
INTRODUCTION
Skin cancers are classified into three types, melanoma, squamous cell carcinoma (SCC) and basal cell carcinoma (BCC), with SCC and BCC known as non-melanoma skin cancer (NMSC) [1, 2]. BCC accounts for approximately 80% of all non-melanoma skin cancers and is the most common skin cancer [3]. Despite the increasing incidence of BCC, metastasis of this type of cancer is rare, occurring in 0.0028% to 0.55% of all BCC cases [4, 5]. Since metastatic BCC (MBCC) is uncommon, the lack of available information regarding this type of cancer is expected. MBCC usually spreads by lymphatic and hematogenous routes [6], and the 5-year survival of patients with metastatic or advanced BCC is only 10% [7], whereas the 10-year survival rate for patients with metastatic melanoma is less than 10% [8]. Therefore, investigating the underlying mechanisms involved in metastasis, invasion and progression of skin cancers is important for the development of novel therapeutic strategies. The vascular endothelial growth factor (VEGF) family consists of VEGF-A, -B, -C, -D, -E, -F and placental growth factor (PlGF), which have been identified as specific angiogenic and lymphangiogenic factors in tumor progression and metastasis [9, 10]. The cellular functions of the VEGFs are mediated by three tyrosine kinase receptors (VEGFRs), VEGFR1, 2 and 3 [11]. Among these VEGF-VEGFR complexes, VEGF-C binds to both VEGFR2 and VEGFR3 and further enhances diverse biological effects, such as cell growth, proliferation, mobility and invasiveness, to promote angiogenesis, metastasis and tumor progression [12–14]. Previous studies reported that activation of the VEGF-C/VEGFR3 signaling pathway increases cell migration and invasion to promote cancer metastasis, and a high expression level of VEGF-C has been correlated with the shortest survival time in lung cancer [10, 15] and esophageal squamous cell carcinoma [16, 17]. Elevated VEGF-C expression is also found in the lymph nodes of patients with metastatic melanoma [18]. In addition, VEGF-C has been recognized as a metastatic tumor promoter by protecting tumor cells against preexisting antitumor immunity and contributes to tumor progression of melanoma cells [19]. However, how VEGF-C is involved in cell mobility and the progression of skin cancer has remained elusive.
Yes-associated protein 1 (YAP1), a downstream molecule of the Hippo signaling pathway, has been identified as an oncogene involved in cancer-promoting processes such as cell proliferation and metastasis [20–22]. YAP1 interacts with various transcription factors, such as RUNX2, p73, p53BP2 and TEAD family members, that are essential for YAP1-mediated tumor growth and metastasis in melanoma and breast cancer [21, 23]. In addition, it has been reported that elevated YAP1 regulates epithelial-mesenchymal transition (EMT) of the atrioventricular cushion [24] and expands multipotent undifferentiated progenitor cells [25], suggesting that YAP1 plays a critical role in promoting cell metastasis, organ growth, cancer stemness and morphological changes. For now, it is still unclear whether YAP1 is involved in the VEGF-C/VEGFR3 signaling pathway in skin cancers.
In this study, we demonstrate that VEGF-C/VEGFR3 signaling affects skin cancer cell mobility by mediating Slug expression. VEGF-C regulates CSC properties and is positively correlated with metastasis and the expression of CSC markers in skin cancer. We further identify VEGF-C/VEGFR3-mediated YAP1 and Slug expression through KRAS/MAPK signaling in skin cancer. By treating skin cancer cells with an anti-VEGFR3 specific peptide [26], we show that suppression of VEGF-C/VEGFR3 signaling significantly reduces YAP1 and Slug expression and diminishes the migration, invasion and cancer stemness of the cells. In conclusion, our findings provide a novel signaling pathway, the VEGF-C/VEGFR3-KRAS/MAPK-YAP1/Slug axis, in skin cancer progression and a potential therapeutic strategy by treatment with an anti-VEGFR3 peptide.
RESULTS
VEGF-C increases the migration and invasion abilities of skin cancer cells
We first determined the activation of VEGFR3 in two skin cancer cell lines, BCC and A2058, by treating the cells with recombinant human VEGF-C protein (rhVEGF-C). We found that rhVEGF-C protein treatment increased VEGFR3 phosphorylation (pVEGFR3) in BCC and A2058 cells after 10 minutes (Figure 1a). To further investigate whether VEGF-C affects skin cancer cell migration and invasion abilities, we established stable VEGF-C-knockdown cells (BCC/shVEGF-C and A2058/shVEGF-C) and control cells (BCC/shCtrl and A2058/shCtrl), and VEGF-C mRNA expression was measured by qRT-PCR analysis (Figure 1b). Knockdown of VEGF-C significantly suppressed the migration and invasion abilities of skin cancer cells (Figure 1c and 1d). Moreover, rhVEGF-C treatment restored the migration and invasion abilities of VEGF-C-knockdown cells (Figure 1c and 1d). An analysis of the Oncomine database also showed that VEGF-C is more highly expressed in metastatic sites of skin cancer patients than in primary sites (Figure 1e), indicating that VEGF-C is crucial for skin cancer progression.
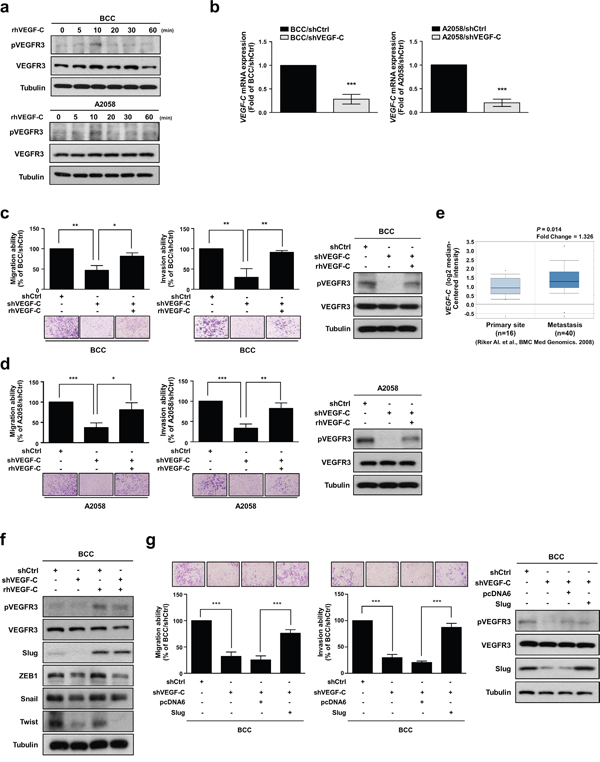
Figure 1: VEGF-C increases the migration and invasion abilities of skin cancer cells. a. BCC and A2058 cells were incubated with 200 ng/ml of VEGF-C for the indicated time points, and phosphorylated VEGFR3 and VEGFR3 were detected by Western blot analysis. Tubulin was used as a loading control. b. The mRNA expression of VEGF-C was analyzed by real-time qRT-PCR. c and d. A transwell assay was performed to examine cell migration and invasion abilities. Cell motility and phosphorylation of VEGFR3 was inhibited by knockdown of VEGF-C and recovered by treatment with rhVEGF-C by transwell assay and Western blot analysis, respectively. e. Oncomine database analysis showed elevated VEGF-C expression in metastatic melanoma tissues. f. Expression of EMT-inducing transcription factors (Slug, ZEB1, Twist and Snail) was determined by Western blot analysis. g. The effects of Slug expression on skin cancer cell motility were determined by transwell assay. Western blot analysis was used to confirm the expression of the indicated proteins. The results are shown as the mean ± SD of three independent experiments, each performed in triplicate. *P < 0.05, **P < 0.001, ***P < 0.0001 (Student’s t test).
