
INTRODUCTION
Cell homeostasis is dependent on the balance between biosynthesis and catabolism of macromolecules. Eukaryotic cells possess two major protein degradation routes: the ubiquitin-proteasome and the lysosomal systems [1]. The proteasome system is responsible for the selective degradation of most short-lived proteins [2], while the lysosomal system degrades and recycles long-lived proteins and defective organelles, these substances from both inside and outside of cell are delivered to the lytic compartments [2, 3]. There are four major pathways to the lysosome for degradation: endocytosis/phagocytosis, microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy [4, 5]. Degradation of exogenous materials and membrane proteins is mediated by the process of endocytosis/phagocytosis, whereas degradation of cytoplasmic component is carried out by microautophagy, CAM and macroautophagy [6].
Macroautophagy (hereafter referred to as “autophagy”) is the main form of autophagy, which is a multi-step process involving at least four stages [7, 8]: (i) autophagy induction: when cells were under the condition of stimulation of autophagy, the type I PI3K-AKT-mTOR signaling is inhibited and type III PI3K mammalian vps34/Beclin-1 (Atg6) is activated. Inhibition of mTOR reassociates dephosphorylated Atg13 with Atg1, which in turn results in redistribution of mAtg9 from trans-Golgi to late endosome and induces autophagy [9, 10]. Simultaneously, the activation of vps34/Beclin-1 generates phosphatidylinositol (3,4,5) P3 (PIP3) on endomembrane, resulting in isolation and decoration with Atg5 and Atg16 of a small template membrane, which designated as phagophore [11]. (ii) vesicle expansion and completion: the structure of phagophore could not get further progression without two ubiquitin-like conjugation systems. One pathway involves the covalent conjugation of Atg12 to Atg5, with the help of Atg7 (E1-like enzyme) and Atg10 (E2-like enzyme). Atg12-Atg5 successionally binds to Atg16 and multi-dimerizes to form a large complex [12–14]. The second pathway involves the conjugation of phosphatidyl ethanolamine (PE) to microtubule-associated protein1 light chain LC3 (homologue of mammalian Atg8) by the sequential action of Atg4, Atg7 and Atg3. Briefly, LC3 is cleaved by Atg4 to produce the cytosolic form LC3-I (non-lipidated, 18KD), which is activated by Atg7 and transferred to Atg3, then modified into autophagic-vesicle-associated form LC3-II (PE-conjugated, 16KD), used as a marker of autophagy [14, 15]. (iii) maturation and fusion: autophagosomes undergo maturation (including the encapsulation of cellular components), and then fuse with lysosomes to become autolysosomes. (iv) degradation: in the autolysosomes, engulfed components are eventually degraded by lysosomal enzymes (Figure 1).
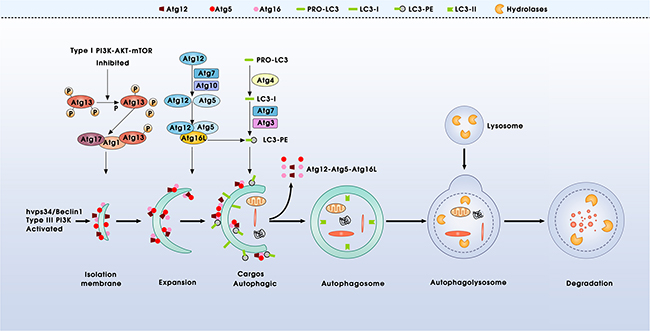
Figure 1: Schematic model of autophagy process. Autophagy is a multi-step process involving at least four main phases, which is controlled by more than 30 autophagy-related (Atg) proteins and mediated by two ubiquitin-like conjugation systems, Atg12-Atg5 and Atg8/LC3: including the initiation, vesicle expansion and completion, maturation and fusion, and ultimate degradation of the membrane and its contents within the lysosomes.
Autophagy is involved in various aspects of biological processes, including cell survival/death, proliferation, differentiation, senescence, and carcinogenesis [16, 17]. The role of autophagy in cancer is controversial. The degradation mechanism enables cells to recycle cytoplasmic constituents and restore metabolic homeostasis, maintaining cells survival under harsh conditions. However, excessive autophagy also induces a non-apoptotic form of programmed cell death, termed as type II programmed cell death [18]. Not surprisingly, aberrant regulation of autophagy is associated with many diseases such as cancer, neurodegenerative disorders, myopathies, cardiovascular diseases and so on. Recently, a series of studies reveal a crucial role of autophagy pathway and its interacting proteins in the regulation of immune response. This review article will focus on autophagy-associated adaptive immune responses and its role in hematologic malignancies because a deeper understanding of the effects of autophagy on immune and autophagy-associated adaptive immune responses allow us to explore potential immunotherapeutic approaches to cure hematologic malignancies.
