
INTRODUCTION
Immune dysfunctions are a key feature of chronic lymphocytic leukemia (CLL) and frequently result in clinically manifest complications contributing to patients’ morbidity and mortality, such as opportunistic infections and autoimmune conditions. The impairment of the host immune system also correlates with disease progression and may reflect the attempt of the leukemic cells to evade immune control [1].
Perturbations of both cellular and humoral immunity are observed in CLL patients. The T-cell compartment is highly abnormal, with changes in the phenotype and absolute number of circulating CD4+ and CD8+ T cells and with the accumulation of terminally differentiated effector memory T cells [2]. T lymphocytes express markers of exhaustion, such as upregulated PD-1, leading to a pronounced Th2 skewing of the T-cell responses [3], have reduced cytotoxic functions and an impaired formation of immune synapses with antigen (Ag) presenting cells [4]. Regulatory T cells are increased in the peripheral blood (PB) of patients with advanced disease [5], and Vγ9Vδ2 T cells display features of exhaustion and are negative prognosticator of a shorter time to first treatment (TTFT) [6].
Quantitative and qualitative changes of the humoral responses are also very common in patients with CLL. Hypogammaglobulinemia affects up to 85% of patients and its severity correlates with stage and duration of the disease and with the susceptibility to recurrent infections [1, 7]. On the other hand, haematopoietic-specific auto-antibodies (Ab) are frequently observed and autoimmune haemolitic anemia (AIHA) and/or immune thrombocytopenia are rather frequent complications. Autoimmunity is usually due to high affinity polyclonal immunoglobulin (Ig) G auto-Ab that are produced by the non-malignant B lymphocytes and recognize red cells- or platelets-derived auto-Ag [4, 8].
So far, little is known on the occurrence of humoral immune responses toward tumor-derived Ag in CLL. In a previous report, Krackhardt et al. detected circulating Ab recognizing 14 Ag derived from primary tumor samples in sera from patients with CLL [9]. However, the biologic functions and the clinical impact of spontaneously occurring humoral responses directed toward tumor-derived Ag have not been investigated.
Serological Proteome Analysis (SERPA) is a valuable method to detect Ag-specific Ab responses in human malignancies. SERPA combines electrophoretic separation of proteins from tumor cells, western blotting and mass spectrometric (MS) identification of Ag recognized by sera. SERPA allowed the identification of tumor-derived Ag able to trigger humoral immune responses in several solid [10–16] and hematologic tumors [17–20]. Dubovsky et al. have recently applied a modified SERPA to CLL to define new membrane-associated targets, and identified lymphocyte cytosolic protein 1 as an important factor in chemokine-induced migration of leukemic cells [21].
The aim of our study was to investigate the occurrence of humoral responses toward Ag derived from primary tumor cells in sera from previously untreated patients with CLL, also evaluating their cytotoxic function and the correlation with parameters of disease evolution.
RESULTS
Immunoreactivity of CLL sera toward CLL-derived Ag and protein identification
Sera obtained from 35 patients with previously untreated CLL were individually screened for the presence of IgG-based immunoreactivity against proteins derived from autologous CLL cells (Figure 1A and 1B). To verify the CLL-specificity of the detection, 8 CLL proteomic maps (patients 05, 06, 10, 11, 12, 13, 16, 29) were individually probed with a different healthy donor’s (HD) serum (data not shown). Overall, the 35 CLL sera recognized 144 Ag (mean:4.1, min:0, max:20), whereas the 8 HD sera recognized only 3 Ag (mean:0.4, min:0, max:1) (p=0.007).
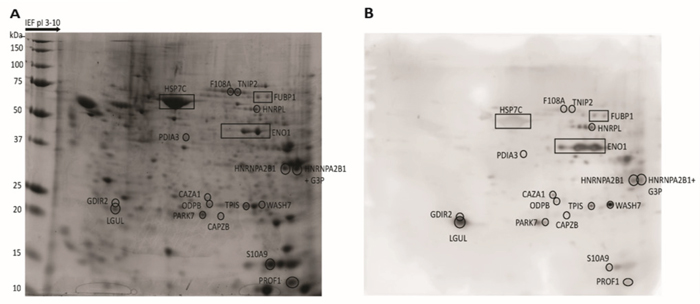
Figure 1: Two-dimensional map of proteins from CLL cells and corresponding immunoblotting map. Total proteins extracted from CLL cells were separated by 2-DE, stained with Coomassie Blue (A), or transferred on nitrocellulose membranes for WB and probed with CLL patient’s autologous serum (B). The proteins that could be assigned to Ag spots in the WB were excised from the gel and treated with trypsin. The resulting fragments were analyzed by MS. The location of proteins recognized by a representative CLL serum (patient 29) are indicated by boxes in the proteomic map and in the corresponding WB membrane.
The MS analyses of the 144 Ag spots produced by CLL sera led to the identification of 47 proteins that were recurrently recognized by patients’ sera. Table 1 summarizes the chemical features, spectrometric data, and frequency of recognition of 18 Ag, which were recognized by at least 3 patients’ sera. Additional proteins recognized by less than 3 patients’ sera are reported in Supplementary Table S1. Only 3 proteins (fructose-bisphosphate aldolase A [ALDOA], 60 kDa heat shock protein [HSP60] and vimentin [VIME]) were also recognized by 1 out of 8 (13%) HD sera. Sera from 29 out of 35 CLL patients (83%) exhibited IgG-based immunoreactivity toward at least 1 Ag (Table 2). Among these, 20 (69%) were polyreactive and recognized a median number of 5 Ag, with a peak of 20 Ag recognized by the serum of patient 22.
Table 1: Proteins recognized by at least 3 CLL sera
MW (kDa)/pI |
Swiss-Prot Accession number |
Protein name |
Sequence coverage (%) |
Metched peptides |
Mascot score |
Recurrence of antibodies (%) |
||
|---|---|---|---|---|---|---|---|---|
CLL patients |
Healthy Donors |
p |
||||||
47,481/7.10 |
Q6GMP2 |
α-Enolase (ENO1) |
53 |
20 |
250 |
19 (54) |
0 |
0.006 |
33,073/5.45 |
P52907 |
F-actin-capping protein subunit alpha-1 (CAZA1) |
32 |
6 |
73 |
9 (26) |
0 |
ns |
36,201/8.57 |
P04406 |
Glyceraldehyde-3-phosphate dehydrogenase (G3P) |
33 |
10 |
125 |
9 (26) |
0 |
ns |
39,550/6.20 |
P11177 |
Pyruvate dehydrogenase E1 component subunit beta, mitochondrial (ODPB) |
43 |
11 |
93 |
8 (23) |
0 |
ns |
23,031/5.10 |
P52566 |
Rho GDP-dissociation inhibitor 2 (GDIR2) |
56 |
9 |
93 |
7 (20) |
0 |
ns |
37,462/8.97 |
P22626 |
Heterogeneous nuclear ribonucleoproteins A2/B1 (HNRNPA2B1) |
39 |
13 |
151 |
6 (17) |
0 |
ns |
53,676/5.06 |
P08670 |
Vimentin (VIME) |
53 |
20 |
250 |
6 (17) |
1 (13) |
ns |
42,052/5.29 42,108/5.31 |
Q96HG5 P63261 |
Actin, cytoplasmic 1 (ACTB) - Actin, cytoplasmic 2 (ACTG) |
39 |
13 |
145 |
5 (14) |
0 |
ns |
31,616/5.36 |
P47756 |
F-actin-capping protein subunit beta (CAPZB) |
36 |
8 |
98 |
5 (14) |
0 |
ns |
45,753/6.86 |
P14902 |
Indoleamine 2,3-dioxygenase 1 (I23O1) |
23 |
8 |
66 |
5 (14) |
0 |
ns |
29,843/5.57 |
P35232 |
Prohibitin (PHB) |
51 |
10 |
148 |
5 (14) |
0 |
ns |
61,187/5,70 |
P10809 |
60 kDa heat shock protein (HSP60) |
37 |
14 |
154 |
4 (11) |
1 (13) |
ns |
39,851/8.30 |
P04075 |
Fructose-bisphosphate aldolase A (ALDOA) |
44 |
13 |
106 |
4 (11) |
1 (13) |
ns |
23,569/5.43 |
P09211 |
Glutathione S-transferase P (GSTP1) |
55 |
7 |
96 |
4 (11) |
0 |
ns |
71,082/8.52 |
P11142 |
Heat shock cognate 71 kDa protein (HSP7C) |
37 |
16 |
197 |
3 (9) |
0 |
ns |
49,240/6.03 |
Q8NFZ5 |
TNFAIP3-interacting protein 2 (TNIP2) |
17 |
8 |
69 |
3 (9) |
0 |
ns |
51,230/5.39 |
P61978 |
Heterogeneous nuclear ribonucleoprotein K (HNRNPK) |
34 |
16 |
109 |
3 (9) |
0 |
ns |
64,720/8.46 |
P14866 |
Heterogeneous nuclear ribonucleoprotein L (HNRPL) |
16 |
8 |
78 |
3 (9) |
0 |
ns |
MW, Molecular weight; pI, isoelectrical point. Sequence coverage (%) is the percentage of the protein primary sequence covered by peptides obtained by tripsyn digestion of the Ag spot. Matched peptides is the rate between the number of peptides corresponding to the primary sequence of the protein and the total number of peptides analyzed. Mascot score is a measure of the correspondence of peptides to the protein primary sequence.
