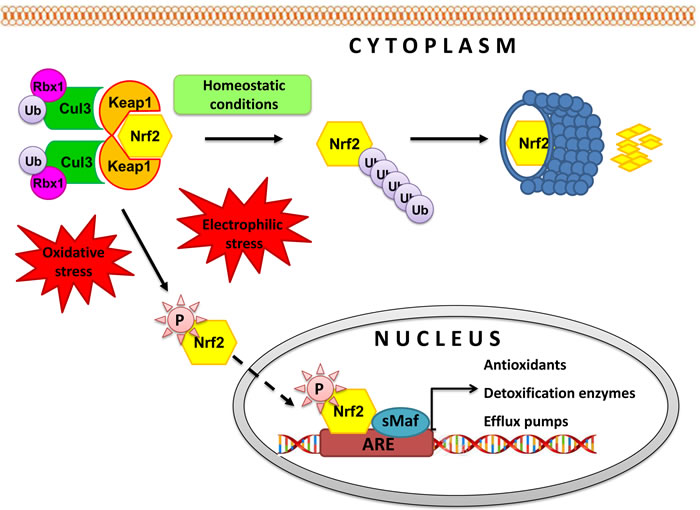
Figure 2: Regulation of Keap1-Nrf2 pathway. Under basal conditions, cytosolic repressor Kelch-like ECH-associated protein 1 (Keap1), a substrate adaptor protein for Cullin 3 (Cul3)/Rbx1 ubiquitin ligase, holds Nrf2 in the cytoplasm and promotes its ubiquitination followed by 26S proteasomal degradation [58,59]. In the presence of electrophilic and/or oxidative stimulus, Nrf2 is released from Keap1 and translocates to the nucleus where it recruits small Maf protein (sMaf) and binds with response element (ARE) in the promoter regions of its target genes, inducing their expression. Activation of Nrf2 pathway allows for cell adaptation and survival by regulating expression of antioxidans, anti-inflammatory and phase II detoxification enzymes such as superoxide dismutase (SOD), gluthatione S-transferase (GST), heme oxygenase-1 (HO-1), NAD(P)H-quinone oxidoreductase (NQO1), UDP-glucuronosyl transferases (UGT), γ-glutamylcysteine synthetase (γGCS) and efflux pumps like multidrug resistance-associated protein 2 (MRP2) and breast cancer resistance protein (BCRP). Proteins transcriptionally controlled by Nrf2 take part in biosynthesis, utilization and regeneration of glutathione, thioredoxin, and NADPH resulting in restoration of cellular redox homeostasis.
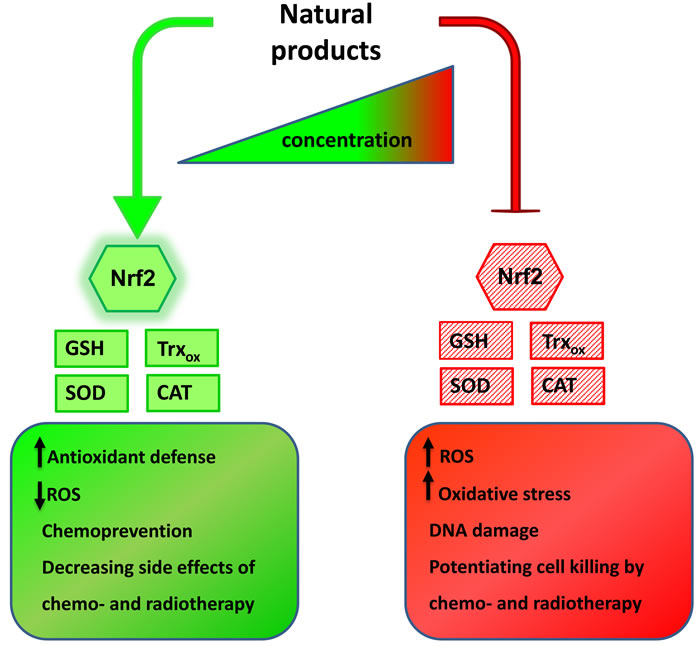
Figure 3: Natural products action on cellular antioxidants is concentration-dependent. Many natural compounds display opposing properties in cancer cells, depending on their concentration. At lower concentrations they often boost cells’ antioxidant capacity by activating Nrf2-dependent signaling and enhancing expression of ROS scavengers, lowering ROS burden. These properties allow for using natural compounds in chemoprevention and as agents decreasing side effects of standard anticancer regimens. On the other hand, same compounds used at higher concentrations inhibit antioxidant defense and induce oxidative stress. By doing that they enhance the effectiveness of chemo- and radiotherapy and allow for lowering their doses.
Natural products targeting Nrf2 pathway
Natural product-derived inhibitors of Nrf2 pathway induce ROS insult in ROS-sensitive cancer cells which might result in cell death. Importantly, they often sensitize cancers to the effects of chemotherapeutics or radiotherapy through the down-regulation of detoxification enzymes and drug excretion transporters [65, 66]. A significant group of Nrf2 inhibitors belongs to polyphenols (see Table 1). Polyphenols are generally recognised as antioxidants and anti-inflammatory agents. At low to micromolar concentrations polyphenols like quercetin, EGCG, resveratrol or curcumin exhibit antioxidant and chemopreventive properties. They can scavenge free radicals either directly, due to the presence of OH groups donating a hydrogen atom to a free radical, or by indirect actions through the induction of Nrf2 pathway or inhibition of ROS generation. Higher doses of polyphenols (>50 µM) and a presence of transition metal ions promote their pro-oxidant actions like suppression of antioxiant systems and inhibition of Nrf2 pathway [49]. Antitumor effects of flavones like apigenin, chrysin, luteolin and wogonin was related to the downregulation of Nrf2 expression mainly by disturbing PI3K/Akt pathway in cell lines and in in vivo mouse models. Nrf2 inhibition sensitized cancer cells to classic chemotherapeutic drugs like doxorubicin, oxaliplatin or paclitaxel both in in vitro and in vivo studies [67-70]. Interestingly, also opposite activity of apigenin, luteolin and chrysin was reported. In rat primary hepatocytes and skin epidermal JB6 P+ cells these flavones induced Nrf2/ARE response and protected against oxidative stress [71, 72]. Differences in their activity between normal and cancer cells and encourage further investigation of their potential in in vivo studies and clinical trials. So far, none of these flavones has been tested clinically for the anticancer activity in combination with chemo- or radiotherapy. Brusatol, a triterpenoid from Brucea javanica - an evergreen shrub grown in Southeast Asia and Northern Australia, was described to inhibit Nrf2 signaling by enhancing ubiquitination and subsequent degradation of Nrf2 in different cancer cell lines and mouse xenograft models [73]. Brusatol sensitized tumors to cisplatin and taxol [73-75]. The bitter coffee alkaloid, trigonelline, inhibited nuclear accumulation of Nrf2 in pancreatic cell lines (Panc1, Colo357 and MiaPaca2) and H6c7 pancreatic duct cells and enhanced their sensitivity to anticancer drugs and TRAIL-induced apoptosis [76]. A naphthoquinone derived from Plumbago species, plumbagin, inhibited nuclear translocation of Nrf2 in human tongue squamous cell carcinoma cells which suppressed the expression of Nrf2 downstream targets resulting in inhibition of epidermal to mesenchymal transition (EMT) and stemness [77]. Parthenolide, a sesquiterpene lactone found in feverfew products, was recently reported to inhibit Nrf2 protein level in breast cancer stem-like cells, derived from dissociation of mammospheres which correlated with an increased ROS production and led to necrosis [78].
Table 1: Natural products inhibiting antioxidant capacity of cancer cells.