Accumulating evidence indicates that the EMT is necessary for tumor progression and malignant transformation, which strongly enhances cancer cell motility [27]. The EMT program is orchestrated by EMT-inducing transcription factors (EMT-TFs) primarily belonging to three families, Snail, Twist and ZEB [28, 29]. Therefore, we investigated whether VEGF-C regulates EMT-TFs and found that VEGF-C depletion decreased the protein expression of Slug, ZEB1 and Twist. Notably, only Slug expression was recovered by treatment with rhVEGF-C (Figure 1f). Slug (also known as Snail2), a member of the Snail family, can repress E-cadherin expression and trigger EMT [30]. Numerous studies have indicated that Slug is involved in malignant transformation and metastatic progression in various cancers [31]. In addition, it has been reported that VEGF-A can induce Slug protein expression in pancreatic carcinoma cells and is positively correlated with Slug in patients with glioblastoma [32, 33]. As shown in Table 1, Slug expression levels were positively correlated with several clinical-pathologic features, such as metastatic event, melanoma and skin squamous cell carcinoma. To further examine the effects of Slug on VEGF-C/VEGFR-mediated cell motility in skin cancer cells, we overexpressed Slug in VEGF-C-knockdown cells and found that Slug re-expression can recover the shVEGF-C-inhibited cell migration and invasion abilities (Figure 1g). Taken together, these results suggest that VEGF-C/VEGFR3 signaling plays an important role in the enhancement of the migration and invasion abilities of skin cancer cells through up-regulation of Slug.
Table 1: Clinicopathological features, clinical outcomes and their association with Slug expression in skin cancer datasets
Datasets |
Clinicopathological features |
*Slug |
†P-value |
|---|---|---|---|
Laurent (n = 63) |
Metastatic Event at 1 Year |
1.606 |
0.025 |
Metastatic Event at 3 Years |
2.420 |
8.22E-4 |
|
Talantov (n = 70) |
Benign Melanocytic Skin Nevus |
2.803 |
6.85E-6 |
Cutaneous Melanoma |
2.075 |
2.68E-4 |
|
Haqq (n = 37) |
Melanoma |
4.491 |
0.004 |
Non-Neoplastic Nevus |
3.302 |
0.011 |
|
Riker (n = 87) |
Skin Squamous Cell Carcinoma |
1.817 |
0.001 |
Skin Basal Cell Carcinoma |
1.448 |
0.013 |
|
Nindl (n = 15) |
Skin Squamous Cell Carcinoma |
1.728 |
0.049 |
*Fold change (Log2 median-centered intensity), †P-value, and ‡Pearson's correlation coefficient of Individual dataset were obtained from the Oncomine database.
VEGF-C enhances the cancer stemness properties in skin cancer
Emerging evidence has suggested that EMT inducers not only contribute cell motility but also regulate stem cell properties and cell survival [34–37]. Cancer stem cells (CSCs) have been described as vital mediators of cell mobility in various cancers including lung, breast and skin [26, 38, 39]. Therefore, we examined whether VEGF-C affects cancer stemness in skin cancer cells and found that knockdown of VEGF-C significantly decreased the expression of the CSC markers SOX2, OCT4, KLF4, NANOG, CD133, CD34 and CD44 as well as ALDH activity, which is a hallmark of CSCs (Figure 2a-2h). As expected, rhVEGF-C treatment restored CSC marker expression and ALDH activity in VEGF-C-knockdown cells (Figure 2a-2h). An Oncomine database analysis also demonstrated that VEGF-C expression was positively correlated with the expression of some CSC markers, such as OCT4, KLF4, NANOG, CD44, CD34 and CD133 (Supplementary Figure S1a-S1f), suggesting that VEGF-C increases the CSC features of skin cancer cells.
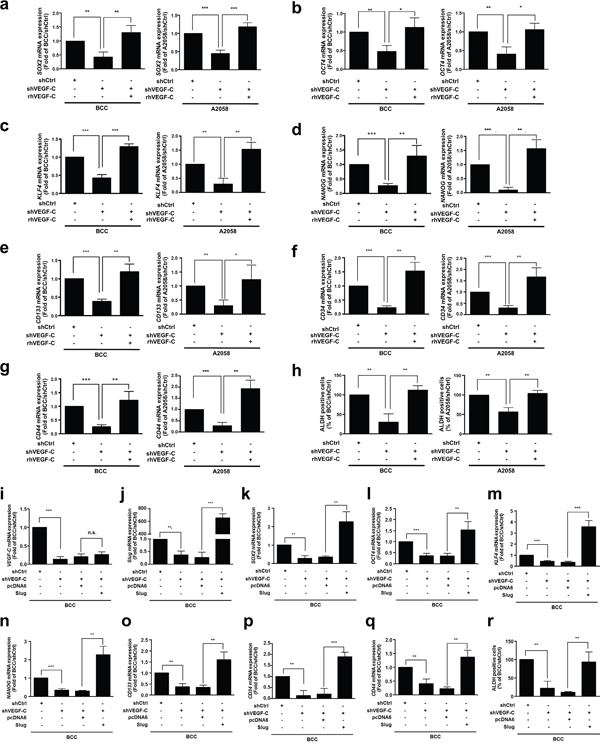
Figure 2: VEGF-C enhances the cancer stemness properties in skin cancer. a-g. QRT-PCR analysis of the SOX2, OCT4, KLF4, NANOG, CD133, CD34 and CD44 mRNA expression in indicated cells with rhVEGF-C (200 ng/ml) treatment. h. ALDH activity of the indicated cells was measured by flow cytometry. i-q. Expression of VEGF-C (i) and Slug (j) was detected, and Slug expression restores the expression of CSC markers (SOX2 (k), OCT4 (l), KLF4 (m), NANOG (n), CD133 (o), CD34 (p) and CD44 (q)) in indicated cells by QRT-PCR analysis. r. Slug expression recovers ALDH activity in indicated cells by flow cytometry.