Table 2: Immunoreactivity of patients with CLL
Patient |
No of recognized proteins |
Anti-ENO1 Ab |
Binet |
Disease state |
IGHV |
V-D-J |
Stereotyped receptor subset |
Cytogenetics by FISH |
Hypogamma-globulinemia |
|---|---|---|---|---|---|---|---|---|---|
01 |
2 |
+ |
A |
stable |
UM |
1-18/6-19/4 |
#1 |
del(17p) |
yes |
02 |
1 |
- |
A |
stable |
UM |
1-18/3-3/6 |
|
del(13q) |
|
03 |
0 |
- |
|
|
UM |
3-23/4-23/4 |
|
|
|
04 |
1 |
- |
A |
stable |
UM |
1-2/1-26/6 |
#28 |
del(11q) |
yes |
05 |
5 |
+ |
B |
progression |
M |
4-34/3-3/4 |
#4 |
normal |
yes |
06 |
6 |
- |
|
stable |
M |
5-51/2-2/5 |
|
del(13q) |
|
07 |
1 |
- |
A |
stable |
n.d. |
|
|
normal |
yes |
08 |
8 |
- |
A |
stable |
M |
3-33/3-22/4 |
|
del(17p) |
yes |
09 |
1 |
+ |
A |
stable |
M |
4-34/5-5/6 |
#4 |
tris(12) |
no |
10 |
6 |
+ |
B |
progression |
M |
3-64/6-6/4 |
|
del(13q) |
yes |
11 |
5 |
+ |
C |
progression |
UM |
3-23/5-5/4 |
|
del(13q) |
|
12 |
4 |
- |
A |
stable |
M |
4-39/6-6/4 |
|
tris(12) |
no |
13 |
2 |
+ |
A |
stable |
M |
5-51/2-2/6 |
|
del(11q) |
yes |
14 |
0 |
- |
|
|
M |
4-34/n.d./6 |
|
normal |
no |
15 |
3 |
+ |
B |
stable |
M |
2-5/6-13/5 |
|
tris(12) |
yes |
16 |
3 |
+ |
A |
progression |
UM |
4-59/3-22/6 |
|
normal |
yes |
17 |
0 |
- |
A |
stable |
UM |
3-33/3-3/6 |
|
normal |
no |
18 |
1 |
- |
A |
progression |
UM |
|
|
normal |
no |
19 |
0 |
- |
A |
stable |
UM |
5-a/6-19/4 |
#1 |
normal |
no |
20 |
0 |
- |
B |
stable |
n.d. |
|
|
del(17p) |
yes |
21 |
1 |
+ |
A |
progression |
M |
2-70/5-24/4 |
|
tris(12) |
yes |
22 |
20 |
+ |
B |
progression |
UM |
3-30/2-2/5 |
|
del(17p) |
no |
23 |
5 |
+ |
B |
progression |
UM |
3-11/3-22/4 |
|
del(11q) |
yes |
24 |
2 |
+ |
B |
stable |
M |
3-66/3-16/4 |
|
del(11q) |
yes |
25 |
12 |
+ |
B |
progression |
M |
3-23/3-22/3 |
|
normal |
no |
26 |
10 |
+ |
A |
stable |
M |
3-7/2-2/4 |
|
normal |
yes |
27 |
1 |
+ |
A |
stable |
UM |
4-59/3-22/6 |
|
normal |
|
28 |
0 |
- |
B |
stable |
UM |
3-33/4-17/4 |
|
del(13q) |
no |
29 |
19 |
+ |
B |
progression |
M |
3-74/3-10/4 |
|
normal |
no |
30 |
1 |
- |
A |
stable |
M |
3-53/6-13/4 |
|
del(13q) |
no |
31 |
1 |
+ |
|
|
UM |
4-39/5-12/5 |
|
normal |
|
32 |
5 |
+ |
A |
stable |
UM |
1-69/3-3/6 |
#7 |
normal |
no |
33 |
9 |
+ |
A |
stable |
M |
7-4/7-27/4 |
|
del(13q) |
yes |
34 |
6 |
- |
A |
stable |
UM |
3-53/n.d./4 |
|
del(13q) |
yes |
35 |
3 |
- |
A |
stable |
UM |
3-9/6-19/6 |
|
tris(12) |
no |
Sera from progressive CLL are more immunoreactive than sera from stable CLL
The immunoreactive status of patients was not associated with gender, age, Rai or Binet stage, lymphocytosis, monocyte and platelet counts, hemoglobin, β2-microglobulin or lactate dehydrogenase values, percentage of CD38+ or ZAP70+ cells, immunoglobulin heavy chain variable regions (IGHV)-mutational status, fluorescence in situ hybridization data, hypogammaglobulinemia, autoimmunity, concurrent infections or allergies. Our cohort included 10 patients in active disease progression and 22 patients with a stable disease. For the remaining 3 patients, the disease status at the time of SERPA was not available. Interestingly, we found that patients with progressive CLL showed a significantly higher number of Ag recognitions compared to patients with stable disease (p=0.01) (Figure 2). Overall, median TTFT of patients was 29 months and median overall survival (OS) was not reached at the median follow-up of 81 months. The immunoreactivity status was not a statistically significant TTFT or OS predictor.
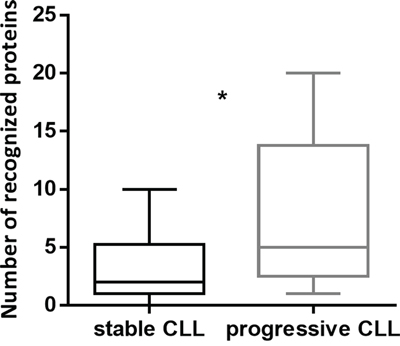
Figure 2: The number of recognized Ag was significantly higher in patients with progressive CLL than in patients with stable disease. The number of recognized Ag was correlated with parameters of disease progression collected at the time of SERPA. Disease progression was evaluated according to IWCLL/NCI-WG 2008 guidelines for CLL. Patients with progressive CLL (n=10) recognized a significantly higher number of spots compared to patients with stable CLL (n=22) (p=0.01). Box and whiskers plots represent median values, first and third quartiles, and minimum and maximum values for each dataset.