BIOACTIVE COMPOUND |
TYPE |
SOURCE |
MECHANISM OF ACTION |
Apigenin |
Polyphenol Flavonoid Flavone |
Fruits and vegetables |
Reduces Nrf2 expression through down-regulation of PI3K/Akt pathway [67] Sensitizes tumor xenografts to doxorubicin [67] Induces glutathione depletion [94] and inhibits mitochondrial complex I activity in rats [151] |
Chaetocin |
Polyphenol thiodioxopiperazine |
Chaetomium spp. fungi |
Inhibits TrxR in vitro [117]; induces oxidative stress-mediated death of myeloma [152] and glioma cells [118] |
Chrysin |
Polyphenol Flavonoid Flavone |
Passion flowers, chamomile, honeycombs, oyster mushrooms |
Reduces Nrf2 expression in hepatocellular carcinoma through down-regulation of PI3K-Akt and ERK pathways re-sensitizing cells to doxorubicin [153] Depletes glutathione and enhances doxorubicin-induced cytotoxicity in epithelial cancer cells [69,94] |
Curcumin |
Polyphenol Curcuminoid |
Rhizomes of Curcuma longa |
Inhibits TrxR required for curcumin-induced radiosensitization [107,119] |
Epigallocatechin gallate (EGCG) |
Polyphenol Flavonoid Falvon-3-ol Catechin |
White, green and black tea (buds and leaves of Camellia sinensis) |
Inhibits TrxR and induces cancer cells death [158] Inhibits catalase, leads to elevated ROS [129] Degrades catalase via JNK in endothelial cells [159] Synergize with luteolin to induce apoptosis and p53 activation in cancer cells, reducing growth of xenografts [113] |
Luteolin |
Polyphenol Flavonoid Flavonol |
Celery, green pepper, parsley, perilla leaf, and chamomile tea |
Reduces Nrf2 expression in non-small-cell lung cancer cells, leading to GSH depletion [160] Sensitizes cells to oxaliplatin, bleomycin, doxorubicin [160,161]. Re-sensitizes oxaliplatin-resistant colorectal cancer cells [68] Inhibits Nrf2 in xenografts [162] |
Myricetin |
Polyphenol Flavonoid Flavonol |
Citrus spp. |
Blocks GST activity in melanoma cells [160] Inhibits TrxR leading to death of lung carcinomas [114] |
Quercetin |
Polyphenol Flavonoid Flavonol |
Citrus spp. |
Inhibits TrxR leading to death of lung carcinomas [114] Inhibits mitochondrial complex I activity in rats [151] |
Resveratrol |
Polyphenol Stilbenoid |
grapes, raspberries, blueberries, mulberries |
Directly binds and inhibits NQO2 and GSTP1 [163-165] Blocks mitochondrial I and III complex activity in colon cancer [166] |
Wogonin |
Polyphenol O-methylated flavone |
roots of Scutellaria baicalensis Georgi |
Down-regulates Nrf2 in resistant myelogenous leukemia cells by modulating PI3K/Akt and DNA-PKcs [167] Inhibits catalase, increasing H2O2. Synergistically sensitizes cancer cells derived from cervix, ovary and lung to TNF-induced apoptosis by blocking TNF-induced NF-κB activation [127] |
Brusatol |
Alkaloid Triterpenoid Quassinoid |
Brucea javanica |
Reduces Nrf2 via Nrf2 ubiquitination and degradation [73] Sensitizes xenografts to cisplatin via Nrf2 inhibition [73] Down-regulates Nrf2, leading to ROS accumulation. Sensitizes mammospheres to taxol and reduces the anchorage-independent growth [75] Inhibits Nrf2 in freshly isolated primary human hepatocytes [74] Enhances efficacy of cisplatin [64] |
Piperlongumine |
Alkaloid Pyridine group |
Fruits and roots of long pepper |
Binds GSH and inhibits its metabolism in leukemias [92] Increases IκBα and suppresses NFκB in human gliomas resulting in ROS-induced apoptosis [93] |
Trigonelline |
Alkaloid Pyridine and piperidine group |
coffee |
Reduces nuclear accumulation of Nrf2 in pancreatic cancer cells and sensitizes them to anticancer drugs and TRAIL via Nrf2 inhibition [76]. Enhances response to chemotherapy in vivo [76] |
Pentyl isothiocyanate (PEITC) |
Glucoside Glucosinolate |
Cruciferous vegetables |
Reacts with glutathione; lowers GSH [168]; inhibits GPX, depletes GSH, disrupts GSSG/GSH ratio [169,170] Decreases SOD in gliomas [171] Inhibits mitochondrial respiratory chain I in leukemias [172] |
Pleurotin |
Quinone |
mushrooms from Pleurotus spp., |
Inhibits TrxR in breast cancer and colon carcinoma lines, leading to HIF-1α downregulation and growth inhibition [173,174] |
Allicin |
Organosulfur compound |
garlic |
Induces GSH depletion in pancreatic cancer cells [96] Inhibits NFκB signaling activation [175] |
Plumbagin |
Naphthoquinone |
Plumbago sp. |
Inhibits Nrf2 signaling in human squamous carcinoma cells [77] Depletes intracellular GSH level and SOD2 in prostate cancer cells [97] Inhibits NFκB activation in human non-small lung cancer cells [176], pancreatic [177] and gastric cancer cells [178] |
EM23 |
Terpene Sesquiterpene lactone |
Elephantopus mollis |
Attenuates TrxR by alkylation of C-terminal redox-active site Ser498; inhibits Trx/TrxR expression facilitating ROS accumulation in human cervical cancer cells [179] Suppresses TNF-α-mediated activation of NFκB in CML cells and AML leukemia cells[180] |
Parthenolide |
Terpene Sesquiterpene lactone |
Tanacetum parthenium |
Downregulates Nrf2 expression in spheroids cultures [78] Activates NADPH oxidase, decreasing reduced thioredoxin and activating PI3K/Akt, inducing FOXO3a phosphorylation and resulting in downregulation of FOXO3a-regulated antioxidants (SOD, CAT) [78] Inhibits NFκB activity by binding and suppressing IκB kinase β [180] |
The cellular antioxidant defense
Increased levels of free radicals enable tumor cells to activate pathways driving proliferation, angiogenesis, metastasis and thrive under hypoxic conditions [79-81]. High levels of ROS create the risk of damage linked to oxidative stress, therefore cancer cells tend to overexpress detoxifying proteins that elevate their antioxidant capacity. Hyper-activation of Nrf2 pathway increases the amount of cellular ROS scavengers. Lowering stress burden by means of enhancing detoxifying force further affects certain pathways that promote growth and proliferation [82-84]. Blocking antioxidant activity in cancer cells decreases their ability to balance oxidative insult and might result in cell death [85]. Below are presented key cellular antioxidant systems and natural compounds disturbing their activity
GSH
One of the major systems involved in response to free radicals relies on a tripeptide - glutathione. The sulfhydryl (SH) group of reduced glutathione accounts for its strong electron-donating properties (Figure 1). Once oxidized, two glutathione molecules form a dimer linked by a disulfide bridge (GSSG). GSH reacts with proteins to form S-glutathionylated proteins, protecting them from further oxidation. Glutathione not only directly scavenges free radicals (hydroxyl radical, singlet oxygen), but also serves as a cofactor of several detoxifying enzymes that require thiol-reducing equivalents (glutathione peroxidase, glutathione transferase). GSH is also involved in recycling other antioxidants by reducing vitamins C and E [86]. Most of cellular GSH content remains in the cytosol, however it can also be found in organelles, including mitochondria, peroxisomes, endoplasmic reticulum and the nucleus [87]. Given the prominent role in keeping cells’ redox homeostasis in check, glutathione metabolism is accelerated in many types of cancer to alleviate oxidative stress and promote proliferation and metastasis [88]. High levels of GSH are associated with apoptosis-resistant phenotypes and its depletion is linked to the early stages of cell death initiation [89-91]. Nuclear and mitochondrial pool of glutathione plays an important role in protecting DNA from oxidative stress-driven lesions. Cell death induced by an intercalating drug doxorubicin was potentiated upon glutathione depletion [89]. This might serve as a rationale to design treatment and boost therapeutic effect of anticancer agents.
Natural products disturbing GSH metabolism
Piperlongumine (PL), an alkaloid derived from long pepper was described to induce ROS in cancer but not in normal cells [92, 93]. Further studies revealed that PL treatment led to a depletion of cellular GSH and promoted ROS. The activity of chrysin and apigenin towards GSH was tested in a number of cancer cell lines, including prostate (PC-3), myeloid (HL-60) and lung (A549) cells. Both flavones proved to be effective glutathione depleting agents. Additionally, chrysin potentiated curcumin cytotoxic effect in PC-3 and HL-60 cells [94]. Doxorubicin and cisplatin cytotoxicity was also strongly induced upon chrysin treatment, which promoted GSH efflux and depletion [69, 94]. Another flavone luteolin attenuated Nrf2 signaling leading to a decreased expression of its target genes and GSH depletion in wild type mouse small intestinal cells. Luteolin sensitizied oxiplatin-resistant colorectal cancer cell lines to cisplatin, doxorubicin and oxiplatin [68] and efficiently inhibited GST leading to GSH depletion in melanoma cells [95]. Allicin, a natural compound derived from garlic, was found to induce ROS in PaCa-2 cells. Oxidative insult was concomitant with depletion of GSH, which facilitated apoptosis [96]. Plumbagin, a ROS-inducing naphthoquinone originally derived from Plumbago plants, was reported to cause GSH depletion and induce death of human prostate cancer cells (PC-3, LNCaP and C4-2) [97]. Phenylethyl iosothiocyanate (PEITC), naturally occurring in cruciferous vegetables, has been widely studied for its biological activity and proved to exert anti-cancer properties. PEITC strongly induced oxidative damage due to the depletion of glutathione and inhibition of GPX in H-Ras transformed ovarian epithelial cells [98]. Depletion of cellular glutathione after PEITC treatment was observed in cancer cells of different origin, including glioma, oral cavity cancer, leukemia, prostate and breast [99-103]. Recent data demonstrate that PEITC caused inhibition of GST in glioma GBM 8401cells, leading to massive ROS induction and causing cell death [104]. PEITC sensitized cancer cells to cisplatin in biliary tract through PEITC-induced depletion of overall GSH, which facilitated Mcl-1 glutathionylation, promoted Mcl-1 degradation and resensitized cells to cisplatin [105]. This data indicate that combined anticancer therapy based on synergistic effect of GSH depletion and strong oxidative stress induction leads to an effective cancer cell killing.
The thioredoxin system
Thioredoxin system includes thioredoxin (Trx), thioredoxin reductase (TrxR) and nicotinamide adenine dinucleotide phosphate (NADPH) (Figure 1). Thioredoxins have a conserved dithiol Cys-Gly-Pro-Cys motif in their catalytic site and participate in the reduction of oxidized proteins. Thioredoxin reductases balance the amount of reduced Trx by transferring electrons from NADPH to oxidized catalytic sites. Humans express three thioredoxin reductase isozymes: TrxR1 (cytosolic), TrxR2 (mitochondrial), TrxR3 (testis specific). Thanks to the oxidoreductase activity of thioredoxins they act as electron carriers for catalytic cycles of enzymes and protect proteins from aggregation or inactivation resulting from their oxidation [106]. Thioredoxins were described as redox regulators of a number of transcription factors like NF-ĸB, HIF1-α, VEGF, modulates matrix metalloproteinase-9 (MMP-9), therefore promoting proliferation, angiogenesis and metastasis. Apart from balancing cell redox state, Trx1 can inhibit apoptosis by binding and blocking the activity of Apoptosis Signal-Regulating Kinase 1 (ASK1), decreasing cell response to anti-cancer drugs [107-110]. Both Trx1 and TrxR1 are highly expressed in malignant cells, maintaining cell viability and protecting from apoptosis [111]. Blocking the activity of thioredoxin system lowers the cell’s detoxifying potential and enhances oxidative insult. Many compounds have been studied for their activity to modulate thioredoxin system in tumor cells.