Next, to determine the effects of Slug on VEGF-C/VEGFR3-mediated cancer stemness in skin cancer cells, we re-expressed Slug in VEGF-C-knockdown BCC cells. The results showed that Slug recovered CSC marker expression and ALDH activity (Figure 2i-2r), suggesting that Slug is a key downstream factor in the VEGF-C/VEGFR pathway.
VEGF-C increases slug expression, cell migration, invasion and stemness through YAP1
According to our analysis of Oncomine datasets, we found that YAP1 was highly expressed in metastatic skin cancer tissues (Supplementary Figure S2a). We next verified the relationship between VEGF-C, YAP1 and Slug by overexpression of YAP1 in VEGF-C-knockdown BCC cells and found that YAP1 restored the shVEGF-C-suppressed Slug expression (Figure 3a). Interestingly, depletion of VEGF-C also decreased YAP1 expression, whereas YAP1 did not affect the VEGF-C expression (Figure 3a and Supplementary Figure S2b). Online datasets further demonstrated a positive correlation between YAP1 and Slug, also known as SNAI2 (Supplementary Figure S2c-S2g). Moreover, YAP1 overexpression improved the migration and invasion abilities that had been abrogated by shVEGF-C in BCC cells (Figure 3b).
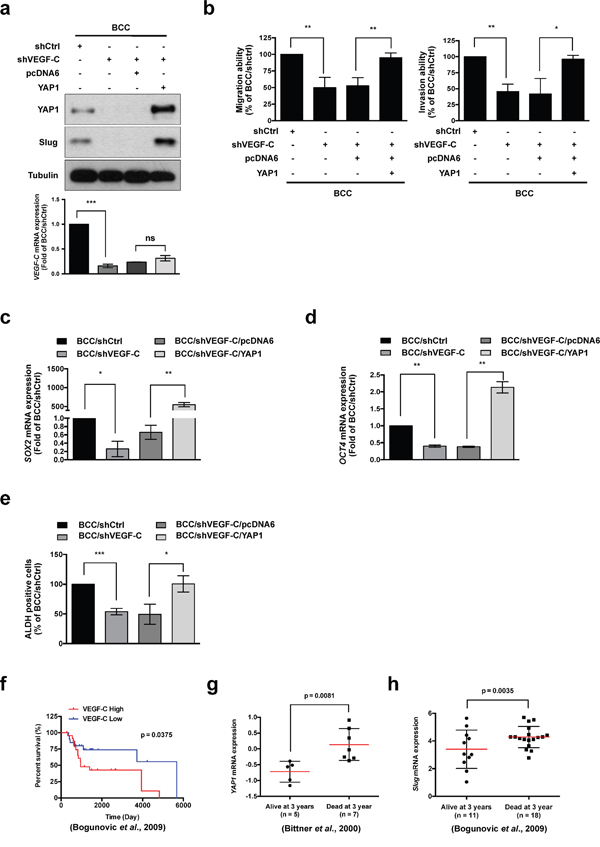
Figure 3: VEGF-C increases Slug expression, cell migration, invasion and stemness through YAP1. a. (Upper panel) Western blot analysis of Slug and YAP1 in indicated cells. Tubulin was used as a loading control. (Lower panel) QRT-PCR analysis of VEGF-C expression in indicated cells. b. A transwell assay demonstrated the migration and invasion abilities of indicated cells. c and d. QRT-PCR analysis of SOX2 and OCT4 mRNA expression in indicated cells. e. ALDH activity of indicated cells was determined by flow cytometry. f-h. The Oncomine database was used to analyze the correlation of VEGF-C, YAP1 and Slug with survival rate in skin cancer patients. The results are shown as the mean ± SD of three independent experiments, each performed in triplicate. *P < 0.05, **P < 0.001, ***P < 0.0001 (Student’s t test).
We further elucidated whether CSC markers and ALDH activity are affected by YAP1 in BCC cells. Loss of VEGF-C expression diminished the expression of the CSC markers SOX2 and OCT4, consistent with a reduction in ALDH activity, whereas YAP1 overexpression abolished these inhibitory effects (Figure 3c-3e). Analysis of the clinical data in the Oncomine datasets also demonstrated a positive correlation between YAP1 and CSC markers such as OCT4, SOX2, NANOG, KLF4, ALDHA1, CD44 and CD133 (Table 2). Consistently, the expression of Slug was positively correlated with the expression of CSC markers such as CD44, NANOG, ALDH1A1 and KLF4 in skin cancer (Table 3). Most importantly, the analysis of online datasets indicated that high expression of VEGF-C, YAP1 and Slug was associated with survival rate in skin cancer patients (Figure 3f-3h). Taken together, these results demonstrated that the VEGF-C/VEGFR3 signaling pathway affects cell mobility, cancer stemness properties and progression by the regulation of Slug through YAP1 in skin cancer.
Table 2: The association between stem cell markers and YAP1 expression in skin cancer datasets
Stem cell markers |
Datasets |
†Pearson correlation, r |
‡P-value |
|---|---|---|---|
OCT4 |
Riker (n = 87) |
0.3445 |
0.0011 |
SOX2 |
Beasley (n = 28) |
0.5504 |
0.0024 |
Nindl (n = 15) |
0.5191 |
0.0474 |
|
NANOG |
Riker (n = 87) |
0.4529 |
< 0.0001 |
KLF4 |
Riker (n = 87) |
0.5593 |
< 0.0001 |
Talantov (n = 70) |
0.3372 |
0.0043 |
|
ALDH1A1 |
Beasley (n = 28) |
0.5799 |
0.0012 |
Bogunovic (n = 44) |
0.3364 |
0.0256 |
|
CD44 |
Bittner (n = 31) |
0.5683 |
0.0009 |
Bogunovic (n = 44) |
0.4901 |
0.0007 |
|
Riker (n = 87) |
0.2940 |
0.0057 |
|
Smith (n = 18) |
0.5956 |
0.0091 |
|
Talantov (n = 70) |
0.4553 |
< 0.0001 |
|
Xu (n = 83) |
0.3048 |
0.0051 |
|
Nindl (n = 15) |
0.7799 |
0.0006 |
|
CD133 |
Smith (n = 18) |
0.6350 |
0.0046 |
†r, Pearson's correlation coefficient and ‡P-value for two-tailed Student’s t test of Individual dataset (Oncomine database). P value < 0.05 was considered statistically significant.