Circulating Ab are produced by the polyclonal B-cell population and not by the leukemic clone
To determine whether the circulating Ab detected by SERPA in patients’ sera were partly produced by a class-switched sub-clone derived from the leukemic clone, we first analyzed the IGHV repertoire of patients who shared the same Ag recognitions. Patients with similar immunoreactivities did not exhibit the same IGHV rearrangements. In addition, the analysis of the complementarity determining region 3 (CDR3) revealed that 6 out of 35 CLL patients displayed stereotyped B-cell receptor (BCR) and belonged to 4 subsets, with no association between stereotypy and Ag recognition. As a confirmatory evidence, we cloned and produced a soluble derivative of the leukemic BCR (ScFv-Fc) from patient 10 and blotted it in parallel with the whole patient’s serum on two identical autologous proteomic maps. The patient’s serum recognized 5 Ag, but none of these was also recognized by the autologous tumor-derived ScFv-Fc (Supplementary Figure S1). Taken together, these results indicate that the Ab detected by SERPA are entirely produced by the normal B-cell population, and do not include a soluble fraction of the leukemic BCR.
Alpha-enolase (ENO1) is the most frequently recognized Ag and is overexpressed by proliferating CLL cells of the LN
ENO1 was the most relevant Ag recognized by patients’ sera, since ENO1-specific circulating Ab were detectable in 19 out of 35 (54%) CLL sera and in none of the HD sera (p=0.006) (Table 1). To confirm the MS identification of the protein, we incubated the proteomic maps obtained from the tumor cells of 10 anti-ENO1 Ab+ (patients 22, 23, 24, 25, 26, 27, 29, 31, 32, 33) and 4 anti-ENO1 Ab- patients (patients 28, 30, 34, 35) with a commercially available anti-ENO1 polyclonal Ab. The anti-ENO1 Ab generated the same Ag spots produced by the sera of patients with CLL (Supplementary Figure S2).
ENO1 expression pattern was investigated by immunohistochemistry and multicolor immunofluorescence confocal microscopy in lymph node (LN) sections obtained from 3 CLL and 3 reactive (R) LN. The CLL LN displayed an almost complete effacement of the normal architecture by CLL cells and evidence of morphologically distinct pseudo-follicles, comprising areas rich in prolymphocytes and paraimmunoblasts. The anti-ENO1 Ab stained all CLL and R LN, and higher magnification indicated an increased expression in correspondence of proliferating cells of both CLL and R LN sections, at least on the basis of cell morphology (Figure 3A and 3B). Multicolor tissue immunofluorescence was then used to determine which cells were mostly ENO1+. The anti-ENO1 Ab was combined with anti-CD2 and anti-CD23 Ab to detect T and B cells, respectively. In the CLL LN ENO1 reactivity was mostly associated to the CD23+ population, while in the R LN it was mostly associated to the CD2+ T cells (Figure 3C–3E). Ki67 was then used to identify proliferating cells in both tissues. Within the CLL LN a significant proportion of proliferating B cells was apparent and these cells were intensely ENO1+. Conversely, in the reactive LN the most of the proliferating cells were CD2+, and they also showed ENO1 reactivity, suggesting that ENO1 is highly expressed by proliferating cells, independently of the lineage (Figure 3D–3K).
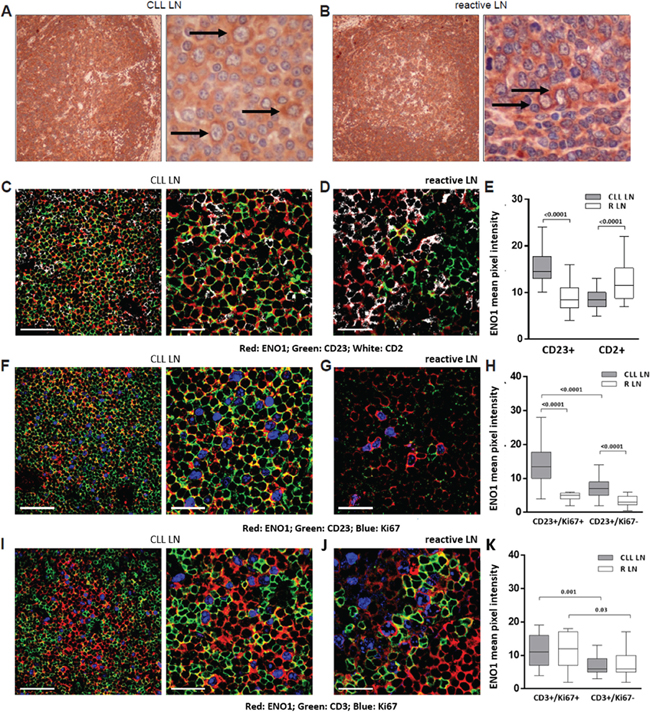
Figure 3: In lymph node ENO1 is significantly more expressed by proliferating CLL cells. ENO1 expression was evaluated by immunohistochemistry and immunofluorescence confocal microscopy on CLL and R LN sections. Anti-ENO1 immunostaining of a representative CLL LN and R LN section was performed using an anti-mouse HRP-conjugated secondary Ab and 3,3′-diaminobenzidine (brown signal). Black arrows indicate paraimmunoblasts (A) and centroblasts (B). Original magnification x4 (left panels) and x40 (right panels). Images were obtained using a Olympus BX41 microscope, equipped with Nikon Coolpix camera. Triple staining of representative CLL and R LN sections with anti-ENO1 (red), anti-CD23 (green) and anti-CD2 (white). Original magnification x63; zoom factor of 2.5 for images on the right. Scale bars represent 50 μm and 25 μm, respectively (C, D). Cumulative data of ENO1 mean pixel intensity (arbitrary units) confirmed a significantly higher intensity of ENO1 expression in close proximity of B cells in CLL LN and T cells in R LN (always p<0.0001) (E). Triple staining of representative CLL and R LN sections with anti-ENO1 (red), anti-CD23 (green) and anti-Ki67 (blue). Original magnification x63; zoom factor of 2.5 for images on the right. Scale bars represent 50 μm and 25 μm, respectively (F, G). Quantitative measurements of ENO1 mean pixel intensity (arbitrary units) confirmed that CD23+/Ki67+ proliferating cells of the CLL LN expressed significantly higher level of ENO1 compared to the CD23+/Ki67- resting fraction (p<0.0001) and to CD23+/Ki67+ proliferating B cells of the R LN (p<0.0001) (H). Triple staining of representative CLL LN and R LN sections with anti-ENO1 (red), anti-CD3 (green) and anti-Ki67 (blue). Original magnification x63; zoom factor of 2.5 for images on the right. Scale bars represent 50 μm and 25 μm, respectively (I, J). Cumulative analysis of ENO1 mean pixel intensity (arbitrary units) confirmed that the CD3+/Ki67+ proliferating fraction of the T-cell compartment expressed significantly higher levels of ENO1 than CD3+/Ki67- resting fraction, in both CLL and R LN (p<0.001 and 0.03 respectively) (K). For cumulative data, at least 3 randomly chosen fields from 3 different samples were analyzed. Samples were analyzed using a TCS SP5 laser scanning confocal microscope (Leica Microsystems) with an oil immersion 63x/1.5 objective lens, images were acquired with LAS AF Version Lite 2.4 software and processed with Photoshop (Adobe Systems). ENO1 mean pixel intensity was calculated with ImageJ software (http://rsbweb.nih.gov/ij/). Box and whiskers plots represent median values, first and third quartiles, and minimum and maximum values for each dataset. Statistical analysis was performed using Mann-Whitney U test.
ENO1 is transferred on the surface of CLL cells undergoing apoptosis
ENO1 expression was also evaluated in the PB compartment. First, we compared the intracellular expression of ENO1 in PB mononuclear cells (PBMC) subpopulations from CLL patients. Cytofluorimetric analysis showed that CD19+/CD5+ CLL cells, CD14+ monocytes, and CD3+ T lymphocytes expressed ENO1 at the intracellular level (Figure 4A).