Natural products targeting thioredoxin system
A study testing tea catechins for their potential to inhibit TrxR1 found that a polyphenol abundant in dried leaves of white, green and black tea, epigallocatechin gallate (EGCG), abrogated TrxR1 activity by direct targeting TrxR1 thiol groups. EGCG led to a significant decrease in HeLa cells viability [112]. EGCG anti-cancer effect was also studied in combination with luteolin in head and neck and lung cancer cell lines and in xenograft models, where they synergistically promoted p53 activation and apoptosis induction, leading to the growth inhibition and reduction of tumor volume [113]. 3-hydroxyl containing flavonoids quercetin and myricetin suppressed growth of A549 cells due to the inhibition of cellular thioredoxins. The observed effect correlated with elevated oxidized thioredoxin levels and reduced TrxR activity [114]. Pleurotin, an irreversible TrxR inhibitor displayed anti-cancer properties in MCF-7 breast cancer and HT-29 colon cancer cell lines. Inhibition of TrxR by pleurotin correlated with decreased protein levels of VEGF, HIF-1α and HIF-1α target genes in studied cell lines and in MCF-7 mouse xenografts [115]. EM23, a natural sesquiterpene lactone isolated from Elephantopus mollis was found to attenuate TrxR activity in CaSki and SiHa cells by direct binding to its selenocysteine site. EM23-mediated inhibition of TrxR was followed by induction of ROS and apoptosis
[116]. Chaetocin, a competitive substrate and inhibitor of TrxR, induced apoptosis in HeLa and glioma cells due to ROS induction [117, 118]. Curcumin, a polyphenol derived from Curcuma longa inhibited TrxR activity, leading to ROS generation in HeLa cells [107]. Javvadi et al. (2010) exploited the potential of curcumin in radiosensitization of squamous carcinoma cells. Thanks to the ability of curcumin to covalently bind to the nucleophilic residues in the C-terminal region of TrxR1, curcumin strongly inhibited its function, enhanced free radicals burst and sensitized cells to radiotherapy [119]. Clinically used inhibitor of thioredoxin reductase, auranofin, displayed synergistic lethality with GSH inhibitor piperlongumine in gastric cancer (GC) suggesting that combined inhibition of different antioxidant systems is more effective in killing cancer cells than abrogation of the activity of single ones. It again emphasizes the role of ROS scavengers as potent anticancer drug targets [120].
Superoxide dismutase
Superoxide dismutase (SOD) drives the reaction of dismutation of superoxide into hydrogen peroxide (Figure 1). There are three types of SOD in cells: CuZnSOD (SOD1) abundant in the cytosol, mitochondrial manganese superoxide dismutase MnSOD (SOD2) and extracellular ECSOD (SOD3). All superoxide dismutases carry metal ions in their active sites: SOD1 and SOD3 have zinc and copper and SOD2 carries manganese. SOD1 is mainly localized in the cytosol, but it has also been found in the outer mitochondrial membrane, where it neutralizes O2.¯ released from Complex III. SOD2 is located in the mitochondria while SOD3 remains in the extracellular matrix and prevents oxidative tissue damage [121]. MnSOD overexpression is common in tumors and contributes to therapy resistance. SOD neutralizes toxic superoxide, but as a consequence creates hydrogen peroxide, which can be further neutralized by catalase and glutathione redox cycle [122].
Natural products blocking SOD activity
Since mitochondria are the primary source of cellular free radicals, decreasing their detoxifying ability by means of blocking SOD2 activity in tumors might contribute to the apoptosis activation. Plumbagin proved to efficiently induce apoptosis in effect in prostate cancer cell lines, partially through decreasing SOD2 expression [97]. PEITC was found to inhibit expression of SOD in LN229 glioma cell line, weakening cellular antioxidant defense and causing apoptosis [99]. Suppression of SOD enzymatic activity by apigenin in combination with ROS-inducing paclitaxel was found to sensitize HeLa cells to apoptosis and allowed to lower paclitaxel doses [123].
Catalase (CAT)
Catalase is a peroxisomal enzyme that neutralizes hydrogen peroxide by its decomposition to water and oxygen (Figure 1). High levels of hydrogen peroxide facilitate DNA mutagenesis, therefore under physiological conditions catalase protects cells from oxidative damage. H2O2 also serves as mediator of apoptosis and can modify regulatory protein complexes, such as Nrf2/Keap1 system. Apart from peroxisomal CAT, malignant cells acquire membrane-associated catalase to survive under oxidative stress [124-126]. Blocking catalase activity can significantly increase oxidative burden through hydrogen peroxide accumulation which triggered tumor cells death.
Natural products inhibiting catalase
Wogonin, a flavonoid isolated from Scutellaria baicalensis was shown to induce cell death in cervix, ovary and lung cancer cells through catalase inhibition that increased hydrogen peroxide levels and facilitated TNF-induced apoptotic signaling [127]. Human hepatoma HepG2 cells subjected to apigenin accumulated H2O2, which correlated with a decrease of catalase mRNA and catalase activity and led to cell death [128]. PEITC treatment lowered catalase protein levels and induced ROS in GBM 8401 glioma cells [104]. EGCG inhibited catalase activity both in vitro and in K562 cells [129] and sensitized cells to arsenite (As) treatment. The proposed mechanism explained that the inhibition of catalase activity upon treatment with As/EGCG occurred via JNK (c-Jun N-terminal kinase) signaling pathway. Genotoxic stress that activated JNK, promoted catalase phosphorylation by c-Abl kinase, marking it for proteasomal degradation. Blocking catalase activity led to high amount of H2O2 and promoted death of epithelial cells subjected to As/EGCG [130].
Exogenous antioxidants
The role of oxidative stress in initiating and promoting cancer on the one hand and in causing oxidative damage on the other justifies two opposite ROS-manipulating strategies against cancer. First is antioxidant approach functional in cancer prevention and therapy. The most important and widespread exogenous dietary antioxidants are vitamins A and E, their analogs carotenoids and tocopherols, vitamin C and polyphenols. Though preventing ROS-induced mutations and subsequent cancer initiation with dietary antioxidants is well documented, their use during anticancer therapy remains controversial. Since cancer therapy highly relies on the production of free radicals, it has been speculated that supplying cells in antioxidants might decrease treatment efficacy. On the other hand, the basic idea behind using antioxidants during therapy is to eliminate excessive oxidative damage and to help alleviate adverse effects. Many patients receiving therapy are taking antioxidants without consulting with a physician. Selenium and vitamin C are widely used in complementary oncology [131]. Radiotherapy trials in head and neck cancers showed that vitamin E reduced the toxicity, however overall recurrence and mortality were raised [132, 133]. Trials on the effect of antioxidants on chemotherapy reported on some benefits of using vitamin E or selenium with cisplatin, taxol and oxiplatin, but the long-term effects were not assessed [134-138]. Decreased recurrence of some cancer types in patients not receiving treatment or after chemotherapy has also been reported [139, 140]. The main conclusion from these trials is that administration of antioxidants to cancer patients in combination with therapy should be taken with great care. Patient phenotype (smoking, alcohol uptake and nutrition), tumor localization (different partial pressures of oxygen among tissues) and type of therapy should be considered in order to choose a suitable antioxidant supplement [141]. Importantly, adverse effects were not reported with antioxidants derived from food. The Women’s Healthy Eating and Living Study (WHELS), where diet composed of high amount of fruit and vegetable, rich in beta-carotene and vitamin C, showed no effect on outcome in patients with early breast cancer [142].
Lessons from clinical trials
Anticancer properties of a few natural products from Table 1 (EGCG, curcumin, resveratrol, PEITC, have been tested clinically mainly in the context of decreasing side effects caused by chemotherapy and radiation therapy or as chemopreventive dietary supplements (Table 2). These trials were basing on ROS scavenging properties of natural compounds. The ability of orally administered EGCG to reduce the incidence and severity of esophagitis was tested in patients with locally advanced stage III non-small-cell lung cancer receiving concurrent chemotherapy and thoracic radiotherapy (phase I, NCT01481818). No dose-limiting toxicity of EGCG was reported. Dramatic regression of esophagitis to grade 0/1 was observed in 22 of 24 patients and the pain score was also reduced [143]. Currently, the EGCG-mediated protection of the esophagus from damage induced by radiotherapy in patients with lung cancer is being tested in phase II (NCT02577393). Also topically administered EGCG was non-toxic and proved effective in decreasing radiation dermatitis in patients with breast cancer after mastectomy receiving adjuvant radiotherapy [144]. Orally administered curcumin significantly reduced the severity of skin reactions (dermatitis) caused by radiation therapy breast cancer patients as shown in phase II/III trial (NCT01246973) [145] and prevented colon cancer by reducing the aberrant crypt foci (ACF) number in smokers at dose 4 g/day [146]. Unfortunately, just a few trials so far addressed a question whether natural compounds could improve the efficacy of the standard chemotherapy or radiation therapy. One such a trial (phase II) tested curcumin ability to potentiate the effect of gemcitabine in patients with advanced pancreatic cancer (NCT00192842). In one out of twenty one patients evaluable for response curcumin caused brief but marked tumor regression (73%) and one patient remained stable for > 18 months. The problem was extremely limited bioavailability of curcumin as only 22 to 41 ng/mL was detectable in plasma when 8 g curcumin/day was given orally. Curcumin levels in the microgram range have been shown to be necessary to show antiproliferative effects in in vitro studies. Therefore, it was suggested to heat-solubilize curcumin before administration to increase its water solubility [147]. Moreover, bioactive compounds of curcumin degradation such as ferulic acid and vanillin also possess strong anticancer properties and can inhibit COX-1, COX-2 and significantly suppress NFκB activation [148-150]. In this way they may contribute to the observed biological activities of curcumin. Awaited are results of ongoing clinical trials on improved formulations of curcumin to enhance chemo- or radiotherapy (see Table 2). There is a strong need for more studies on different natural compounds as growing evidence is emerging for their benefits in improving results of standard anticancer treatments.