Table 3: The association between stem cell markers and Slug expression in skin cancer datasets
Stem cell markers |
Datasets |
†Pearson correlation, r |
‡P-value |
|---|---|---|---|
CD44 |
Beasley (n = 28) |
0.6761 |
< 0.0001 |
Bogunovic (n = 44) |
0.4893 |
0.0007 |
|
Haqq (n = 28) |
0.4985 |
0.0069 |
|
Harlin (n = 44) |
0.3852 |
0.0098 |
|
Riker (n = 86) |
0.2308 |
0.0325 |
|
Xu (n = 82) |
0.3626 |
0.0008 |
|
Talantov (n = 70) |
0.6651 |
< 0.0001 |
|
NANOG |
Beasley (n = 28) |
0.4171 |
0.0272 |
Harlin (n = 44) |
0.3198 |
0.0343 |
|
Riker (n = 86) |
0.3112 |
0.0035 |
|
ALDH1A1 |
Bogunovic (n = 44) |
0.3042 |
0.0447 |
KLF4 |
Riker (n = 86) |
0.4082 |
< 0.0001 |
†r, Pearson's correlation coefficient and ‡P-value for two-tailed Student’s t test of Individual dataset (Oncomine database). P value < 0.05 was considered statistically significant.
YAP1 is regulated by VEGF-C via the RAS/MAPK pathway
YAP1 was previously defined as a key effector of oncogenic RAS/MAPK signaling, which promotes cell proliferation and motility [24, 40]. We next assessed whether RAS is involved in VEGF-C-regulated YAP1 expression. The phosphorylation of ERK was decreased in VEGF-C-knockdown BCC cells, but recovered by overexpression of constitutively activated KRASG12V [41, 42] (Figure 4a). Moreover, KRASG12V abrogated the reduction of YAP1 and Slug expression driven by VEGF-C knockdown (Figure 4a). To address the clinical relevance of VEGF-C, YAP1, Slug and the RAS/MAPK pathway in clinical samples, the public database showed that VEGF-C was positively correlated with KRAS, MAPK1 and YAP1 expression (Figure 4b-4d). Furthermore, expression of Slug was positively correlated with KRAS and MAPK1 expression in skin cancer (Supplementary Figure S3 and S4). KRAS expression was also significantly correlated with clinical-pathologic features such as metastatic event and skin cancer (Table 4). Consequently, these results indicate that VEGF-C regulates YAP1 and Slug expression via the RAS/MAPK pathway to enhance cell mobility and cancer stemness in skin cancer cells.
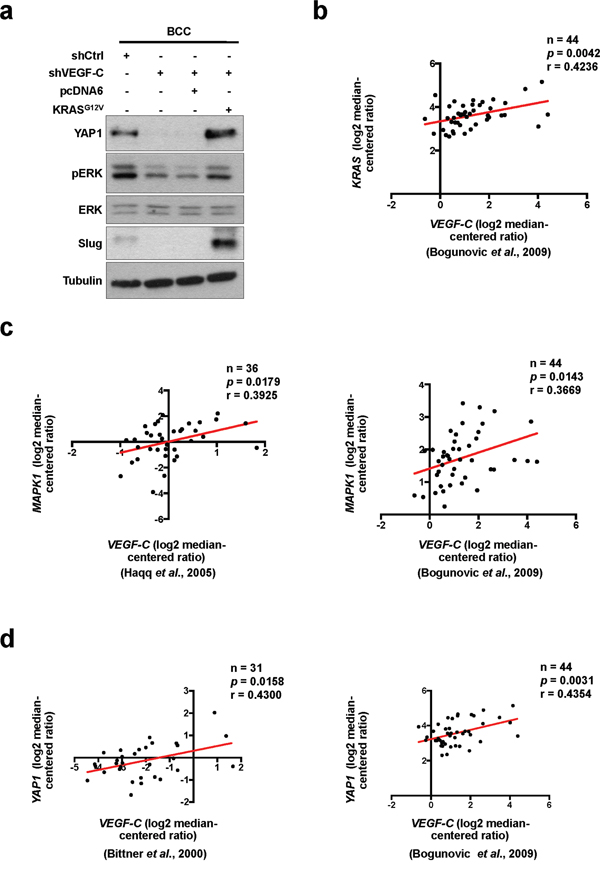
Figure 4: YAP1 is regulated by VEGF-C via the RAS/MAPK pathway. a. Western blot analysis of indicated proteins in VEGF-C-knockdown cells with or without KRASG12V overexpression. b-d. The Oncomine dataset analysis showed a positive correlation of VEGF-C with KRAS (b), MAPK1 (c) and YAP1 (d) in skin cancer tissues.
Table 4: Clinicopathological features, clinical outcomes and their association with KRAS expression in skin cancer datasets
Datasets |
Clinicopathological features |
*KRAS expression |
†P-value |
|---|---|---|---|
Xu (n = 154) |
Metastasis |
1.527 |
1.67E-7 |
Laurent (n = 63) |
Metastatic Event at 1 Year |
1.511 |
0.041 |
Talantov (n = 70) |
Benign Melanocytic Skin Nevus |
1.650 |
4.32E-4 |
Cutaneous Melanoma |
1.619 |
6.51E-4 |
|
Nindl (n = 15) |
Skin Squamous Cell Carcinoma |
3.039 |
0.020 |
Riker (n = 87) |
Skin Squamous Cell Carcinoma |
1.641 |
0.002 |
Skin Basal Cell Carcinoma |
1.380 |
0.010 |
*Fold change (Log2 median-centered intensity), †P-value, and ‡Pearson's correlation coefficient of Individual dataset were obtained from the Oncomine database.
Anti-VEGFR3 peptide represses VEGF-C-induced signaling, cell mobility and cancer stemness in skin cancer
In our previous study, we demonstrated that a small peptide specifically inhibits VEGF-C/VEGFR3-mediated signaling, migration and invasion in cancer cells [26]. We observed that the anti-VEGFR3 peptide significantly blocked the rhVEGF-C-induced migration and invasion abilities of BCC cells (Figure 5a). We further analyzed the effects of the anti-VEGFR3 peptide on VEGF-C induced signaling and found that treatment with the anti-VEGFR3 peptide abolished the rhVEGF-C-induced phosphorylation of VEGFR3 and ERK. VEGF-C-induced YAP1 and Slug expression were also repressed by the anti-VEGFR3 peptide (Figure 5b). Since YAP1 acts as a critical downstream mediator of VEGF-C/VEGFR3 signaling, we overexpressed YAP1 in BCC cells and found that YAP1 restored anti-VEGFR3 peptide-inhibited SOX2 and OCT4 expression and ALDH activity, as well as migration and invasion abilities (Figure 5c-5e). These data confirm that the anti-VEGFR3 peptide abolishes activation of the VEGF-C/VEGFR3-KRAS/MAPK signaling pathway to decrease the YAP1-mediated Slug expression, cancer stemness, migration and invasion of skin cancer cells.