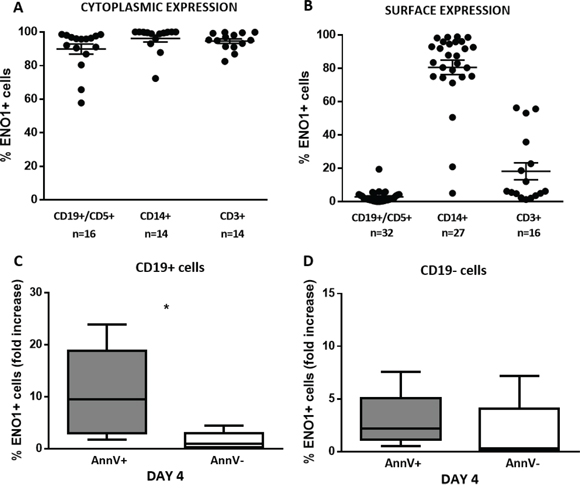
Figure 4: ENO1 is translocated on the surface of apoptotic CD19+ cells. ENO1 expression was evaluated in PBMC isolated from patients with CLL, at baseline conditions and after 4-day in vitro culture by flow cytometry. A. ENO1 cytoplasmic expression. High percentages of ENO1 were expressed in the cytoplasm of CD19+/CD5+ CLL cells, CD14+ monocytes and CD3+ T cells. B. ENO1 surface expression. ENO1 was not expressed on the surface of CD19+/CD5+ CLL cells, whereas it was widely expressed by CD14+ monocytes and in a proportion of CD3+ lymphocytes. C, D. Surface ENO1 fold increase expression in CD19+ and CD19- cell fractions. Fold increase was calculated for each patient as a ratio between the percentage of CD19+/ENO1+ detected after 4-day in vitro culture and the percentage of CD19+/ENO1+ cells at baseline. After 4 days of culture, the fold increase in ENO1 expression was significantly higher in the apoptotic (AnnV+) than in the viable (AnnV-) fraction of CD19+ cells (p=0.02) (C). By contrast, there was no difference in the fold increase of ENO1 expression between the apoptotic (AnnV+) and the viable (AnnV-) fraction of CD19- cells (D). Box and whiskers plots represent median values, first and third quartiles, and minimum and maximum values for each dataset.
We then evaluated the membrane expression of ENO1. ENO1 was not expressed on the surface of CD19+/CD5+ leukemic cells, whereas it was expressed to higher extent by CD14+ monocytes, and in a proportion of CD3+ T lymphocytes (Figure 4B). After 4 days of in vitro culture, the mean fold increase in the percentage of cells expressing ENO1 on the surface was significantly higher in the apoptotic (Annexin-V+ [AnnV+]) than in the viable (AnnV-) fraction of CD19+ cells (p=0.02) (Figure 4C). This difference in ENO1 surface expression (mean fold increase) was not observed between the apoptotic and viable fraction of CD19- cells (Figure 4D).
Immunoreactivity toward ENO1 associates with CLL progression and shorter TTFT
The presence of circulating anti-ENO1 Ab was not associated with the occurrence of clinically evident autoimmune manifestations. By contrast, ENO1 reactivity was significantly associated to parameters of disease progression such as higher lymphocyte counts (p=0.02) and lower platelet numbers (p=0.007) (Supplementary Figure S3A and S3B). Similarly, anti-ENO1 Ab (Figure 5A) were more frequently detected in sera from patients with progressive disease than in sera from patients with stable disease (p<0.0001). As expected, log-rank test indicated that the presence of anti-ENO1 Ab in patients’ sera was a negative TTFT predictor in univariate analysis: the median TTFT of anti-ENO1 Ab+ CLL patients was 7 months, whereas that of anti-ENO1 Ab- CLL patients was 107 months (p=0.01) (Figure 5B). Lastly, we observed that the total level of serum IgG was significantly lower in anti-ENO1 Ab+ compared to anti-ENO1 Ab- CLL sera (p=0.02) (Supplementary Figure S4).
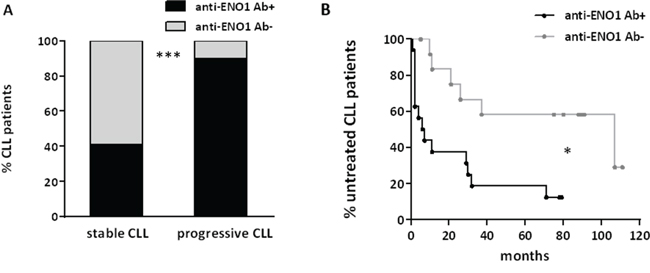
Figure 5: Anti-ENO1 Ab reactivity is associated with parameters of progressive disease and with a shorter TTFT in patients with CLL. The presence of circulating anti-ENO1 Ab in sera of patients with CLL was correlated with parameters of disease progression and TTFT. Anti-ENO1 Ab were more frequently detected in sera from patients with progressive CLL than in sera from patients with stable CLL (p<0.0001) (A). Kaplan-Meier survival curves for TTFT in our cohort of patients stratified on the basis of anti-ENO1 reactivity are shown. The presence of anti-ENO1 Ab correlated with shorter TTFT (ENO1 Ab+, n=19; ENO1 Ab-, n=16) (p=0.01) (B).
Serum Ab are not able to trigger complement dependent cytotoxicity (CDC) and antibody dependent cellular cytotoxicity (ADCC)
Next, we asked whether the circulating Ab identified in patients sera were effective in triggering CDC. We first tested the efficacy of our samples of CLL sera as source of complement. As expected, the viability of CLL cells exposed to the monoclonal Ab alemtuzumab was significantly reduced compared to untreated controls (p<0.01). By contrast, we did not observe any significant decrease in cell viability when CLL cells were exposed to patients’ sera, in the absence of alemtuzumab (Figure 6A–6D). Flow cytometry revealed that there was no deposition of the complement component 4 on the surface of CLL cells after incubation with patients’ sera (data not shown). Overall, these data show that the circulating Ab detected by SERPA in CLL sera are not able to induce CDC toward CLL cells.
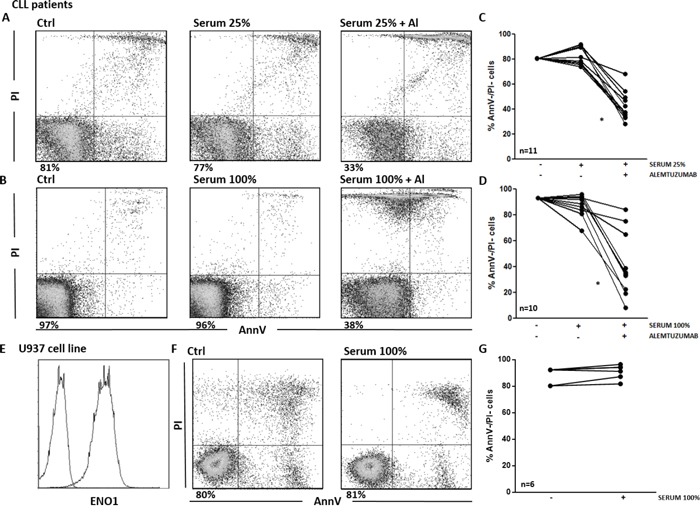
Figure 6: Serum Ab are not able to trigger CDC toward tumor cells. A-D. Viability of CLL cells incubated for 1 hour with different concentrations of CLL serum, in the presence or in the absence of alemtuzumab. Cell viability was determined by Ann-V/PI staining and cytofluorimetric analysis on CLL cells purified from patients’ samples. A, B. Representative analyses of AnnV/PI expression on CLL cells left untreated or incubated with patient’s serum at 25% (A) and 100% (B) concentration, in the presence or in the absence of alemtuzumab. Indicated is the percentage of AnnV/PI double negative (AnnV-/PI-) cells. C, D. Percentage of AnnV-/PI- viable CLL cells. Line plots show that cell viability was significantly reduced when CLL cells were exposed to alemtuzumab (p<0.01). By contrast, there was no significant decrease in cell viability when CLL cells were exposed to sera alone, both at the 25% (C) and 100% (D) concentrations. E-G. CDC assay on the U937 cell line. E. ENO1 surface expression. Representative analyses of ENO1 expression on U937 cells. F. Representative analyses of AnnV/PI expression on U937 cells left untreated or incubated with ENO1-Ab+ patients’ sera at 100% concentration. Indicated is the percentage of AnnV-/PI- cells. G. Percentage of viable AnnV-/PI- U937 cells. Line plot shows no significant decrease in cell viability after exposure of U937 cells to 6 ENO1-Ab+ sera from CLL patients.