Table 2: Representative clinical trials on natural compounds modifying antioxidant response (from www.clinicaltrials.gov)
Compound/dose |
Clinical trial number/phase |
Purpose |
Results/Status |
EGCG 40 to 440 µmol/l |
NCT01481818 Phase I |
To evaluate safety and efficiency of EGCG in eosophagus protection in patients with locally advanced stage III non-small-cell lung cancer |
No dose-limiting toxicity of EGCG was reported. Regression of esophagitis to grade 0/1 was observed in 22 of 24 patients at the end of radiotherapy. The pain score was reduced [143] |
EGCG 40 to 660 μmol/l spray in the radiation field |
NCT01481818 Phase I |
To assess safety, tolerability and preliminary effectiveness of topical EGCG for radiation dermatitis in patients with breast cancer receiving adjuvant radiotherapy |
The topical administration of EGCG was well tolerated and the maximum tolerated dose was not found. Patient-reported symptom scores were significantly decreased at 2 weeks after the end of radiotherapy in pain, burning, itching and tenderness [144] |
EGCG 10 ml solution/day (440 µmol/l) |
NCT02577393 Phase II |
To evaluate the protection of the esophagus from damage induced by radiotherapy in patients with lung cancer |
enrolling participants |
Polyphenon E (PolyE, a defined green tea polyphenol extract with high EGCG content) 4 x 200 mg/day |
NCT00676793 Phase II |
To evaluate the short-term effects of PolyE administered during the interval between breast biopsy and surgery in women with recently diagnosed breast cancer: determination if EGCG inhibits c-Met signaling and activation of pathways contributing to breast cancer progression |
completed, no results published |
Polyphenon E, 4 x 200 mg/day |
NCT00676780 Phase II |
To evaluate the short-term effects of PolyE administered during the interval between prostate biopsy and radical prostatectomy in men with recently diagnosed prostate cancer |
A significant reduction in serum levels of prostate-specific antigen (PSA), hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF) was observed [181] |
Polyphenon E, 2 x 200 mg/day |
NCT00596011 Phase II |
To determine if PolyE reduces the rate of progression to prostate cancer (PCa) in men diagnosed with high-grade prostatic intraepithelial neoplasia (HGPIN) or atypical small acinar proliferation (ASAP) |
No differences in the number of prostate cancer (PCa) cases were observed but there was a decrease in a cumulative rate of progression to PCa or ASAP in a PolyE group vs. placebo group [182] |
curcumin, 6 g/day during radiotherapy |
NCT01246973 Phase II/III |
To determine whether curcumin can prevent or reduce the severity of dermatitis caused by radiation therapy in breast cancer patients |
Curcumin reduced the severity of radiation dermatitis in breast cancer patients [145]. |
curcumin 2 or 4 g/day for 30 days |
NCT00365209 Phase IIa |
To evaluate how well curcumin works in preventing colon cancer in smokers with aberrant crypt foci (ACF) |
A significant 40% reduction in ACF number was observed with the 4 g dose, whereas in the 2 g group ACF were not reduced. Curcumin was well tolerated at both doses [146] |
nanostructured lipid curcumin particle 2 x 100 mg/day |
NCT02439385 Phase II |
To evaluate progression-free survival in colorectal cancer patients with unresectable metastasis after treatment with Avastin/FOLFIRI in combination with a nanostructured lipid curcumin particle which improved biotransformation and bioavailability of curcumin. |
This study is not yet open for participant recruitment. |
Meriva (lecithinized curcumin delivery system) 2 x 500 mg/day |
NCT01740323 Phase II |
To determine if curcumin reduces NF-ĸB DNA binding in patients receiving radiotherapy for their breast cancer after having completed chemotherapy |
This study is currently recruiting participants |
curcumin 8 g/day along the chemotherapeutic protocol of weekly gemcitabine |
NCT00192842 Phase II |
To assess if curcumin can improve the efficacy of the standard chemotherapy gemcitabine in patients with advanced pancreatic cancer. |
5 out of 17 patients (29%) discontinued curcumin due to intractable abdominal fullness or pain, and the dose of curcumin was reduced to 4 mg/day because of abdominal complaints in 2 other patients. One of 11 evaluable patients (9%) had partial response, 4 (36%) had stable disease, and 6 (55%) had tumor progression. [183] |
curcumin, dosage not provided |
NCT02095717 Phase II |
To assess taxotere plus curcumin combination in first-line treatment of prostate cancer metastatic castration resistant. |
study is ongoing |
nanocurcumin SinaCurcumin® 3 x 40 mg/day 3 days before and during radiotherapy |
NCT02724618 Phase II |
To determine the role of curcumin as a radioprotector against radiation-induced injury in normal tissues as well as a radiosensitizer in tumor in prostate cancer patients undergoing radiotherapy |
recruiting participants |
curcumin capsules dosage not provided |
NCT00852332 phase II |
To study how well giving docetaxel together with a curcumin works compared with giving docetaxel alone as first- or second-line therapy in treating patients with breast cancer. |
recruiting participants |
Isoquercetin 2 x 225 or 2 x: 450 mg/day along the chemotherapy with Sunitinib |
NCT02446795 Phase I/II |
A trial of isoquercetin as an adjunct therapy in patients with kidney cancer receiving first-line Sunitinib |
This study is not yet open for participant recruitment. |
Quercetin 2 x 250 mg / day for 3 weeks |
NCT01732393 |
To evaluate the effect of quercetin on prevention and treatment of chemotherapy-induced oral mucositis in patients with blood malignancies. |
This study has been completed, |
SRT501 (micronized resveratrol) 5 g/day for 14 days |
NCT00920803 Phase I |
To determine safety and tolerability of SRT501 in subjects with colorectal cancer and hepatic metastases |
SRT501 was well tolerated. Mean plasma resveratrol levels following a single dose of SRT501 administration were exceeding those for equivalent doses of non-micronized resveratrol by 3.6-fold. Resveratrol was detectable in hepatic tissue. Cleaved caspase-3 was significantly increased [184]. |
PEITC (dosage not provided) |
NCT00691132 Phase II |
PEITC in preventing lung cancer in people who smoke |
The recruitment status unknown |
Conclusions
The power of natural products lies in using them as adjuvants to standard anticancer therapies but the struggle is that they often exhibit contrary actions, depending on concentration. At high doses ( > 50 µM) natural compounds presented in this article have pro-oxidant properties by limiting antioxidant capacity of cancer cells (Figure 3). Direct inhibition of cellular antioxidants or suppression of pathways leading to their expression can sensitize cancer cells to chemo- and radiotherapy. Normal cells are not that sensitive to the manipulations in redox homeostasis as their growth and proliferation are not that much ROS-dependent. Contrarily, cancer cells operate under constant oxidative stress and are very sensitive to the disruption of their enhanced ability to scavenge free radicals. Therefore, impairing antioxidant capacity of tumors emerges as a good strategy to target them. Especially inhibition of Nrf2 pathway seems a very promising approach as Nrf2 controls expression of crucial cellular antioxidants, drug efflux pumps and detoxification enzymes. Simultaneous inhibition of Nrf2 and prosurvival NFĸB signaling is even more effective in promoting death of tumor cells. Therefore, natural products that suppress Nrf2 and NFĸB pathways are promising candidates for adjuvants to chemo- and radiotherapy allowing for lowering their doses. It is nevertheless essential to bear in mind that the effect they induce in cells depends on the applied dose, cell type, exposure time and environmental conditions. The same natural product in different concentrations often possesses contrary properties. This is why it is so challenging to translate results from in vitro models to in vivo conditions. Concentrations used in cell lines experiments are often very hard to achieve in patients. Given the poor plasmatic bioavailability of active compounds and biotransformation processes they undergo in the body, the circulating concentration of natural compounds administered orally are rather low. Moreover, the biological effects they produce do not necessarily need to be a consequence of the action of only the parent compound, but might also be assigned to its metabolites. Therefore, the effects natural products present in vivo might be different or even opposite to expected and instead of potentiating the effect of chemo- or radiotherapy, they might weaken their action. The majority of clinical trials test ROS scavenging properties of natural compounds in the context of cancer chemoprevention or their ability to alleviate side effects of chemo- and radiotherapy. Just a few addressed a question of synergistic effects of natural products with classic anticancer therapies and the results so far warrant further investigation. There is a strong need for clinical studies testing these combination treatments in defined cancer types with special focus on bioavailability and stability of natural products.
Abbreviations
ABCC2, ABC transporters MRP2; Akt, Protein Kinase B;AP-1, activator protein 1; ARE, antioxidant response element; As, arsenite; ASK1, apoptosis signal-regulating kinase 1; BCRP, breast cancer resistance protein; B-Raf(V619E), mutation in B-Raf that replaces amino acid valine with glutamic acid at the position 619; c-Abl, Abelson tyrosine kinase; CAT, catalase; COX1, cyclooxygenase 1; COX2, cyclooxygenase 2; Cul, cullin; EGCG, epigallocatechin gallate; EMT, epidermal to mesenchymal transition; EGFR, Epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; ETC, electron transport chain; GC, gastric cancer; GSH, glutathione; GSSG, oxidized glutathione; GST, glutathione S-transferase; GST1α/β, Glutathione S-transferase 1α/β; H2O2, hydrogen peroxide; HIF-1α, Hypoxia Inducible Factor 1α; HNSCC, head and neck squamous cell carcinoma; H-Ras(G12V), mutation in H-Ras that replaces amino acid glycine with valine at the position 12; JNK, c-Jun N-terminal kinase; Keap1, Kelch-like ECH-associated protein 1; K-Ras(G12D), mutation in K-Ras that replaces amino acid glycine with aspartic acid at position 12; K-Ras, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; Mcl-1, Induced myeloid leukemia cell differentiation protein; MEFs, murine embryonic fibroblasts; MMP-9, matrix metalloproteinase-9; mtp53, mutant p53; Myc, myelocytomatosis oncogene; Myr-Akt, myristoylated form of Akt kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NCI, National Cancer Institute; NFĸB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOX, NADPH oxidases; NOX1, NADPH oxidase 1; NOX5-L, NADPH oxidase 5; NQO1, NAD(P)H-quinone oxidoreductase; Nrf2, nuclear factor (erythroid-derived 2)-like 2; O2.-, superoxide anion; OH., hydroxyl radical; OONO-, peroxynitrite; p38, MAPK14, Mitogen Activated Protein Kinase 14; p47phox, the phagocyte NADPH oxidase/NOX2 organizer; PDPK1, 3-Phosphoinositide Dependent Protein Kinase 1; PEITC, phenylethyl iosothiocyanate; PI3K, phosphoinositide 3-kinase; PKCδ, Protein Kinase C δ; PL, piperlongumine; ROS, reactive oxygen species; SH, sulfhydryl group; SOD, superoxide dismutase; SOD1, superoxide dismutase 1; SOD2, superoxide dismutase 2; SOD3, superoxide dismutase 3; STAT3, signal transducer and activator of transcription 3; TRAIL, TNF-related apoptosis-inducing ligand; Trx, thioredoxin; Trx1, thioredoxin 1; TrxR, thioredoxin reductase; TrxR1, thioredoxin reductase 1; UGT, UDP-glucuronosyl transferases; VEGF, Vascular Endothelial Growth Factor; V-Ras, Neuroblastoma RAS viral (V-Ras) oncogene homolog; WHELS, Women’s Healthy Eating and Living Study; γGCS, γ-glutamylcysteine synthetase;
ACKNOWLEDGMENTS
We would like to express gratitude to dr M.Rybicka for critical reading of the manuscript. This work was supported by National Science Centre PRELUDIUM 9 grant no 2015/17/N/NZ3/03773.