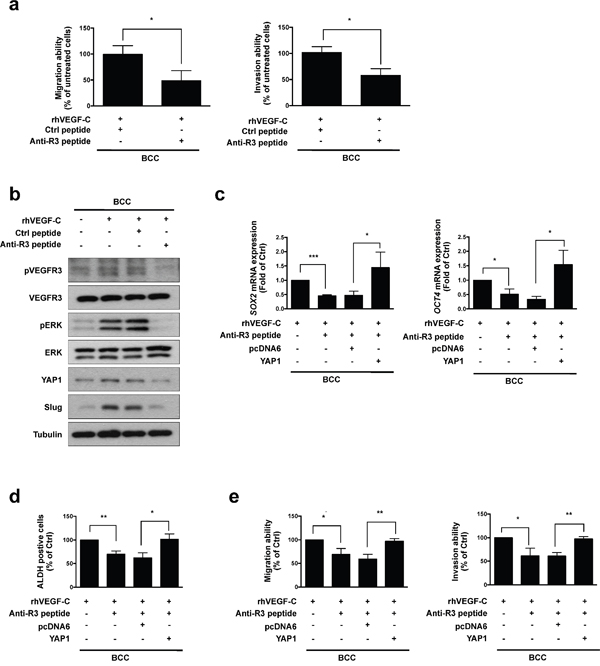
Figure 5: Anti-VEGFR3 peptide represses VEGF-C-induced signaling, cell mobility and cancer stemness in skin cancer. a. A transwell assaydemonstrated the migration and invasion abilities in BCC cells pre-incubated with rhVEGF-C followed by anti-VEGFR3 (anti-R3) or control (Ctrl) peptide treatment. b. Western blot analysis of indicated proteins in rhVEGF-C-pre-incubated cells treated with anti-R3 or Ctrl peptide. c. qRT-PCR analysisof SOX2 and OCT4 mRNA expression in indicated cells. d. ALDH activity in indicated cells was analyzed by flow cytometry. e. A transwell assay showed migration and invasion abilities in indicated cells. The results are shown as the mean ± SD of three independent experiments, each performed in triplicate. *P < 0.05, **P < 0.001, ***P < 0.0001 (Student’s t test).
DISCUSSION
Elevated YAP1 expression has been shown to induce cell proliferation and ECM remodeling through CCN1 and CCN2 expression in BCC [43]. Consistent with previous findings, we further demonstrated that VEGF-C-mediated YAP1 expression increases Slug expression and contributes to the migration and invasion abilities of skin cancer cells. Previous reports have indicated that YAP1 expression not only positively correlates with the expression of OCT4 and SOX2 and is involved in self-renewal properties and differentiation of embryonic stem cells [44] but also promotes oncogenic transformation by interfering with the Hippo signaling pathway [45]. It has recently been proposed that migrating cancer cells that contain stem-like properties might divide asymmetrically; one daughter cell starts proliferation and differentiation, the remaining cell can either undergoes a new asymmetric division or produces metastasis [46]. In the present study, we showed that VEGF-C-induced YAP1 expression modulates Slug expression to enhance cell migration and invasion, which further increases cancer stemness to promote skin cancer progression (Figure 6), suggesting that regulation of YAP1 is crucial for studying tumorigenicity of skin cancer and is a potential target of therapeutic strategies.
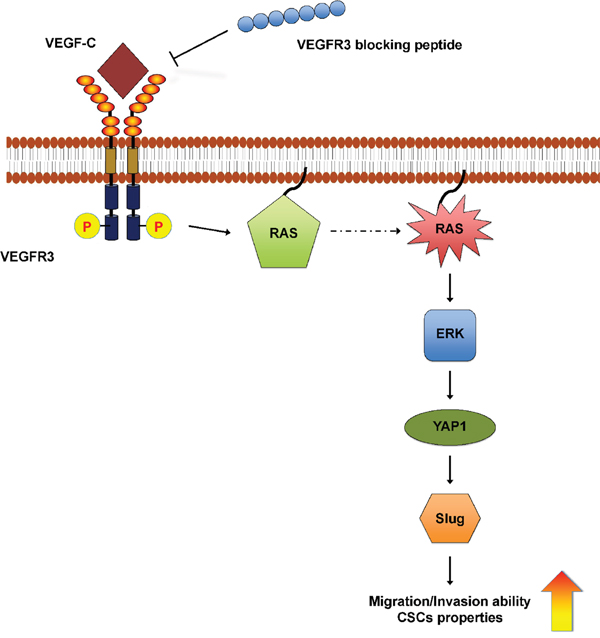
Figure 6: A schematic model of the VEGF-C/VEGFR3-mediated KRAS/MAPK-YAP1/Slug pathway in skin cancer progression. In skin cancer cells, VEGF-C/VEGFR3 signaling activates the KRAS/MAPK axis to up-regulate YAP1 and Slug expression, further enhancing cell mobility and cancer stemness. Anti-VEGFR3 peptide functions as a repressor and could be a therapeutic strategy.
We have successfully developed an anti-VEGFR3 peptide that blocks VEGF-C/VEGFR3 axis-induced tumor initiation, migration, invasion and chemoresistance [26], suggesting that the anti-VEGFR3 peptide has the potential to serve as a novel therapy for skin cancer. In a previous study, high VEGF-C expression in patients with metastatic melanoma was shown to correlate with lymph node localization [18]. Elevated VEGF-C leads to enhanced tumor lymphangiogenesis and metastasis in SCC [47], suggesting that VEGF-C may be a biomarker of tumor metastasis and serve as a novel therapeutic target in the treatment of skin cancer. A recent study indicates that VEGF-C/VEGF-D regulate the early stages of skin cancer progression and inhibition of VEGF-C/VEGF-D by a soluble Fc-fused form of VEGFR3 significantly suppressed chemical-induced SCC carcinogenesis in an animal model in vivo [48]. Consistently, our findings demonstrate that the novel and simply prepared anti-VEGFR3 peptide is able to significantly block the VEGF-C/VEGFR3 pathway, which further inhibits cancer stemness, migration and invasion in skin cancer cells.
BCC is the most common non-melanoma skin cancer in humans, which morbidity is growing annually worldwide. In some cases, BCC can be highly invasive due to genetic alterations in the Hedgehog (HH) signaling pathway [3]. Although several pathways associated with BCC are known, the mechanisms involved in the morphology, aggressiveness and response to treatment in BCC still need to be elucidated. KRAS activation regulates many cell properties, such as cell growth, division and survival, through many critical signaling pathways [49]. Significant evidence suggests that mutation of KRAS causes constitutive activation and persistent stimulation of downstream signaling pathways, and this phenomenon can be found in many types of cancer [50–52]. An increasing number of studies also indicate that KRAS mutations (G12C or G12D) are required for BCC, SCC and melanoma development [53–55]. Bonilla et al. showed that mutations in the RTK-RAS-PI3K pathway are involved in BCC tumor progression [56]. Moreover, KRAS is a critical regulator for the maintenance of mesenchymal phenotypes and metastatic ability in skin, breast, colon and pancreatic cancers [40, 57–59]. KRAS-mediated Slug expression is necessary for the EMT processes [40, 58]. Interestingly, KRAS has been reported to regulate the activity of YAP1 through the MAPK pathway [24]; however, a link between VEGF-C, KRAS and YAP1 has not been established. Our study confirms that VEGF-C is a critical inducer of cancer stemness, metastasis and invasion in skin cancer through regulating YAP1 expression via KRAS/MAPK signaling.