We then performed CDC assays with sera from anti-ENO1 Ab+ patients and the U937 cell line, which expresses high levels of surface ENO1 (Figure 6E). Again, no modification of cell viability was observed when U937 cells were treated with anti-ENO1 Ab+ sera, even when sera were used at 100% concentration (Figure 6F and 6G). These data demonstrate that the circulating Ab do not trigger CDC at the in vivo concentrations, even when the cognate Ag is highly expressed on the surface of the target cells.
In the last set of experiments, we tested the ability of anti-ENO1 Ab+ sera to trigger ADCC toward both the U937 cell line and primary CLL cells. We found that the % of ADCC of U937 cells cultured for 18 hours in the presence of 10% diluted anti-ENO1 Ab+ serum and HD PBMC, used as effector cells, was not significantly increased compared to target cells cultured with PBMC alone, in the absence of serum. Similar results were obtained using CLL cells as targets (Figure 7A–7D). Therefore, we can conclude that the circulating Ab detected by SERPA are unable to trigger ADCC against leukemic cells.
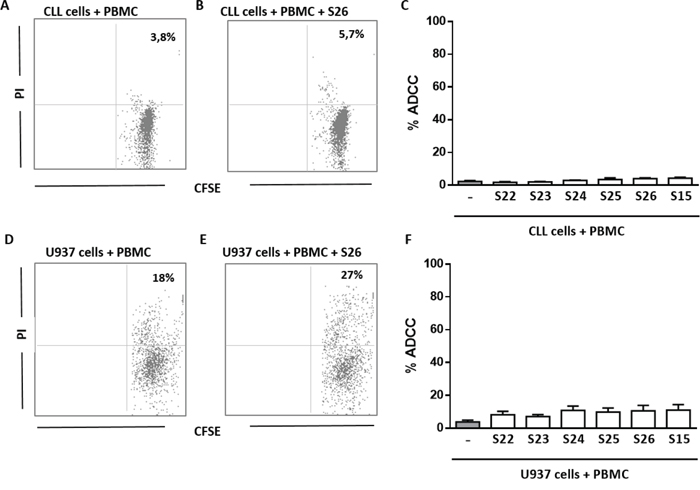
Figure 7: Anti-ENO1 Ab are not able to trigger ADCC against leukemic cells. A-C. % of ADCC of CFSE labelled CLL cells co-cultured for 18 hours with HD PBMC, in presence or absence of 10% diluted anti-ENO1 Ab+ serum. Cell lysis was determined by PI staining and flow cytometry. % of ADCC was calculated by the formula % PI (T + E + serum) - % PI (T)/% PI (triton X treated T) – % PI (T). A, B. Representative analysis of CFSE/PI expression on CLL cells (gated on CFSE+ cells) purified from one patient’s sample co-cultured with HD PBMC, used as effector cells, in the absence (A) or presence (B) of anti-ENO1 Ab+ serum at 10% concentration. Indicated is the % of CFSE/PI double positive cells. C. Bar graph show no significant differences in the % of ADCC of primary CLL cells cultured with PBMC and 10% diluted anti-ENO1 Ab+ serum (n=6) compared to target cells cultured with PBMC alone, in the absence of serum. D-F. ADCC assay on U937 cell line. D, E. Representative analysis of CFSE/PI expression on U937 cells (gated on CFSE+ cells) co-cultured with HD PBMC, used as effector cells, in the absence (D) or presence (E) of anti-ENO1 Ab+ serum at 10% concentration. Indicated is the % of CFSE/PI double positive cells. F. Bar graph show no significant differences in the % of ADCC of U937 cells cultured with PBMC and 10% diluted anti-ENO1 Ab+ serum (n=6) compared to target cells cultured with PBMC alone, in the absence of serum. Mean results from three different experiments.
DISCUSSION
In CLL, cellular immune responses are unable to respond to the tumor and they can even support disease evolution [22]. The impact of humoral responses toward tumor-derived Ag in CLL is largely unexplored. Through the use of SERPA we show that 83% of CLL sera have circulating Ab toward at least one CLL-derived Ag. Interestingly, we observe that sera from progressive CLL are significantly more immunoreactive than sera from stable CLL.
The circulating Ab detected by SERPA are expression of an immune response generated by the residual polyclonal B-cell population toward self-Ag. We do not observe shared Ag recognition between a representative CLL serum and the ScFv-Fc derived from the autologous leukemic BCR. On the other hand, patients who share the same Ag recognitions do not show recurrent IGHV rearrangements or BCR stereotypies. Autoimmune phenomena are rather frequent complications of CLL and mostly result in autoimmune cytopenias [23]. A clear association between autoimmune cytopenias and poor prognostic variables, such as high β2-microglobulin, CD38 and ZAP70 expression, and unmutated IGHV status, has already been described [24–26]. Moreover, direct antiglobulin test positivity, even in the absence of overt AIHA, associates with adverse prognostic factors and shorter OS [27]. In our cohort, we observe a significantly higher degree of immunoreactivity in patients with progressive CLL than in patients with stable disease, even though the immunoreactive status is not related to adverse prognostic factors.
MS analyses show that the Ab detected by SERPA mostly recognize intracellular Ag, such as glycolytic enzymes, cytoskeletal elements and ribonucleoproteins. Among others, ENO1, a glycolytic enzyme, is the most relevant Ag, detected in 19 out of 35 (54%) CLL sera. The overexpression of ENO1 is associated with tumor development through a process known as Warburg effect [28]. The Warburg effect is a metabolic reprogramming, which consists in a positive regulation exerted by the hypoxia-inducible factor (HIF) on the expression of glycolytic enzymes, such as ENO1, occurring when cancer cells are exposed to hypoxic conditions [29, 30]. Interestingly, CLL cells express the oxygen-regulated HIF-1α subunit even under normoxia [31], and the expression and the transcriptional activity of HIF-1α are further upregulated by exposure of CLL cells to stromal cells [32]. In the LN, proliferating CLL cells are confined to specialized structures called pseudo-follicles, where they interact with T lymphocytes and stromal cells. Our data show that ENO1 is more expressed in the leukemic cells of the CLL LN than in the normal B cells of the R LN, and confirm that it is more expressed by the proliferating B cells of the pseudo-follicles than by the resting B-cell fraction.
Due to its tumor-related overexpression and ability to induce humoral and/or cellular immune responses, ENO1 has already been classified as a tumor-associated Ag in solid cancers, such as pancreatic ductal adenocarcinoma (PDA) [12, 33]. Cappello et al. have reported that a DNA vaccine coding for ENO1 can elicit anti-ENO1 IgG Ab which are able to induce the killing of murine PDA cells by CDC [33]. Unexpectedly, in our cohort of CLL patients, the presence of anti-ENO1 Ab is prevalent in sera from patients with progressive CLL and is predictive of a shorter TTFT. One possible reason of the different clinical impact exerted by anti-ENO1 Ab in solid tumors and in CLL is that ENO1 is highly expressed at the intracellular level, but is not expressed on the cell membrane. It is not clear how intracellular self-Ag can become immunogenic and trigger humoral responses. One possible explanation is that overexpressed self-proteins are exposed during the apoptotic turnover of the leukemic cells, thus becoming visible and capable of inducing autoreactive immune responses. This hypothesis is corroborated by our data showing that after 4 days of in vitro culture, the mean fold increase in the percentage of cells expressing ENO1 on the surface is significantly higher in the apoptotic than in the viable fraction of CD19+ cells. The higher frequency of immunoreactivities detected by SERPA in the sera of patients with progressive CLL can be explained by the observation that in advanced-stage CLL the malignant clone, which in early-stage is arrested in G0/G1, evolve into an autonomously proliferating cell population showing a greater ability to enter spontaneous apoptosis [34]. In the appropriate context, such as in the spleen and LN, where T cells may induce the activation and Ag presentation by B cells, this turnover potentially leads to a greater exposition of intracellular overexpressed Ag and to the production of autoreactive Ab.