CONFLICTs OF INTEREST
Authors declare no conflict of interest regarding this article.
REFERENCES
1. D’Arpa P, Liu LF. Topoisomerase-targeting antitumor drugs. Biochim Biophys Acta. 1989; 989: 163–77.
2. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014; 25: 2677–81.
3. Zhou S, Palmeira CM, Wallace KB. Doxorubicin-induced persistent oxidative stress to cardiac myocytes. Toxicol Lett. 2001; 121: 151–7.
4. Jang HJ, Hwang S, Cho KY, Kim DK, Chay K-O, Kim J-K. Taxol induces oxidative neuronal cell death by enhancing the activity of NADPH oxidase in mouse cortical cultures. Neurosci Lett. 2008; 443: 17–22.
5. Yadav N, Kumar S, Marlowe T, Chaudhary AK, Kumar R, Wang J, O’Malley J, Boland PM, Jayanthi S, Kumar TKS, Yadava N, Chandra D. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015;
6. Liou G-Y, Storz P. Reactive oxygen species in cancer. Free Radic Res. NIH Public Access; 2010; 44: 479–96.
7. Rotblat B, Grunewald TGP, Leprivier G, Melino G, Knight RA. Anti-oxidative stress response genes: bioinformatic analysis of their expression and relevance in multiple cancers. Oncotarget. 2013; 4: 2577–90. doi: 10.18632/oncotarget.1658.
8. Chen PM, Lin CH, Li NT, Wu YM, Lin MT, Hung SC, Yen ML. c-Maf regulates pluripotency genes, proliferation/self-renewal, and lineage commitment in ROS-mediated senescence of human mesenchymal stem cells. Oncotarget. 2015; 6: 35404–18. doi: 10.18632/oncotarget.6178.
9. Zhou S, Palmeira CM, Wallace KB. Doxorubicin-induced persistent oxidative stress to cardiac myocytes. Toxicol Lett. 2001; 121: 151–7.
10. Adão R, de Keulenaer G, Leite-Moreira A, Brás-Silva C. Cardiotoxicity associated with cancer therapy: Pathophysiology and prevention. Rev Port Cardiol. 2013; 32: 395–409.
11. Zhao H, Ning S, Scicinski J, Oronsky B, Knox SJ, Peehl DM. Epigenetic effects of RRx-001: a possible unifying mechanism of anticancer activity. Oncotarget. 2015; 6: 43172–81. doi: 10.18632/oncotarget.6526.
12. Crawford S. Anti-inflammatory/antioxidant use in long-term maintenance cancer therapy: a new therapeutic approach to disease progression and recurrence. Ther Adv Med Oncol. 2014; 6: 52–68.
13. Palikaras K, Lionaki E, Tavernarakis N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015; 22: 1399–401.
14. Roy K, Wu Y, Meitzler JL, Juhasz A, Liu H, Jiang G, Lu J, Antony S, Doroshow JH. NADPH oxidases and cancer. Clin Sci. 2015; 128: 863–75.
15. Maniam S, Coutts AS, Stratford MR, McGouran J, Kessler B, La Thangue NB. Cofactor Strap regulates oxidative phosphorylation and mitochondrial p53 activity through ATP synthase. Cell Death Differ. 2015; 22: 156–63.
16. Li H-Y, Zhang J, Sun L-L, Li B-H, Gao H-L, Xie T, Zhang N, Ye Z-M. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis. 2015; 6: e1604.
17. Oberley LW, Oberley TD, Buettner GR. Cell division in normal and transformed cells: the possible role of superoxide and hydrogen peroxide. Med Hypotheses. 1981; 7: 21–42.
18. Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, Gorgoulis VG, d’Adda di Fagagna F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014; 21: 998–1012.
19. Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014; 2: 17.
20. Park M-T, Kim M-J, Suh Y, Kim R-K, Kim H, Lim E-J, Yoo K-C, Lee G-H, Kim Y-H, Hwang S-G, Yi J-M, Lee S-J. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014; 21: 1185–97.
21. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GRS, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010; 107: 8788–93.
22. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, Joshi PA, Wakeham A, Molyneux SD, Martin B, Bouwman P, Cescon DW, Elia AJ, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013; 210: 1529–44.
23. Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P, Goldstein I, Madar S, Rotter V. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J Cell Sci. 2012; 125: 5578–86.
24. Wardman P. Electron transfer and oxidative stress as key factors in the design of drugs selectively active in hypoxia. Curr Med Chem. 2001; 8: 739–61.
25. Kovacic P, Osuna JA. Mechanisms of anti-cancer agents: emphasis on oxidative stress and electron transfer. Curr Pharm Des. 2000; 6: 277–309.
26. Hasanain M, Bhattacharjee A, Pandey P, Ashraf R, Singh N, Sharma S, Vishwakarma AL, Datta D, Mitra K, Sarkar J. α-Solanine induces ROS-mediated autophagy through activation of endoplasmic reticulum stress and inhibition of Akt/mTOR pathway. Cell Death Dis. 2015; 6: e1860.
27. Lee S-J, Jung YH, Oh SY, Song EJ, Choi SH, Han HJ. Vibrio vulnificus VvhA induces NF-κB-dependent mitochondrial cell death via lipid raft-mediated ROS production in intestinal epithelial cells. Cell Death Dis. 2015; 6: 1655.
28. Kluckova K, Sticha M, Cerny J, Mracek T, Dong L, Drahota Z, Gottlieb E, Neuzil J, Rohlena J. Ubiquinone-binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 2015; 6: e1749.
29. Gomez L, Thiebaut P-A, Paillard M, Ducreux S, Abrial M, Crola Da Silva C, Durand A, Alam MR, Van Coppenolle F, Sheu S-S, Ovize M. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2015; 22: 1890.
30. Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002; 3: 221–7.
31. Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004; 14: 64–9.
32. Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. 2015; 79: 253–63.
33. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J-I. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008; 320: 661–4.
34. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, Witkiewicz AK, Flomenberg N, Howell A, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010; 9: 3515–33.
35. Dho SH, Kim JY, Kwon ES, Lim JC, Park SS, Kwon KS. NOX5-L can stimulate proliferation and apoptosis depending on its levels and cellular context, determining cancer cell susceptibility to cisplatin. Oncotarget. 2015;6:39235-46. doi: 10.18632/oncotarget.5743.
36. Gonzalez Y, Aryal B, Chehab L, Rao VA, Gonzalez Y, Aryal B, Chehab L, Rao VA. Atg7- and Keap1-dependent autophagy protects breast cancer cell lines against mitoquinone-induced oxidative stress. Oncotarget. 2014; 5: 1526–37. doi: 10.18632/oncotarget.1715.
37. Fang Y, Wang J, Xu L, Cao Y, Xu F, Yan L, Nie M, Yuan N, Zhang S, Zhao R, Wang H, Wu M, Zhang X, et al. Autophagy maintains ubiquitination-proteasomal degradation of Sirt3 to limit oxidative stress in K562 leukemia cells. Oncotarget. 2016; 7: 35692–702. doi: 10.18632/oncotarget.9592.
38. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015; 22: 377–88.
39. Itsumi M, Inoue S, Elia AJ, Murakami K, Sasaki M, Lind EF, Brenner D, Harris IS, Chio IIC, Afzal S, Cairns RA, Cescon DW, Elford AR, et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ . 2015; 22: 1837–45.
40. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475: 106–9.
41. Rotblat B, Melino G, Knight RA. NRF2 and p53: Januses in cancer? Oncotarget. 2012; 3: 1272–83. doi: 10.18632/oncotarget.754.
42. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, Elia A, Berger T, Cescon DW, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015; 27: 211–22.
43. Jacquemin G, Margiotta D, Kasahara A, Bassoy EY, Walch M, Thiery J, Lieberman J, Martinvalet D. Granzyme B-induced mitochondrial ROS are required for apoptosis. Cell Death Differ. 2015; 22: 862–74.
44. Marini ES, Giampietri C, Petrungaro S, Conti S, Filippini A, Scorrano L, Ziparo E. The endogenous caspase-8 inhibitor c-FLIPL regulates ER morphology and crosstalk with mitochondria. Cell Death Differ. 2015; 22: 1131–43.
45. Tian Y, Kuo C-F, Sir D, Wang L, Govindarajan S, Petrovic LM, Ou J-HJ. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015; 22: 1025–34.
46. Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang M, Zhang B, Yang B, Li B, Yang H, Wu Y. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget. 2015; 6: 39839–54. doi: 10.18632/oncotarget.5674.
47. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015; 22: 248–57.
48. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013; 12: 931–47.
49. Stepanic V, Gasparovic AC, Troselj KG, Amic D, Zarkovic N. Selected attributes of polyphenols in targeting oxidative stress in cancer. Curr Top Med Chem. 2015; 15: 496–509.
50. Calderon-Aparicio A, Strasberg-Rieber M, Rieber M. Disulfiram anti-cancer efficacy without copper overload is enhanced by extracellular H2O2 generation: antagonism by tetrathiomolybdate. Oncotarget. 2015; 6: 29771–81. doi: 10.18632/oncotarget.4833.
51. Lamb R, Fiorillo M, Chadwick A, Ozsvari B, Reeves KJ, Smith DL, Clarke RB, Howell SJ, Cappello AR, Martinez-Outschoorn UE, Peiris-Pagès M, Sotgia F, Lisanti MP. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget. 2015; 6: 14005–25. doi: 10.18632/oncotarget.4159.
52. Managò A, Leanza L, Carraretto L, Sassi N, Grancara S, Quintana-Cabrera R, Trimarco V, Toninello A, Scorrano L, Trentin L, Semenzato G, Gulbins E, Zoratti M, et al. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 2015; 6: e1930.
53. Zhang C, Liu K, Yao K, Reddy K, Zhang Y, Fu Y, Yang G, Zykova TA, Shin SH, Li H, Ryu J, Jiang Y-N, Yin X, et al. HOI-02 induces apoptosis and G2-M arrest in esophageal cancer mediated by ROS. Cell Death Dis. 2015; 6: e1912.
54. Guzzo G, Sciacovelli M, Bernardi P, Rasola A. Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget. 2014; 5: 11897–908. doi: 10.18632/oncotarget.2472.
55. Fitzgerald AL, Osman AA, Xie T-X, Patel A, Skinner H, Sandulache V, Myers JN. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 2015; 6: e1678.
56. Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. 2010; 3: 23–34.
57. Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, Leprivier G, Somasekharan SP, Barokas A, Deng Y, Tang T, Mathers J, Cetinbas N, et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A. 2014; 111: 3032–7.
58. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999; 13: 76–86.
59. Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004; 24: 8477–86.
60. Kumar H, Kim I-S, More SV, Kim B-W, Choi D-K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat Prod Rep. 2014; 31: 109–39.
61. Geismann C, Arlt A, Sebens S, Schäfer H. Cytoprotection “gone astray”: Nrf2 and its role in cancer. Onco Targets Ther. 2014; 7: 1497–518.
62. Syu JP, Chi JT, Kung HN. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget. 2016; 7: 14659–72. doi: 10.18632/oncotarget.7406.
63. Rocha CRR, Kajitani GS, Quinet A, Fortunato RS, Menck CFM, Ribeiro Reily Rocha C, Satoru Kajitani G, Quinet A, Soares Fortunato R, Frederico Martins Menck C. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget; 2016; 7: 48081–92. doi: 10.18632/oncotarget.10129.
64. Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, Zhang DD. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014; 74: 7430–41.
65. Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, Blackford A, Goodman SN, Bunz F, Watson WH, Gabrielson E, Feinstein E, Biswal S. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008; 68: 7975–84.
66. Wang X-J, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008; 29: 1235–43.
67. Gao A-M, Ke Z-P, Wang J-N, Yang J-Y, Chen S-Y, Chen H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis. 2013; 34: 1806–14.
68. Chian S, Li Y-Y, Wang X-J, Tang X-W. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac J Cancer Prev. 2014; 15: 2911–6.
69. Brechbuhl HM, Kachadourian R, Min E, Chan D, Day BJ. Chrysin enhances doxorubicin-induced cytotoxicity in human lung epithelial cancer cell lines: the role of glutathione. Toxicol Appl Pharmacol. 2012; 258: 1–9.
70. Gong T, Wang C-F, Yuan J-R, Li Y, Gu J-F, Zhao B-J, Zhang L, Jia X-B, Feng L, Liu S-L. Inhibition of Tumor Growth and Immunomodulatory Effects of Flavonoids and Scutebarbatines of Scutellaria barbata D. Don in Lewis-Bearing C57BL/6 Mice. Evid Based Complement Alternat Med. 2015; 2015: 630760.
71. Huang C-S, Lii C-K, Lin A-H, Yeh Y-W, Yao H-T, Li C-C, Wang T-S, Chen H-W. Protection by chrysin, apigenin, and luteolin against oxidative stress is mediated by the Nrf2-dependent up-regulation of heme oxygenase 1 and glutamate cysteine ligase in rat primary hepatocytes. Arch Toxicol. 2013; 87: 167–78.
72. Paredes-Gonzalez X, Fuentes F, Su Z-Y, Kong A-NT. Apigenin reactivates Nrf2 anti-oxidative stress signaling in mouse skin epidermal JB6 P + cells through epigenetics modifications. AAPS J. 2014; 16: 727–35.
73. Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, Zhang DD. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011; 108: 1433–8.
74. Olayanju A, Copple IM, Bryan HK, Edge GT, Sison RL, Wong MW, Lai Z-Q, Lin Z-X, Dunn K, Sanderson CM, Alghanem AF, Cross MJ, Ellis EC, et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med. 2015; 78: 202–12.
75. Wu T, Harder BG, Wong PK, Lang JE, Zhang DD. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol Carcinog. 2015; 54: 1494–502.
76. Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse M-L, Schreiber S, Schäfer H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013; 32: 4825–35.
77. Pan S-T, Qin Y, Zhou Z-W, He Z-X, Zhang X, Yang T, Yang Y-X, Wang D, Zhou S-F, Qiu J-X. Plumbagin suppresses epithelial to mesenchymal transition and stemness via inhibiting Nrf2-mediated signaling pathway in human tongue squamous cell carcinoma cells. Drug Des Devel Ther. 2015; 9: 5511–51.
78. Carlisi D, Buttitta G, Di Fiore R, Scerri C, Drago-Ferrante R, Vento R, Tesoriere G. Parthenolide and DMAPT exert cytotoxic effects on breast cancer stem-like cells by inducing oxidative stress, mitochondrial dysfunction and necrosis. Cell Death Dis. 2016; 7: e2194.
79. Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014; 39: 191–8.
80. Amelio I, Melino G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem Sci. 2015; 40: 425–34.
81. Amelio I, Inoue S, Markert EK, Levine AJ, Knight RA, Mak TW, Melino G. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1α degradation. Proc Natl Acad Sci U S A. 2015; 112: 226–31.
82. Cho J, Seo J, Lim CH, Yang L, Shiratsuchi T, Lee M-H, Chowdhury RR, Kasahara H, Kim J-S, Oh SP, Lee YJ, Terada N. Mitochondrial ATP transporter Ant2 depletion impairs erythropoiesis and B lymphopoiesis. Cell Death Differ. 2015; 22: 1437–50.
83. Yadav N, Kumar S, Marlowe T, Chaudhary AK, Kumar R, Wang J, O’Malley J, Boland PM, Jayanthi S, Kumar TKS, Yadava N, Chandra D. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015; 6: e1969.
84. Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, McAllister S, Soroceanu L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015; 6: e1601.
85. Mei H, Sun S, Bai Y, Chen Y, Chai R, Li H. Reduced mtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell Death Dis. 2015; 6: e1710.
86. Jones DP. Redefining Oxidative Stress. Antioxid Redox Signal. 2006; 8: 1865–79.
87. Lash LH. Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem Biol Interact. 2006; 163: 54–67.
88. Franco R, Cidlowski J a. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ . Nature Publishing Group; 2009; 16: 1303–14.
89. Friesen C, Kiess Y, Debatin K-M. A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ. 2004; 11 Suppl 1: S73-85.
90. Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 30: 1–12.
91. Martinez BA, Kim H, Ray A, Caldwell GA, Caldwell KA. A bacterial metabolite induces glutathione-tractable proteostatic damage, proteasomal disturbances, and PINK1-dependent autophagy in C. elegans. Cell Death Dis. 2015; 6: e1908.
92. Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011; 475: 231–4.
93. Chen Y, Liu JM, Xiong XX, Qiu XY, Pan F, Liu D, Lan SJ, Jin S, Yu S Bin, Chen XQ. Piperlongumine selectively kills hepatocellular carcinoma cells and preferentially inhibits their invasion via ROS-ER-MAPKs-CHOP. Oncotarget. 2015; 6: 6406–21. doi: 10.18632/oncotarget.3444.
94. Kachadourian R, Day BJ. Flavonoid-induced glutathione depletion: potential implications for cancer treatment. Free Radic Biol Med. 2006; 41: 65–76.
95. Balyan R, Kudugunti SK, Hamad HA, Yousef MS, Moridani MY. Bioactivation of luteolin by tyrosinase selectively inhibits glutathione S-transferase. Chem Biol Interact. 2015; 240: 208–18.
96. Chhabria S V, Akbarsha MA, Li AP, Kharkar PS, Desai KB. In situ allicin generation using targeted alliinase delivery for inhibition of MIA PaCa-2 cells via epigenetic changes, oxidative stress and cyclin-dependent kinase inhibitor (CDKI) expression. Apoptosis. 2015; 20: 1388–409.
97. Powolny AA, Singh S V. Plumbagin-induced apoptosis in human prostate cancer cells is associated with modulation of cellular redox status and generation of reactive oxygen species. Pharm Res. 2008; 25: 2171–80.
98. Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by ??-phenylethyl isothiocyanate. Cancer Cell. 2006; 10: 241–52.
99. Su JC, Lin K, Wang Y, Sui SH, Gao ZY, Wang ZG. In vitro studies of phenethyl isothiocyanate against the growth of LN229 human glioma cells. Int J Clin Exp Pathol. 2015; 8: 4269–76.
100. Yeh Y-T, Yeh H, Su S-H, Lin J-S, Lee K-J, Shyu H-W, Chen Z-F, Huang S-Y, Su S-J. Phenethyl isothiocyanate induces DNA damage-associated G2/M arrest and subsequent apoptosis in oral cancer cells with varying p53 mutations. Free Radic Biol Med. 2014; 74: 1–13.
101. Chen G, Chen Z, Hu Y, Huang P. Inhibition of mitochondrial respiration and rapid depletion of mitochondrial glutathione by β-phenethyl isothiocyanate: mechanisms for anti-leukemia activity. Antioxid Redox Signal. 2011; 15: 2911–21.
102. Chaiswing L, Zhong W, Oberley TD. Distinct Redox Profiles of Selected Human Prostate Carcinoma Cell Lines: Implications for Rational Design of Redox Therapy. Cancers (Basel). 2011; 3: 3557–84.
103. Syed Alwi SS, Cavell BE, Donlevy A, Packham G. Differential induction of apoptosis in human breast cancer cell lines by phenethyl isothiocyanate, a glutathione depleting agent. Cell Stress Chaperones. 2012; 17: 529–38.
104. Chou Y-C, Chang M-Y, Wang M-J, Harnod T, Hung C-H, Lee H-T, Shen C-C, Chung J-G. PEITC induces apoptosis of Human Brain Glioblastoma GBM8401 Cells through the extrinsic- and intrinsic -signaling pathways. Neurochem Int. 2015; 81: 32–40.
105. Li Q, Zhan M, Chen W, Zhao B, Yang K, Yang J, Yi J, Huang Q, Mohan M, Hou Z, Wang J. Phenylethyl isothiocyanate reverses cisplatin resistance in biliary tract cancer cells via glutathionylation-dependent degradation of Mcl-1. Oncotarget. 2016; 7:10271-82. doi: 10.18632/oncotarget.7171.
106. Arnér ESJ, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000; 267: 6102–9.
107. Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem. 2005; 280: 25284–90.
108. Farina AR, Cappabianca L, DeSantis G, Di Ianni N, Ruggeri P, Ragone M, Merolle S, Tonissen KF, Gulino A, Mackay AR. Thioredoxin stimulates MMP-9 expression, de-regulates the MMP-9/TIMP-1 equilibrium and promotes MMP-9 dependent invasion in human MDA-MB-231 breast cancer cells. FEBS Lett. 2011; 585: 3328–36.