VEGF-A is reported to be an inducer of EMT through enhancing Snail expression in breast cancer cells, partly by inhibiting GSK3β activity [60, 61]. VEGF-C can induce vimentin and N-cadherin expression and suppress cytokeratins and E-cadherin expression, which are common markers used for the detection of EMT [62]. In the present study, we demonstrate that VEGF-C induces Slug expression to increase EMT in skin cancer. EMT is also suggested to confer CSC qualities. In the tumor microenvironment, several soluble factors are indicated to orchestrate EMT and induce CSC production from differentiated cancer cells [63]. For example, HGF in colon cancer cells [64] and TGF-β in mammary epithelial cells [65] can induce the EMT-CSC program. Our findings exhibit that VEGF-C can induce CSC features in skin cancer cells (Figure 2). However, it is not clear whether VEGF-C induces EMT and cancer stemness through identical or similar molecular pathway, or if the regulation of EMT-TFs modulates CSC features.
In summary, our study reveals a novel mechanism by which the VEGF-C/VEGFR3 axis regulates Slug expression in skin cancer through the KRAS/MAPK-YAP1 pathway and contributes to the cancer stemness and metastasis of skin cancer. Moreover, these findings suggest that targeting the VEGFR3 signaling pathway with an anti-VEGFR3 peptide may be a potential therapeutic strategy for skin cancer treatment.
MATERIALS AND METHODS
Antibodies and reagents
Recombinant human VEGF-C protein (rhVEGF-C) was purchased from Sino Biological (Beijing, China). Western blot analyses were performed with the following antibodies: VEGFR3 (LifeSpan BioSciences, WA, USA), p-VEGFR3 (Y1063/1068, Cell Applications, CA, USA), YAP1 (#4912, Cell signaling, Beverly, MA, USA), p44/p42 MAP Kinase (ERK) (#9102, Cell signaling, Beverly, MA, USA), Phospho-p44/p42 MAPK (p-ERK) (Thr202/Tyr204, #4377, Cell signaling, Beverly, MA, USA), Vimentin (Neomarkers, Fremont, CA, USA), Snail (GTX81086, GeneTex, San Antonio, TX, USA), Slug (#9585, Cell signaling, Beverly, MA, USA), Twist (sc-81417, Santa Cruz Biotechnology, Inc.) and α-Tubulin (T5168, Sigma-Aldrich, St. Louis, MO, USA). The sequences of the peptides selected by binding to VEGFR3 are as follows: Ctrl peptide (peptide 4: CNDSHMKLC) and anti-VEGFR3 (anti-R3) peptide (peptide 5: CNYSHLFYC) [26].
Cell culture and transfection
The human basal cell carcinoma cell line BCC was a kind gift from Dr. Min-Liang Kuo [66], the human melanoma cell line A2058 (ATCC CRL-11147) was kindly provided by Dr. Wun-Shaing Chang [67], and the human embryonic kidney cell line 293T (ATCC CRL-3216) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). BCC cells were cultured in RPMI/1640 medium, A2058 cells were cultured in DMEM/F12 (1:1) medium, and 293T cells were cultured in high-glucose/DMEM medium. All cells were incubated at 37°C in a humidified 5% CO2 atmosphere, and the media were supplemented with 10% fetal bovine serum (FBS, (Carlsbad, CA, USA) and 1% penicillin/streptomycin (Carlsbad, CA, USA).
Plasmid and shRNA clones
The YAP1 expression plasmid was purchased from Bioresource Collection and Research Center, Hsinchu, Taiwan. For establishment of the VEGF-C-knockdown skin cancer cells, lentiviral VEGF-C shRNA clones TRCN0000058505 and TRCN0000058507 were purchased from National RNAi Core Facility at Academia Sinica, Taipei, Taiwan.
Lentivirus production and infection
Recombinant lentiviruses were packaged according to a previously described method [39]. Briefly, HEK293T cells were co-transfected with mixtures of the indicated expressing plasmid, the pCMV plasmid and the packaging plasmid (pVSVG) using PEI (Sigma-Aldrich). The viruses were collected from the culture medium on day 2 after transfection and filtered with a 0.45-μm filter. For lentivirus infection, the BCC and A2058 cells were infected with 3 ml lentivirus supernatant and 2 μl of polybrene (Hexadimethrine bromide, Sigma) and after 48 hours the stably transduced cells were selected by treatment with 5 μg/ml puromycin.
RNA Isolation and reverse transcription and quantitative RT-PCR
Total RNA was isolated using Trizol (Invitrogen) and cDNAs were reverse transcribed by M-MLV reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Quantitative real-time PCR was performed for at least three independent experiments, each performed in triplicate, by the LightCycler 480 (Roche). For mRNA detection, PCR reactions contained 2 μl of cDNA, 0.5 μM of each forward and reverse primer, 1 μM Universal ProbeLibrary Probe (Roche) and 1× LightCycler TaqMan Master mix. Amplification curves were generated with an initial denaturing step at 95°C for 10 minutes, followed by 50 cycles of 95°C for 5 seconds, 60°C for 10 seconds, and 72°C for 1 second. The GAPDH gene was used as an internal control. The PCR primers for the gene expression analysis are as follows: VEGF-C (forward primer: tgccagcaacactaccacag and reverse primer: gtgattattccacatgtaattggtg), SOX2 (forward primer: atgggttcggtggtcaagt and reverse primer: actctggggctccttcttg), OCT4 (forward primer: gaggcaacctggagaatttg and reverse primer: cagaaccacactcggacca), and GAPDH (forward primer: agccacatcgctcagacac and reverse primer: gcccaatacgaccaaatcc). The relative levels of gene expression were calculated by 2-ΔCT = CT of the target gene – CT of the GAPDH gene.
Western blotting analysis
Western blot analysis was performed as previously described [68]. Briefly, cells were washed twice with PBS and lysed with RIPA lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA, 10 mM Na3VO4 and 1× protease inhibitor cocktail (Sigma)). Cell lysates were sonicated for 1 minute and then centrifuged at 13,000 ×g for 30 minutes. An equal amount of total protein was separated by SDS-PAGE and transferred to PVDF membranes. Primary antibodies were incubated overnight at 4°C and followed by the appropriate secondary antibodies conjugated to horseradish peroxidase. The indicated proteins were visualized using ECL reagents (Millipore Forte (WBLUF0500); Millipore Crescendo (WBLUR0500)).
Transwell migration and invasion assays
Transwell assays were performed as described previously [26]. A2058 cells (2 × 104 cells) or BCC cells (3 × 104 Cells) were seeded in the upper well (Corning Costar; Lowell, MA, USA) with serum-free medium for the migration assay or a matrigel-coated membrane for the invasion assay, followed by addition of 10% FBS-containing medium in the lower well. The images of cells migrating or invading through the lower membrane were taken by light microscope.