To functionally characterize the humoral responses detected by SERPA we evaluated the in vitro ability of patients’ sera to kill primary CLL cells by CDC or ADCC. Deficiencies or reduced levels in one or more complement components have already been reported in CLL patients [35, 36]. However, we demonstrated that CLL sera behave as proper source of complement, in inducing alemtuzumab-mediated CDC as effectively as HD sera. By contrast, in the absence of alemtuzumab, polyreactive CLL sera are not able to induce complement deposition on the surface of CLL cells and do not trigger CDC toward primary tumor cells. Similarly, CLL sera do not trigger ADCC against CLL cells, in the presence of PBMC isolated from HD used as effector cells. A further demonstration of the functional incompetence of circulating Ab detected by SERPA is the observation that CLL patients with immunoreacitive sera, even those with polyreactive sera, lack clinically manifest autoimmune phenomena. A number of reasons may underlie this functional incompetence. First of all, the tumor Ag recognized by the circulating Ab are mainly intracellular proteins and therefore are not optimal targets for an Ab-mediated immune response. However, we do not observe any cytotoxicity even when the U937 cell line, which expresses high levels of surface ENO1, is used as target for the CDC and ADCC assays. In line with these results is the observation that the absolute number of monocytes, which also express high levels of surface ENO1, is not decreased in the PB of CLL patients with anti-ENO1 circulating Ab (data not shown). These findings show that Ab-related issues, such as low sera concentrations of the circulating Ab and/or low binding affinity toward the corresponding Ag, may also contribute to the functional incompetence of the humoral responses detected by SERPA.
Overall, our results show that the Ab responses detected by SERPA are an epiphenomenon of a disrupted immune system, which is unable to control disease evolution. It has already been reported that the chronic inflammation state determined by an enhanced and persistent activation of humoral immune responses, in combination with the suppressed cellular anti-tumor immunity, may favour the tumor progression and support disease evolution [37]. In this context, it will be of interest to determine the immunomodulatory properties of the new BCR inhibitors, which have now been introduced in the clinical practice for the treatment of CLL. Current studies already show a recovery of humoral immunity and normal B-cell numbers in patients on ibrutinib, leading to a decrease in the rate of infections [38]. This observation, together with the promising results obtained in experimental models of autoimmune diseases [39, 40], support the hypothesis of an ibrutinib-induced immunomodulation which may contribute to tumor control.
MATERIALS AND METHODS
Patient population
PB samples were collected from a total of 86 patients with untreated CLL, after their informed consent, in accordance with the Declaration of Helsinki and approval by the local institutional review board. Sections of LN infiltrated by CLL cells and reactive R LN were obtained from the Department of Medical Sciences of the University of Torino, Italy. The diagnosis and progression of CLL were defined according to International Workshop on CLL-National Cancer Institute (IWCLL/NCI-WG) guidelines [41]. The disease was defined stable in absence of signs of progression. A cohort of 35 patients with CLL was analyzed by SERPA. Patients’ biological and clinical features were collected, when available, by chart review (Supplementary Table S2). The median follow-up of all patients was 81 months. The IGVH mutational status and the CDR3 clustering analyses were performed as previously reported [42, 43]. The control group consisted of 12 HD kindly provided by the local blood bank.
Cells and serum samples
PBMC and purified B lymphocytes were prepared as described [32]. Serum samples were isolated from PB by centrifugation and stored at -80°C until use. In selected experiments, the human cell line U937 (ATCC, CRL 1593.2) was used.
Sample preparation and 2-Dimension Electrophoresis (2-DE)
Pellets obtained from at least 107 purified CLL cells were solubilized in lysis buffer (urea 9M, CHAPS 4%, Na3VO4 1mM, DTT 80mM, protease inhibitors and nuclease). Samples were spun down at 13800 g for 10 minutes at 4°C. The clear supernatant was quantified with DC Protein assay kit (Bio-Rad, Hercules, CA, USA). 2-DE on ready-made IPG strip (7-cm IPG strips, pH 3-10NL; Bio-Rad) was performed essentially as described [44]. The 2-DE maps were obtained in duplicate and stained with Coomassie Blue or transferred onto a nitrocellulose membrane (GE Healthcare Biosciences GmbH, Uppsala, Sweden) for serological analysis. The 2-D gel images were acquired using the ChemiDoc MP system (Bio-Rad).
Western blot analysis
The membrane was incubated overnight at 4°C with serum, as primary Ab at working dilution of 1:100, with a single chain variable fragment-fragment crystallisable region (ScFv-Fc) or with an anti-ENO1 Ab (Santa Cruz Biotechnology, Inc; CA, USA). Then the membrane was incubated with an anti-human (Santa Cruz Biotechnology, Inc) or anti-mouse (GE Healthcare Biosciences GmbH) IgG HRP-conjugated Abs. Images were acquired by the ChemiDoc MP system (Bio-Rad).
Protein identification by MS
MS analysis was performed as described [44]. The 25 most intense masses were used for database searches against the Swiss-Prot database using the free search program Mascot (http://www.matrixscience.com). Only proteins with a Mascot score greater than 56 were taken into consideration.
ScFv-Fc production
VH and VL genes were amplified with specific primers from CLL derived cDNA as already described [45]. V genes were assembled as ScFv and cloned in fusion with the human Fc region (as scFv-Fc) in the pUCOE vector [46]. CHO-S cell line was transfected and stable clones was obtained for ScFv-Fc expression. Ab produced in cell culture supernatants were purified using a protein A/G column.
Flow cytometry
ENO1 surface and intracytoplasmic expression was evaluated cytofluorimetrically on B cells, T cells and monocytes in PB samples as reported in Supplemental Methods.
Cell culture and viability assay
PBMC from patients with CLL were cultured as described [32]. The percentage of viable and apoptotic cells was determined by Annexin-V (AnnV) or AnnV/Propidium Iodide (PI) staining with the MEBCYTO-Apoptosis Kit (MBL Medical and Biological Laboratories, Naka-ku Nagoya, Japan).
Immunohistochemistry and confocal immunofluorescent microscopy
Cell morphology and numbers were studied by Giemsa staining. For immunocytochemistry, coverslips were stained as detailed in Supplemental Methods. Formalin-fixed, paraffin-embedded sections of CLL or R LN were stained and analyzed by light microscopy as already described [47] (details in Supplemental Methods).
CDC and ADCC assays
For CDC, purified CLL cells (2 x 105) were incubated in presence or absence of alemtuzumab (10 μg/ml) for 30 minutes at room temperature. CLL cells were then washed with PBS and incubated with selected patients’ sera at different dilutions for 60 minutes at 37°C. U937 cells were incubated only with CLL patients’ sera. Cell viability was evaluated using AnnV/PI staining (details in Supplemental Methods). For ADCC, purified CLL cells or U937 cells, used as target cells (T), were labeled with CFSE (5 μM) for 30 minutes at 37 °C. CLL cells and U937 cells were then incubated with PBMC from healthy donors, used as effector cells (E) for 18 hours at 30:1 E:T ratio, in the presence or absence of 10% diluted CLL patients’ sera. At the end of the co-culture target cells viability was evaluated by PI staining. % of ADCC was calculated as follow: % PI (T + E + serum) - % PI (T)/% PI (triton X treated T) – % PI (T).
Statistical analysis
Statistical analyses were performed with GraphPad Prism (San Diego, CA, USA). Continuous variables were compared by Mann-Whitney U (unpaired data) or Wilcoxon signed rank (paired data) tests. The χ2 test was used in case of dichotomous variables. OS and TTFT were defined as the time between the date of SERPA and, respectively, the date of death or last follow-up and the date of first-line treatment or the last follow-up. OS and TTFT were estimated by the Kaplan-Meier method and the difference between groups was assessed by log-rank test. A p value <.05 was considered significant.