109. Streicher KL, Sylte MJ, Johnson SE, Sordillo LM. Thioredoxin reductase regulates angiogenesis by increasing endothelial cell-derived vascular endothelial growth factor. Nutr Cancer. 2004; 50: 221–31.
110. Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002; 62: 5089–95.
111. Saccoccia F, Angelucci F, Boumis G, Carotti D, Desiato G, Miele AE, Bellelli A. Thioredoxin reductase and its inhibitors. Curr Protein Pept Sci. 2014; 15: 621–46.
112. Wang Y, Zhang H, Holmgren A, Tian W, Zhong L. Inhibitory effect of green tea extract and (-)-epigallocatechin-3-gallate on mammalian thioredoxin reductase and HeLa cell viability. Oncol Rep. 2008; 20: 1479–87.
113. Ruhul Amin ARM, Wang D, Zhang H, Peng S, Shin HJC, Brandes JC, Tighiouart M, Khuri FR, Chen ZG, Shin DM. Enhanced anti-tumor activity by the combination of the natural compounds (-)-epigallocatechin-3-gallate and luteolin: Potential role of p53. J Biol Chem. 2010; 285: 34557–65.
114. Lu J, Papp L V, Fang J, Rodriguez-Nieto S, Zhivotovsky B, Holmgren A. Inhibition of Mammalian thioredoxin reductase by some flavonoids: implications for myricetin and quercetin anticancer activity. Cancer Res. 2006; 66: 4410–8.
115. Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther. 2003; 2: 235–43.
116. Li H, Li M, Wang G, Shao F, Chen W, Xia C, Wang S, Li Y, Zhou G, Liu Z. EM23, A Natural Sesquiterpene Lactone from Elephantopus mollis, Induces Apoptosis in Human Myeloid Leukemia Cells through Thioredoxin- and Reactive Oxygen Species-Mediated Signaling Pathways. Front Pharmacol. 2016; 7: 77.
117. Tibodeau JD, Benson LM, Isham CR, Owen WG, Bible KC. The anticancer agent chaetocin is a competitive substrate and inhibitor of thioredoxin reductase. Antioxid Redox Signal. 2009; 11: 1097–106.
118. Dixit D, Ghildiyal R, Anto NP, Sen E. Chaetocin-induced ROS-mediated apoptosis involves ATM-YAP1 axis and JNK-dependent inhibition of glucose metabolism. Cell Death Dis. 2014; 5: e1212.
119. Javvadi P, Hertan L, Kosoff R, Datta T, Kolev J, Mick R, Tuttle SW, Koumenis C. Thioredoxin reductase-1 mediates curcumin-induced radiosensitization of squamous carcinoma cells. Cancer Res. 2010; 70: 1941–50.
120. Zou P, Chen M, Ji J, Chen W, Chen X, Ying S, Zhang J, Zhang Z, Liu Z, Yang S, Liang G. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget. 2015; 6: 36505–21. doi: 10.18632/oncotarget.5364.
121. Indo HP, Yen H-C, Nakanishi I, Matsumoto K-I, Tamura M, Nagano Y, Matsui H, Gusev O, Cornette R, Okuda T, Minamiyama Y, Ichikawa H, Suenaga S, et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr. 2015; 56: 1–7.
122. Buettner GR. Superoxide dismutase in redox biology: the roles of superoxide and hydrogen peroxide. Anticancer Agents Med Chem. 2011; 11: 341–6.
123. Xu Y, Xin Y, Diao Y, Lu C, Fu J, Luo L, Yin Z. Synergistic effects of apigenin and paclitaxel on apoptosis of cancer cells. PLoS One. 2011; 6: e29169.
124. Bechtel W, Bauer G. Catalase protects tumor cells from apoptosis induction by intercellular ROS signaling. Anticancer Res. 2009; 29: 4541–57.
125. Bai J, Rodriguez AM, Melendez JA, Cederbaum AI. Overexpression of Catalase in Cytosolic or Mitochondrial Compartment Protects HepG2 Cells against Oxidative Injury. J Biol Chem. 1999; 274: 26217–24.
126. Bauer G, Zarkovic N. Revealing mechanisms of selective, concentration-dependent potentials of 4-hydroxy-2-nonenal to induce apoptosis in cancer cells through inactivation of membrane-associated catalase. Free Radic Biol Med. 2015; 81: 128–44.
127. Yang L, Zheng XL, Sun H, Zhong YJ, Wang Q, He HN, Shi XW, Zhou B, Li JK, Lin Y, Zhang L, Wang X. Catalase suppression-mediated H(2)O(2) accumulation in cancer cells by wogonin effectively blocks tumor necrosis factor-induced NF-κB activation and sensitizes apoptosis. Cancer Sci. 2011; 102: 870–6.
128. Valdameri G, Trombetta-Lima M, Worfel PR, Pires ARA, Martinez GR, Noleto GR, Cadena SMSC, Sogayar MC, Winnischofer SMB, Rocha MEM. Involvement of catalase in the apoptotic mechanism induced by apigenin in HepG2 human hepatoma cells. Chem Biol Interact. 2011; 193: 180–9.
129. Pal S, Dey SK, Saha C. Inhibition of catalase by tea catechins in free and cellular state: a biophysical approach. PLoS One. 2014; 9: e102460.
130. Kim J-Y, Choi J-Y, Lee H-J, Byun CJ, Park J-H, Park JH, Cho H-S, Cho S-J, Jo SA, Jo I. The Green Tea Component (-)-Epigallocatechin-3-Gallate Sensitizes Primary Endothelial Cells to Arsenite-Induced Apoptosis by Decreasing c-Jun N-Terminal Kinase-Mediated Catalase Activity. PLoS One. 2015; 10: e0138590.
131. Gröber U. Antioxidants and Other Micronutrients in Complementary Oncology. Breast Care (Basel). 2009; 4: 13–20.
132. Bairati I, Meyer F, Jobin E, Gélinas M, Fortin A, Nabid A, Brochet F, Têtu B. Antioxidant vitamins supplementation and mortality: a randomized trial in head and neck cancer patients. Int J cancer. 2006; 119: 2221–4.
133. Ferreira PR, Fleck JF, Diehl A, Barletta D, Braga-Filho A, Barletta A, Ilha L. Protective effect of alpha-tocopherol in head and neck cancer radiation-induced mucositis: a double-blind randomized trial. Head Neck. 2004; 26: 313–21.
134. Argyriou AA, Chroni E, Koutras A, Iconomou G, Papapetropoulos S, Polychronopoulos P, Kalofonos HP. A randomized controlled trial evaluating the efficacy and safety of vitamin E supplementation for protection against cisplatin-induced peripheral neuropathy: final results. Support Care Cancer. 2006; 14: 1134–40.
135. Sieja K, Talerczyk M. Selenium as an element in the treatment of ovarian cancer in women receiving chemotherapy. Gynecol Oncol. 2004; 93: 320–7.
136. Weijl NI, Elsendoorn TJ, Lentjes EGWM, Hopman GD, Wipkink-Bakker A, Zwinderman AH, Cleton FJ, Osanto S. Supplementation with antioxidant micronutrients and chemotherapy-induced toxicity in cancer patients treated with cisplatin-based chemotherapy: a randomised, double-blind, placebo-controlled study. Eur J Cancer. 2004; 40: 1713–23.
137. Afonseca SO de, Cruz FM, Cubero D de IG, Lera AT, Schindler F, Okawara M, Souza LF de, Rodrigues NP, Giglio A del. Vitamin E for prevention of oxaliplatin-induced peripheral neuropathy: a pilot randomized clinical trial. Rev Paul Med. 2013; 131: 35–8.
138. Kottschade LA, Sloan JA, Mazurczak MA, Johnson DB, Murphy BP, Rowland KM, Smith DA, Berg AR, Stella PJ, Loprinzi CL. The use of vitamin E for the prevention of chemotherapy-induced peripheral neuropathy: results of a randomized phase III clinical trial. Support Care Cancer. 2011; 19: 1769–77.
139. Hoenjet KMJLF, Dagnelie PC, Delaere KPJ, Wijckmans NEG, Zambon J V, Oosterhof GON. Effect of a nutritional supplement containing vitamin E, selenium, vitamin c and coenzyme Q10 on serum PSA in patients with hormonally untreated carcinoma of the prostate: a randomised placebo-controlled study. Eur Urol. 2005; 47: 433-9-40.
140. Karp DD, Lee SJ, Keller SM, Wright GS, Aisner S, Belinsky SA, Johnson DH, Johnston MR, Goodman G, Clamon G, Okawara G, Marks R, Frechette E, et al. Randomized, double-blind, placebo-controlled, phase III chemoprevention trial of selenium supplementation in patients with resected stage I non-small-cell lung cancer: ECOG 5597. J Clin Oncol. 2013; 31: 4179–87.
141. Harvie M. Nutritional supplements and cancer: potential benefits and proven harms. Am Soc Clin Oncol Educ Book. 2014; e478-86.
142. Pierce JP, Natarajan L, Caan BJ, Parker BA, Greenberg ER, Flatt SW, Rock CL, Kealey S, Al-Delaimy WK, Bardwell WA, Carlson RW, Emond JA, Faerber S, et al. Influence of a diet very high in vegetables, fruit, and fiber and low in fat on prognosis following treatment for breast cancer: the Women’s Healthy Eating and Living (WHEL) randomized trial. JAMA. 2007; 298: 289–98.
143. Zhao H, Zhu W, Xie P, Li H, Zhang X, Sun X, Yu J, Xing L. A phase I study of concurrent chemotherapy and thoracic radiotherapy with oral epigallocatechin-3-gallate protection in patients with locally advanced stage III non-small-cell lung cancer. Radiother Oncol . 2014; 110: 132–6.
144. Zhao H, Zhu W, Jia L, Sun X, Chen G, Zhao X, Li X, Meng X, Kong L, Xing L, Yu J. Phase I study of topical epigallocatechin-3-gallate (EGCG) in patients with breast cancer receiving adjuvant radiotherapy. Br J Radiol. 2016; 89: 20150665.
145. Ryan JL, Heckler CE, Ling M, Katz A, Williams JP, Pentland AP, Morrow GR. Curcumin for radiation dermatitis: a randomized, double-blind, placebo-controlled clinical trial of thirty breast cancer patients. Radiat Res. 2013; 180: 34–43.