ALDH activity assay
ALDH enzymatic activity was determined by the Aldefluor kit (StemCell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. ALDH-positive cells were detected in the green fluorescence channel (FL1) by flow cytometry analysis. The specific ALDH inhibitor (diethylaminobenzaldehyde (DEAB))-treated cells were used as the control to set the gates defining the ALDH-positive region. The results are representative of three independent experiments, each performed in triplicate.
Cell proliferation assay
Cell proliferation was examined by MTT assay. Cells (5 × 103) were seeded in 96-well culture plates for the indicated time points, and then incubated with 5 mg/ml MTT reagent. After 4 hours, the supernatants were removed and 100 μl DMSO was added to dissolve the crystals. The absorbance in each well was measured at a wavelength of 570 nm with background subtraction at 630 nm by an ELISA reader.
Statistical analysis
All statistical analyses were performed with Prism 6 software (La Jolla, CA). Data were analyzed as the mean ± SD. The two-tailed Student’s t test was used to analyze the means of the different two groups. A P value of less than 0.05 was considered statistically significant.
Abbreviations
VEGF-C, Vascular endothelial growth factor-C; EMT, epithelial-mesenchymal transition; MAPK, mitogen-activated protein kinases; YAP1, yes-associated protein 1; VEGFR3, VEGF receptor 3; SCC, squamous cell carcinoma; BCC, basal cell carcinoma; MBCC, metastatic BCC; PlGF, placental growth factor; rhVEGF-C, recombinant human VEGF-C; EMT-TFs, EMT-inducing transcription factors; CSCs, cancer stem cells; HH, Hedgehog.
ACKNOWLEDGMENTS
This work was supported by Tri-Service General Hospital Songshan Branch, National Defense Medical Center from Taiwan (No. 10307 and 10408); Ministry of Science and Technology (National Science Council) grants from Taiwan (NSC 101-2320-B-400-016-MY3, NSC 102-2314-B-038-028-MY3, NSC 103-2314-B-038-059); National Health Research Institutes grant from Taiwan (CA-102-PP-41, CA-104-SP-01, CA-104-PP-12, MOHW104-TDU-B-212-124-008).
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Author contributions
Conception and design: YWY and JLS; Development of methodology: YWY, CCC, EF, CPC and JLS; Acquisition of data: YWY, CCC and STY; Analysis and interpretation of data: CCC, STY and SYT; Writing, review, and/or revision of the manuscript: CCC, STY, CFT, TYC and JLS; Administrative, technical, or material support: LCL, PWT, YLY and JLS; Study supervision: YLY and JLS.
REFERENCES
1. Khavari PA. Modelling cancer in human skin tissue. Nature reviews Cancer. 2006; 6:270-280.
2. Yeh Y-W, Chen S-Y, Wu B-Y, Gao H-W, Liu C-Y, Chien W-C, Chiang C-P. Epidemiologic and pathologic characteristics of basal cell carcinoma in northern Taiwan: Experience from a medical center. Journal of Medical Sciences. 2014; 34:98-103.
3. Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, Low JA, Mackey HM, Yauch RL, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. The New England journal of medicine. 2012; 366:2171-2179.
4. Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. The New England journal of medicine. 2005; 353:2262-2269.
5. Conde DM, Kashimoto E, Torresan RZ, Alvarenga M. Pseudomamma on the foot: an unusual presentation of supernumerary breast tissue. Dermatology online journal. 2006; 12:7.
6. Ganti AK, Kessinger A. Systemic therapy for disseminated basal cell carcinoma: an uncommon manifestation of a common cancer. Cancer treatment reviews. 2011; 37:440-443.
7. Walling HW, Fosko SW, Geraminejad PA, Whitaker DC, Arpey CJ. Aggressive basal cell carcinoma: presen ation, pathogenesis, and management. Cancer Metastasis Rev. 2004; 23:389-402.
8. Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology (Williston Park). 2009; 23:488-496.
9. Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003; 9:685-693.
10. Su JL, Yen CJ, Chen PS, Chuang SE, Hong CC, Kuo IH, Chen HY, Hung MC, Kuo ML. The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br J Cancer. 2007; 96:541-545.
11. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nature reviews Cancer. 2008; 8:579-591.
12. Dias S, Choy M, Alitalo K, Rafii S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002; 99:2179-2184.
13. Su JL, Chen PS, Chien MH, Chen PB, Chen YH, Lai CC, Hung MC, Kuo ML. Further evidence for expression and function of the VEGF-C/VEGFR-3 axis in cancer cells. Cancer cell. 2008; 13:557-560.
14. Sun P, Gao J, Liu YL, Wei LW, Wu LP, Liu ZY. RNA interference (RNAi)-mediated vascular endothelial growth factor-C (VEGF-C) reduction interferes with lymphangiogenesis and enhances epirubicin sensitivity of breast cancer cells. Mol Cell Biochem. 2008; 308:161-168.
15. Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, Hung MC, Kuo ML. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer cell. 2006; 9:209-223.
16. Hong CC, Chen PS, Chiou J, Chiu CF, Yang CY, Hsiao M, Chang YW, Yu YH, Hung MC, Hsu NW, Shiah SG, Hsu NY, Su JL. miR326 maturation is crucial for VEGF-C-driven cortactin expression and esophageal cancer progression. Cancer research. 2014; 74:6280-6290.
17. Su CM, Su YH, Chiu CF, Chang YW, Hong CC, Yu YH, Ho YS, Wu CH, Yen CS, Su JL. Vascular endothelial growth factor-C upregulates cortactin and promotes metastasis of esophageal squamous cell carcinoma. Ann Surg Oncol. 2014; 21:S767-775.
18. Schietroma C, Cianfarani F, Lacal PM, Odorisio T, Orecchia A, Kanitakis J, D'Atri S, Failla CM, Zambruno G. Vascular endothelial growth factor-C expression correlates with lymph node localization of human melanoma metastases. Cancer. 2003; 98:789-797.
19. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, Issa A, Hugues S, Swartz MA. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012; 1:191-199.
20. Kang W, Tong JH, Chan AW, Lee TL, Lung RW, Leung PP, So KK, Wu K, Fan D, Yu J, Sung JJ, To KF. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin Cancer Res. 2011; 17:2130-2139.
21. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:E2441-2450.
22. Liu R, Huang S, Lei Y, Zhang T, Wang K, Liu B, Nice EC, Xiang R, Xie K, Li J, Huang C. FGF8 promotes colorectal cancer growth and metastasis by activating YAP1. Oncotarget. 2015; 6:935-952. doi: 10.18632/oncotarget.2822.
23. Saucedo LJ, Edgar BA. Filling out the Hippo pathway. Nature reviews Molecular cell biology. 2007; 8:613-621.
24. Zhang H, von Gise A, Liu Q, Hu T, Tian X, He L, Pu W, Huang X, He L, Cai CL, Camargo FD, Pu WT, Zhou B. Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. The Journal of biological chemistry. 2014; 289:18681-18692.
25. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007; 17:2054-2060.
26. Chang YW, Su CM, Su YH, Ho YS, Lai HH, Chen HA, Kuo ML, Hung WC, Chen YW, Wu CH, Chen PS, Su JL. Novel peptides suppress VEGFR-3 activity and antagonize VEGFR-3-mediated oncogenic effects. Oncotarget. 2014; 5:3823-3835. doi: 10.18632/oncotarget.1709.
27. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. 2005; 24:7443-7454.
28. Ansieau S, Collin G, Hill L. EMT or EMT-Promoting Transcription Factors, Where to Focus the Light? Frontiers in oncology. 2014; 4:353.
29. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nature cell biology. 2014; 16:488-494.
30. Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, Li KC, Hong TM, Yang PC. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nature cell biology. 2009; 11:694-704.
31. Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front Biosci (Landmark Ed). 2009; 14:3035-3050.
32. Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ, Ellis LM. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer research. 2006; 66:46-51.
33. Yang HW, Menon LG, Black PM, Carroll RS, Johnson MD. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer. 2010; 10:301.
34. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715.
35. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS one. 2008; 3:e2888.
36. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nature cell biology. 2009; 11:1487-1495.
37. Preca BT, Bajdak K, Mock K, Sundararajan V, Pfannstiel J, Maurer J, Wellner U, Hopt UT, Brummer T, Brabletz S, Brabletz T, Stemmler MP. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. International journal of cancer. 2015; 137:2566-2577.
38. Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annual review of cell and developmental biology. 2007; 23:675-699.
39. Chang YW, Chiu CF, Lee KY, Hong CC, Wang YY, Cheng CC, Jan YH, Huang MS, Hsiao M, Ma JT, Su JL. CARMA3 Represses Metastasis Suppressor NME2 to Promote Lung Cancer Stemness and Metastasis. Am J Respir Crit Care Med. 2015; 192:64-75.
40. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T, Hahn WC. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014; 158:171-184.
41. Zhou Y, Rideout WM, 3rd, Zi T, Bressel A, Reddypalli S, Rancourt R, Woo JK, Horner JW, Chin L, Chiu MI, Bosenberg M, Jacks T, Clark SC, Depinho RA, Robinson MO, Heyer J. Chimeric mouse tumor models reveal differences in pathway activation between ERBB family- and KRAS-dependent lung adenocarcinomas. Nature biotechnology. 2010; 28:71-78.
42. Fatrai S, van Gosliga D, Han L, Daenen SM, Vellenga E, Schuringa JJ. KRAS(G12V) enhances proliferation and initiates myelomonocytic differentiation in human stem/progenitor cells via intrinsic and extrinsic pathways. The Journal of biological chemistry. 2011; 286:6061-6070.
43. Quan T, Xu Y, Qin Z, Robichaud P, Betcher S, Calderone K, He T, Johnson TM, Voorhees JJ, Fisher GJ. Elevated YAP and its downstream targets CCN1 and CCN2 in basal cell carcinoma: impact on keratinocyte proliferation and stromal cell activation. Am J Pathol. 2014; 184:937-943.
44. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, Ding S, Guan KL. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes & development. 2010; 24:1106-1118.
45. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010; 19:491-505.
46. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nature reviews Cancer. 2005; 5:744-749.
47. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. 2007; 109:1010-1017.
48. Alitalo AK, Proulx ST, Karaman S, Aebischer D, Martino S, Jost M, Schneider N, Bry M, Detmar M. VEGF-C and VEGF-D blockade inhibits inflammatory skin carcinogenesis. Cancer research. 2013; 73:4212-4221.
49. Jancik S, Drabek J, Radzioch D, Hajduch M. Clinical relevance of KRAS in human cancers. Journal of biomedicine & biotechnology. 2010; 2010:150960.
50. Downward J. Targeting RAS signalling pathways in cancer therapy. Nature reviews Cancer. 2003; 3:11-22.
51. Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, Luoh SM, Anderson DJ, Ludlam MJ, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013; 501:232-236.
52. Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014; 111:817-822.
53. Milagre C, Dhomen N, Geyer FC, Hayward R, Lambros M, Reis-Filho JS, Marais R. A mouse model of melanoma driven by oncogenic KRAS. Cancer research. 2010; 70:5549-5557.
54. Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, Blanpain C. Identifying the cellular origin of squamous skin tumors. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:7431-7436.
55. van der Schroeff JG, Evers LM, Boot AJ, Bos JL. Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. The Journal of investigative dermatology. 1990; 94:423-425.
56. Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, Ribaux PG, Popadin K, Basset-Seguin N, Ben Chaabene R, Santoni FA, Andrianova MA, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nature genetics. 2016; 48:398-406.
57. Wong CE, Yu JS, Quigley DA, To MD, Jen KY, Huang PY, Del Rosario R, Balmain A. Inflammation and Hras signaling control epithelial-mesenchymal transition during skin tumor progression. Genes & development. 2013; 27:670-682.
58. Kim RK, Suh Y, Yoo KC, Cui YH, Kim H, Kim MJ, Gyu Kim I, Lee SJ. Activation of KRAS promotes the mesenchymal features of basal-type breast cancer. Experimental & molecular medicine. 2015; 47:e137.
59. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell. 2005; 7:469-483.
60. Wanami LS, Chen HY, Peiro S, Garcia de Herreros A, Bachelder RE. Vascular endothelial growth factor-A stimulates Snail expression in breast tumor cells: implications for tumor progression. Experimental cell research. 2008; 314:2448-2453.
61. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014; 15:178-196.
62. Khromova N, Kopnin P, Rybko V, Kopnin BP. Downregulation of VEGF-C expression in lung and colon cancer cells decelerates tumor growth and inhibits metastasis via multiple mechanisms. Oncogene. 2012; 31:1389-1397.
63. Medema JP. Cancer stem cells: the challenges ahead. Nature cell biology. 2013; 15:338-344.
64. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G, Medema JP. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature cell biology. 2010; 12:468-476.
65. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011; 145:926-940.
66. Jee SH, Shen SC, Chiu HC, Tsai WL, Kuo ML. Overexpression of interleukin-6 in human basal cell carcinoma cell lines increases anti-apoptotic activity and tumorigenic potency. Oncogene. 2001; 20:198-208.
67. Chang YP, Mahadeva R, Chang WS, Lin SC, Chu YH. Small-molecule peptides inhibit Z alpha1-antitrypsin polymerization. J Cell Mol Med. 2009; 13:2304-2316.
68. Yu YL, Su KJ, Chen CJ, Wei CW, Lin CJ, Yiang GT, Lin SZ, Harn HJ, Chen YL. Synergistic anti-tumor activity of isochaihulactone and paclitaxel on human lung cancer cells. J Cell Physiol. 2012; 227:213-222.