CONFLICTS OF INTEREST
M.B.: Consulting or Advisory Role: Celgene, Janssen-Cilag, Onyx, Sanofi. M.M.: research support Roche, Novartis; advisory board Janssen, Roche. M.Coscia.: honoraria and advisory board Roche; honoraria Mundipharma, Janssen, Gilead; research support Karyopharm Therapeutics Inc., Janssen. The remaining authors declare no conflicts of interest.
FUNDING
Work supported by grants from Regione Piemonte (Ricerca Sanitaria Finalizzata 2008 to Marta Coscia; Progetto Immonc to Massimo Massaia); Italian Ministry of University and Research (www.miur.it; PRIN 2010NECHBX_002 to Massimo Massaia and FIRB 2012 to Chiara Riganti); Italian Ministry of Health (Progetto Giovani Ricercatori bando 2012 to Silvia Deaglio); Italian Association for Cancer Research (AIRC, www.airc.it; IG 16985 to Massimo Massaia, IG 12023 to Roberto Chiarle, IG 12754 to Silvia Deaglio, 5 x mille n.12182 and IG 15257 to Francesco Novelli); Compagnia San Paolo, Torino, Italy (Progetto Ateneo 2014 S1315_RIC14_L2_CAP_01 to Francesco Novelli); Fondazione Neoplasie Sangue (Fo.Ne.Sa), Torino, Italy; University of Torino - Progetti di Ateneo 2011 (grant Rethe-ORTO11RKTW to Francesco Novelli) local funds of the University of Torino (ex-60%); Fondazione Ricerca Molinette. Valentina Griggio was the recipient of a fellowship from Fondazione Cassa di Risparmio di Torino (CRT), a “Giorgio Bissolotti e Teresina Bosio” fellowship from Fondazione “Angela Bossolasco”, Torino, Italy and currently is the recipient of a “Anna Nappa” fellowship from the Italian Association for Cancer Research (AIRC, Ref 16343). Barbara Castella and Myriam Foglietta are post-doc research fellows supported by PRIN (B.C.), and AIRC (M.F.).
Authorship contributions
V.G. contributed to the research design, performed the experiments, analyzed the data and wrote the paper. G.M. performed the experiments and analyzed the data. C.V. provided patients’ care and contributed to data analyses. M.Capello, P.M., S.S., B.C., S.P., M.F., D.D., P.O., D.S. and R.C. assisted with the experiments. P.M., D.D., P.O., D.S., P.C. and R.C. revised the paper. M.B. reviewed the manuscript. M.M, S.D. and F.N. contributed to the research design and revised the paper. M.Coscia contributed to the research design, supervised the study, wrote and revised the paper. All authors approved final manuscript.
REFERENCES
1. Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015; 126: 573-81. doi: 10.1182/blood-2015-03-567388.
2. Nunes C, Wong R, Mason M, Fegan C, Man S, Pepper C. Expansion of a CD8(+)PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin Cancer Res. 2012; 18: 678-87. doi: 10.1158/1078-0432.CCR-11-2630.
3. Brusa D, Serra S, Coscia M, Rossi D, D'Arena G, Laurenti L, Jaksic O, Fedele G, Inghirami G, Gaidano G, Malavasi F, Deaglio S. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica. 2013; 98: 953-63. doi: 10.3324/haematol.2012.077537.
4. Cutucache CE. Tumor-induced host immunosuppression: special focus on CLL. Int Immunopharmacol. 2013; 17: 35-41. doi: 10.1016/j.intimp.2013.05.021.
5. D'Arena G, Laurenti L, Minervini MM, Deaglio S, Bonello L, De Martino L, De Padua L, Savino L, Tarnani M, De Feo V, Cascavilla N. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res. 2011; 35: 363-8. doi: 10.1016/j.leukres.2010.08.010.
6. Coscia M, Vitale C, Peola S, Foglietta M, Rigoni M, Griggio V, Castella B, Angelini D, Chiaretti S, Riganti C, Guarini A, Drandi D, Ladetto M, et al. Dysfunctional Vgamma9Vdelta2 T cells are negative prognosticators and markers of dysregulated mevalonate pathway activity in chronic lymphocytic leukemia cells. Blood. 2012; 120: 3271-9. doi: 10.1182/blood-2012-03-417519.
7. Dhalla F, Lucas M, Schuh A, Bhole M, Jain R, Patel SY, Misbah S, Chapel H. Antibody deficiency secondary to chronic lymphocytic leukemia: Should patients be treated with prophylactic replacement immunoglobulin? J Clin Immunol. 2014; 34: 277-82. doi: 10.1007/s10875-014-9995-5.
8. Ghia P, Scielzo C, Frenquelli M, Muzio M, Caligaris-Cappio F. From normal to clonal B cells: Chronic lymphocytic leukemia (CLL) at the crossroad between neoplasia and autoimmunity. Autoimmun Rev. 2007; 7: 127-31. doi: 10.1016/j.autrev.2007.02.014.
9. Krackhardt AM, Witzens M, Harig S, Hodi FS, Zauls AJ, Chessia M, Barrett P, Gribben JG. Identification of tumor-associated antigens in chronic lymphocytic leukemia by SEREX. Blood. 2002; 100: 2123-31. doi: 10.1182/blood-2002-02-0513.
10. Unwin RD, Harnden P, Pappin D, Rahman D, Whelan P, Craven RA, Selby PJ, Banks RE. Serological and proteomic evaluation of antibody responses in the identification of tumor antigens in renal cell carcinoma. Proteomics. 2003; 3: 45-55. doi: 10.1002/pmic.200390008.
11. Gnjatic S, Ritter E, Buchler MW, Giese NA, Brors B, Frei C, Murray A, Halama N, Zornig I, Chen YT, Andrews C, Ritter G, Old LJ, et al. Seromic profiling of ovarian and pancreatic cancer. Proc Natl Acad Sci U S A. 2010; 107: 5088-93. doi: 10.1073/pnas.0914213107.
12. Tomaino B, Cappello P, Capello M, Fredolini C, Sperduti I, Migliorini P, Salacone P, Novarino A, Giacobino A, Ciuffreda L, Alessio M, Nistico P, Scarpa A, et al. Circulating autoantibodies to phosphorylated alpha-enolase are a hallmark of pancreatic cancer. J Proteome Res. 2011; 10: 105-12. doi: 10.1021/pr100213b.
13. Hamrita B, Chahed K, Kabbage M, Guillier CL, Trimeche M, Chaieb A, Chouchane L. Identification of tumor antigens that elicit a humoral immune response in breast cancer patients' sera by serological proteome analysis (SERPA). Clin Chim Acta. 2008; 393: 95-102. doi: 10.1016/j.cca.2008.03.017.
14. Li L, Chen SH, Yu CH, Li YM, Wang SQ. Identification of hepatocellular-carcinoma-associated antigens and autoantibodies by serological proteome analysis combined with protein microarray. J Proteome Res. 2008; 7: 611-20. doi: 10.1021/pr070525r.
15. Forgber M, Trefzer U, Sterry W, Walden P. Proteome serological determination of tumor-associated antigens in melanoma. PLoS One. 2009; 4: e5199. doi: 10.1371/journal.pone.0005199.
16. Yang F, Xiao ZQ, Zhang XZ, Li C, Zhang PF, Li MY, Chen Y, Zhu GQ, Sun Y, Liu YF, Chen ZC. Identification of tumor antigens in human lung squamous carcinoma by serological proteome analysis. J Proteome Res. 2007; 6: 751-8. doi: 10.1021/pr0602287.
17. Cui JW, Li WH, Wang J, Li AL, Li HY, Wang HX, He K, Li W, Kang LH, Yu M, Shen BF, Wang GJ, Zhang XM. Proteomics-based identification of human acute leukemia antigens that induce humoral immune response. Mol Cell Proteomics. 2005; 4: 1718-24. doi: 10.1074/mcp.M400165-MCP200.