146. Carroll RE, Benya R V, Turgeon DK, Vareed S, Neuman M, Rodriguez L, Kakarala M, Carpenter PM, McLaren C, Meyskens FL, Brenner DE. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res (Phila). 2011; 4: 354–64.
147. Kurien BT, Scofield RH. Heat-solubilized curcumin should be considered in clinical trials for increasing bioavailability. Clin Cancer Res. 2009; 15: 747; author reply 747.
148. Jayaprakasam B, Vanisree M, Zhang Y, Dewitt DL, Nair MG. Impact of alkyl esters of caffeic and ferulic acids on tumor cell proliferation, cyclooxygenase enzyme, and lipid peroxidation. J Agric Food Chem. 2006; 54: 5375–81.
149. Murakami Y, Hirata A, Ito S, Shoji M, Tanaka S, Yasui T, Machino M, Fujisawa S. Re-evaluation of cyclooxygenase-2-inhibiting activity of vanillin and guaiacol in macrophages stimulated with lipopolysaccharide. Anticancer Res. 2007; 27: 801–7.
150. Jung KJ, Go EK, Kim JY, Yu BP, Chung HY. Suppression of age-related renal changes in NF-kappaB and its target gene expression by dietary ferulate. J Nutr Biochem. 2009; 20: 378–88.
151. Lagoa R, Graziani I, Lopez-Sanchez C, Garcia-Martinez V, Gutierrez-Merino C. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochim Biophys Acta. 2011; 1807: 1562–72.
152. Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC. Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood. 2007; 109: 2579–88.
153. Gao A-M, Ke Z-P, Shi F, Sun G-C, Chen H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem Biol Interact. 2013; 206: 100–8.
154. Singh S, Aggarwal BB. Activation of Transcription Factor NF- B Is Suppressed by Curcumin (Diferuloylmethane). J Biol Chem. 1995; 270: 24995–5000.
155. Marín YE, Wall BA, Wang S, Namkoong J, Martino JJ, Suh J, Lee HJ, Rabson AB, Yang CS, Chen S, Ryu J-H. Curcumin downregulates the constitutive activity of NF-kappaB and induces apoptosis in novel mouse melanoma cells. Melanoma Res. 2007; 17: 274–83.
156. Marquardt JU, Gomez-Quiroz L, Arreguin Camacho LO, Pinna F, Lee Y-H, Kitade M, Domínguez MP, Castven D, Breuhahn K, Conner EA, Galle PR, Andersen JB, Factor VM, et al. Curcumin effectively inhibits oncogenic NF-κB signaling and restrains stemness features in liver cancer. J Hepatol. 2015; 63: 661–9.
157. Pfeffer U, Amaro A, Bachmeier B, Angelini G. Curcumin: Towards molecularly targeted chemoprevention of cancer. New Horizons Transl Med. 2015; 2: 60.
158. Wang Y, Zhang H, Holmgren A, Tian W, Zhong L. Inhibitory effect of green tea extract and (-)-epigallocatechin-3-gallate on mammalian thioredoxin reductase and HeLa cell viability. Oncol Rep. 2008; 20: 1479–87.
159. Kim J-Y, Choi J-Y, Lee H-J, Byun CJ, Park J-H, Park JH, Cho H-S, Cho S-J, Jo SA, Jo I. The Green Tea Component (-)-Epigallocatechin-3-Gallate Sensitizes Primary Endothelial Cells to Arsenite-Induced Apoptosis by Decreasing c-Jun N-Terminal Kinase-Mediated Catalase Activity. PLoS One. 2015; 10: e0138590.
160. Tang X, Wang H, Fan L, Wu X, Xin A, Ren H, Wang XJ. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med. 2011; 50: 1599–609.
161. Sabzichi M, Hamishehkar H, Ramezani F, Sharifi S, Tabasinezhad M, Pirouzpanah M, Ghanbari P, Samadi N. Luteolin-loaded phytosomes sensitize human breast carcinoma MDA-MB 231 cells to doxorubicin by suppressing Nrf2 mediated signalling. Asian Pac J Cancer Prev . 2014; 15: 5311–6. Available from http://www.ncbi.nlm.nih.gov/pubmed/25040994
162. Chian S, Thapa R, Chi Z, Wang XJ, Tang X. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochem Biophys Res Commun. 2014; 447: 602–8.
163. Buryanovskyy L, Fu Y, Boyd M, Ma Y, Hsieh T, Wu JM, Zhang Z. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry. 2004; 43: 11417–26.
164. Wang Z, Hsieh T, Zhang Z, Ma Y, Wu JM. Identification and purification of resveratrol targeting proteins using immobilized resveratrol affinity chromatography. Biochem Biophys Res Commun. 2004; 323: 743–9.
165. Hsieh T-C, Wang Z, Deng H, Wu JM. Identification of glutathione sulfotransferase-pi (GSTP1) as a new resveratrol targeting protein (RTP) and studies of resveratrol-responsive protein changes by resveratrol affinity chromatography. Anticancer Res. 2008; 28: 29–36.
166. Sassi N, Mattarei A, Azzolini M, Szabo’ I, Paradisi C, Zoratti M, Biasutto L. Cytotoxicity of mitochondria-targeted resveratrol derivatives: interactions with respiratory chain complexes and ATP synthase. Biochim Biophys Acta. 2014; 1837: 1781–9.
167. Xu X, Zhang Y, Li W, Miao H, Zhang H, Zhou Y, Li Z, You Q, Zhao L, Guo Q. Wogonin reverses multi-drug resistance of human myelogenous leukemia K562/A02 cells via downregulation of MRP1 expression by inhibiting Nrf2/ARE signaling pathway. Biochem Pharmacol. 2014; 92: 220–34.
168. Brown KK, Eriksson SE, Arnér ESJ, Hampton MB. Mitochondrial peroxiredoxin 3 is rapidly oxidized in cells treated with isothiocyanates. Free Radic Biol Med . 2008; 45: 494–502.
169. Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ, Arlinghaus RB, Huang P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia. 2008; 22: 1191–9.
170. Trachootham D, Zhang H, Zhang W, Feng L, Du M, Zhou Y, Chen Z, Pelicano H, Plunkett W, Wierda WG, Keating MJ, Huang P. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood. 2008; 112: 1912–22.
171. Su J-C, Lin K, Wang Y, Sui S-H, Gao Z-Y, Wang Z-G. In vitro studies of phenethyl isothiocyanate against the growth of LN229 human glioma cells. Int J Clin Exp Pathol. 2015; 8: 4269–76.
172. Chen G, Chen Z, Hu Y, Huang P. Inhibition of mitochondrial respiration and rapid depletion of mitochondrial glutathione by β-phenethyl isothiocyanate: mechanisms for anti-leukemia activity. Antioxid Redox Signal. 2011; 15: 2911–21.
173. Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther. 2003; 2: 235–43.
174. Kunkel MW, Kirkpatrick DL, Johnson JI, Powis G. Cell line-directed screening assay for inhibitors of thioredoxin reductase signaling as potential anti-cancer drugs. Anticancer Drug Des . 1997; 12: 659–70.
175. Dorai T, Aggarwal BB. Role of chemopreventive agents in cancer therapy. Cancer Lett. 2004; 215: 129–40.
176. Xu T-P, Shen H, Liu L-X, Shu Y-Q. Plumbagin from Plumbago Zeylanica L induces apoptosis in human non-small cell lung cancer cell lines through NF- κB inactivation. Asian Pac J Cancer Prev. 2013; 14: 2325–31.
177. Li J, Shen L, Lu F, Qin Y, Chen R, Li J, Li Y, Zhan H, He Y. Plumbagin inhibits cell growth and potentiates apoptosis in human gastric cancer cells in vitro through the NF-κB signaling pathway. Acta Pharmacol Sin. 2012; 33: 242–9.
178. Hafeez B Bin, Jamal MS, Fischer JW, Mustafa A, Verma AK. Plumbagin, a plant derived natural agent inhibits the growth of pancreatic cancer cells in in vitro and in vivo via targeting EGFR, Stat3 and NF-κB signaling pathways. Int J cancer. 2012; 131: 2175–86.
179. Shao F-Y, Wang S, Li H-Y, Chen W-B, Wang G-C, Ma D-L, Wong NS, Xiao H, Liu Q-Y, Zhou G-X, Li Y-L, Li M-M, Wang Y-F, et al. EM23, a natural sesquiterpene lactone, targets thioredoxin reductase to activate JNK and cell death pathways in human cervical cancer cells. Oncotarget. 2016; 7: 6790–808. doi: 10.18632/oncotarget.6828.
180. Li H, Li M, Wang G, Shao F, Chen W, Xia C, Wang S, Li Y, Zhou G, Liu Z. EM23, A Natural Sesquiterpene Lactone from Elephantopus mollis, Induces Apoptosis in Human Myeloid Leukemia Cells through Thioredoxin- and Reactive Oxygen Species-Mediated Signaling Pathways. Front Pharmacol. 2016; 7: 77.
181. McLarty J, Bigelow RLH, Smith M, Elmajian D, Ankem M, Cardelli JA. Tea Polyphenols Decrease Serum Levels of Prostate-Specific Antigen, Hepatocyte Growth Factor, and Vascular Endothelial Growth Factor in Prostate Cancer Patients and Inhibit Production of Hepatocyte Growth Factor and Vascular Endothelial Growth Factor In v. Cancer Prev Res. 2009; 2: 9.
182. Kumar NB, Pow-Sang J, Egan KM, Spiess PE, Dickinson S, Salup R, Helal M, McLarty J, Williams CR, Schreiber F, Parnes HL, Sebti S, Kazi A, et al. Randomized, Placebo-Controlled Trial of Green Tea Catechins for Prostate Cancer Prevention. Cancer Prev Res (Phila) . 2015 [cited 2016 Aug 15]; 8: 879–87. doi: 10.1158/1940-6207.CAPR-14-0324
183. Epelbaum R, Schaffer M, Vizel B, Badmaev V, Bar-Sela G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr Cancer. 2010; 62: 1137–41.
184. Howells LM, Berry DP, Elliott PJ, Jacobson EW, Hoffmann E, Hegarty B, Brown K, Steward WP, Gescher AJ. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases--safety, pharmacokinetics, and pharmacodynamics. Cancer Prev Res (Phila). 2011; 4: 1419–25.