18. Zou L, Wu Y, Pei L, Zhong D, Gen M, Zhao T, Wu J, Ni B, Mou Z, Han J, Chen Y, Zhi Y. Identification of leukemia-associated antigens in chronic myeloid leukemia by proteomic analysis. Leuk Res. 2005; 29: 1387-91. doi: 10.1016/j.leukres.2005.04.021.
19. Zappasodi R, Bongarzone I, Ghedini GC, Castagnoli L, Cabras AD, Messina A, Tortoreto M, Tripodo C, Magni M, Carlo-Stella C, Gianni AM, Pupa SM, Di Nicola M. Serological identification of HSP105 as a novel non-Hodgkin lymphoma therapeutic target. Blood. 2011; 118: 4421-30. doi: 10.1182/blood-2011-06-364570.
20. Forgber M, Gellrich S, Sharav T, Sterry W, Walden P. Proteome-based analysis of serologically defined tumor-associated antigens in cutaneous lymphoma. PLoS One. 2009; 4: e8376. doi: 10.1371/journal.pone.0008376.
21. Dubovsky JA, Chappell DL, Harrington BK, Agrawal K, Andritsos LA, Flynn JM, Jones JA, Paulaitis ME, Bolon B, Johnson AJ, Byrd JC, Muthusamy N. (2013). Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing.
22. Scrivener S, Goddard RV, Kaminski ER, Prentice AG. Abnormal T-cell function in B-cell chronic lymphocytic leukaemia. Leuk Lymphoma. 2003; 44: 383-9. doi: 10.1080/1042819021000029993.
23. Hamblin TJ. Non-hemic autoimmunity in CLL. Leuk Res. 2009; 33: 366-7. doi: 10.1016/j.leukres.2008.09.011.
24. Moreno C, Hodgson K, Ferrer G, Elena M, Filella X, Pereira A, Baumann T, Montserrat E. Autoimmune cytopenia in chronic lymphocytic leukemia: prevalence, clinical associations, and prognostic significance. Blood. 2010; 116: 4771-6. doi: 10.1182/blood-2010-05-286500.
25. Zanotti R, Frattini F, Ghia P, Visco C, Zamo A, Perbellini O, Stella S, Facco M, Giaretta I, Chilosi M, Pizzolo G, Ambrosetti A. ZAP-70 expression is associated with increased risk of autoimmune cytopenias in CLL patients. Am J Hematol. 2010; 85: 494-8. doi: 10.1002/ajh.21737.
26. Visco C, Novella E, Peotta E, Paolini R, Giaretta I, Rodeghiero F. Autoimmune hemolytic anemia in patients with chronic lymphocytic leukemia is associated with IgVH status. Haematologica. 2010; 95: 1230-2. doi: 10.3324/haematol.2010.022079.
27. Quinquenel A, Al Nawakil C, Baran-Marszak F, Eclache V, Letestu R, Khalloufi M, Boubaya M, Le Roy C, Varin-Blank N, Delmer A, Levy V, Ajchenbaum-Cymbalista F. Old DAT and new data: positive direct antiglobulin test identifies a subgroup with poor outcome among chronic lymphocytic leukemia stage A patients. Am J Hematol. 2015; 90: E5-8. doi: 10.1002/ajh.23861.
28. Warburg O. The Metabolism of Tumours. Constable: London. 1930.
29. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324: 1029-33. doi: 10.1126/science.1160809.
30. Capello M, Ferri-Borgogno S, Cappello P, Novelli F. alpha-Enolase: a promising therapeutic and diagnostic tumor target. FEBS J. 2011; 278: 1064-74. doi: 10.1111/j.1742-4658.2011.08025.x.
31. Shachar I, Cohen S, Marom A, Becker-Herman S. Regulation of CLL survival by hypoxia-inducible factor and its target genes. FEBS Lett. 2012; 586: 2906-10. doi: 10.1016/j.febslet.2012.07.016.
32. Rigoni M, Riganti C, Vitale C, Griggio V, Campia I, Robino M, Foglietta M, Castella B, Sciancalepore P, Buondonno I, Drandi D, Ladetto M, Boccadoro M, et al. Simvastatin and downstream inhibitors circumvent constitutive and stromal cell-induced resistance to doxorubicin in IGHV unmutated CLL cells. Oncotarget. 2015; 6: 29833-46. doi: 10.18632/oncotarget.4006.
33. Cappello P, Tomaino B, Chiarle R, Ceruti P, Novarino A, Castagnoli C, Migliorini P, Perconti G, Giallongo A, Milella M, Monsurro V, Barbi S, Scarpa A, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer. 2009; 125: 639-48. doi: 10.1002/ijc.24355.
34. Sieklucka M, Pozarowski P, Bojarska-Junak A, Hus I, Dmoszynska A, Rolinski J. Apoptosis in B-CLL: the relationship between higher ex vivo spontaneous apoptosis before treatment in III-IV Rai stage patients and poor outcome. Oncol Rep. 2008; 19: 1611-20.
35. Garcia-Munoz R, Galiacho VR, Llorente L. Immunological aspects in chronic lymphocytic leukemia (CLL) development. Ann Hematol. 2012; 91: 981-96. doi: 10.1007/s00277-012-1460-z.
36. Middleton O, Cosimo E, Dobbin E, McCaig AM, Clarke C, Brant AM, Leach MT, Michie AM, Wheadon H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia. 2015; 29: 107-14. doi: 10.1038/leu.2014.146.
37. Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol. 2007; 19: 209-16. doi: 10.1016/j.coi.2007.01.001.
38. Sun C, Tian X, Lee YS, Gunti S, Lipsky A, Herman SE, Salem D, Stetler-Stevenson M, Yuan C, Kardava L, Moir S, Maric I, Valdez J, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015. doi: 10.1182/blood-2015-04-639203.
39. Hutchinson CV, Dyer MJ. Breaking good: the inexorable rise of BTK inhibitors in the treatment of chronic lymphocytic leukaemia. Br J Haematol. 2014; 166: 12-22. doi: 10.1111/bjh.12895.
40. Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, Robinson WH, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011; 13: R115. doi: 10.1186/ar3400.
41. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008; 111: 5446-56. doi: 10.1182/blood-2007-06-093906.
42. Ricca I, Rocci A, Drandi D, Francese R, Compagno M, Lobetti Bodoni C, De Marco F, Astolfi M, Monitillo L, Vallet S, Calvi R, Ficara F, Omede P, et al. Telomere length identifies two different prognostic subgroups among VH-unmutated B-cell chronic lymphocytic leukemia patients. Leukemia. 2007; 21: 697-705. doi: 10.1038/sj.leu.2404544.
43. Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, Belhoul L, Stella S, Stavroyianni N, Crespo M, Hadzidimitriou A, Sutton L, Bosch F, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. 2007; 109: 259-70. doi: 10.1182/blood-2006-03-012948.
44. Mandili G, Marini C, Carta F, Zanini C, Prato M, Khadjavi A, Turrini F, Giribaldi G. Identification of Phosphoproteins as Possible Differentiation Markers in All-Trans-Retinoic Acid-Treated Neuroblastoma Cells. PLoS ONE. 2011; 6: e18254. doi: 10.1371/journal.pone.0018254.
45. Sblattero D, Bradbury A. A definitive set of oligonucleotide primers for amplifying human V regions. Immunotechnology. 1998; 3: 271-8.
46. Boscolo S, Mion F, Licciulli M, Macor P, De Maso L, Brce M, Antoniou MN, Marzari R, Santoro C, Sblattero D. Simple scale-up of recombinant antibody production using an UCOE containing vector. N Biotechnol. 2012; 29: 477-84. doi: 10.1016/j.nbt.2011.12.005.
47. Serra S, Horenstein AL, Vaisitti T, Brusa D, Rossi D, Laurenti L, D'Arena G, Coscia M, Tripodo C, Inghirami G, Robson SC, Gaidano G, Malavasi F, et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood. 2011; 118: 6141-52. doi: 10.1182/blood-2011-08-374728.