AUTOPHAGY REGULATIONG SIGNALING PATHWAYS
It is well known that autophagy is regulated by many signaling pathways (Figure 2). The PI3K- AMPK-mTOR signaling pathways play a central role in the regulation of autophagy [19, 20]. Among them, class I PI3Ks phosphorylatePI(4)P and PI(4,5)P2, which bind to the pleckstrin homology domain of AKT and its activator 3-phosphoinositide-dependent protein kinase-1 (PDK-1), as a result, activation of AKT attenuates autophagy [21–24]. Conversely, a dominant negative form of AKT enhances autophagy. AKT-PDK-1 signaling pathway activates a series of downstream signals, including mTOR that has been considered as a “gate keeper” of the autophagic pathway for it is a sensor for amino acids and ATP, two metabolites known to regulate autophagy [20, 25, 26]. In contrast to class I PI3Ks, the class III PI3Ks stimulate autophagy, which phosphorylate PI to generate PIP3 and participate in sequestration of cytoplasmic material in autophagic vacuoles [27]. An integral protein in the class III PI3Ks pathway is Beclin-1, a mammalian ortholog of yeast Atg6/Vps30, which is required for autophagosome formation. Knockdown or antisense Beclin-1 inhibits autophagy [28, 29]. The AMP-dependent protein kinase (AMPK) is activated during hypoxia, metabolic stress or ATP consumption by increased ratios of AMP to ATP and stimulates autophagy through inhibiting mTOR pathway [30, 31].
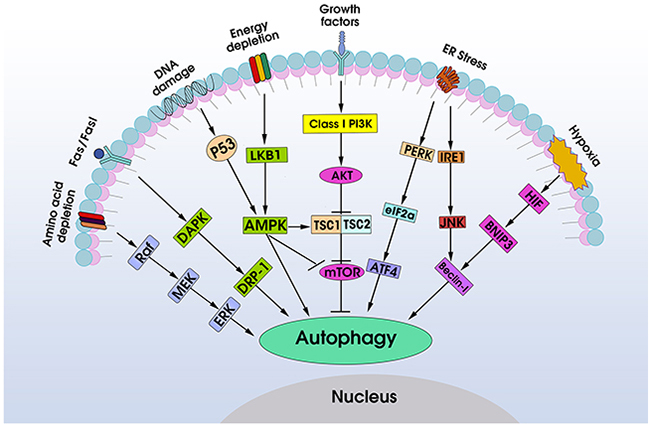
Figure 2: Schematic overview of autophagy-associated signaling pathways in cancer. Autophagy can be activated under multiple stress situations during cancer progression, including amino acid depletion, hyper-expression of death-associated protein kinase, DNA damage, energy depletion, endoplasmic reticulum (ER) stress, hypoxia and other diverse stresses. The key signaling molecules are the PI3K-AMPK-mTOR signaling pathways, which determinate the levels of autophagy in cancer. Other autophagy-associated signaling pathways, including DAPK-DRP1, PERK-eIF2α-ATF4, IRE1-ASK1-JNK1-Beclin-1, p53/LKB1-AMPK, Raf-MEK-ERK, HIF1-BNPI3-Beclin-1 pathways, which are activated by one or more stresses, also play an important role in regulation of autophagy in cancer.
Endoplasmic reticulum (ER) stress response can induce autophagy through the PERK-eIF2α-ATF4 and IRE1-ASK1-JNK1 pathways [32, 33]. The hyper-expression of death-associated protein kinase (DAPK) and DAPK related protein kinase (DRP-1) trigger cell membrane blebbing and extend autophagy continually [34]. DNA damage promotes autophagy through p53 signal [35]. Raf/MEK/ERK pathways stimulate autophagy in amino acid depletion condition [36]. Hypoxia irritates autophagy by up-regulation of hypoxia inducible factor-1(HIF-1) [37]. Together, these signaling pathways not only control autophagy but also are involved in cancerogenesis, and modulation of autophagy by targeting these pathways may affect autophagy-associated adaptive immune responses.
AUTOPHAGY AFFECTS ADAPTIVE IMMUNITY
Recent accumulating evidences have shown that autophagy is also related to regulation of innate and adaptive immunity. Immune system utilized autophagy as an instrument to detect invading pathogens or monitor transforms in the status of self [38, 39]. Specific roles of autophagy in innate immunity, which is regulated by pathogen-recognition receptors (PRRs) signaling, include the regulation of the inflammasome and the clearance of apoptotic corpses to prevent either insufficient inflammatory or excessive inflammatory responses [17, 39]. In adaptive immunity, autophagy is essential to antigen presentation, thymus selection, lymphocyte development and homeostasis, which participates in anti-cancer effects. In this part, we therefore summarize current understanding of roles of autophagy in adaptive immune regulation.
Autophagy in antigen presentation
T cells recognize intra- and extra-cellular antigen peptides that are presented to on major histocompatibility complex (MHC) molecules at cell surface, which is crucial for activation of CD4+ or CD8+ T cells, respectively. In generally, MHC-I molecules present antigenic peptides derived from intracellular proteins. For this purpose, MHC-I molecules are loaded mainly with proteasomal products for their recognition by CD8+ T cells, while MHC-II molecules receive antigenic peptides from extracellular antigens processed via lysosomal degradation for their recognition by CD4+ T cells [40]. However, there is an unconventional pathway named “cross-presentation”, which allows dendritic cells (DCs) to present extracellular antigenic peptides after lysosomal degradation through MHC-I molecules [41, 42]. Similarly, intracellular peptides can be loaded onto MHC-II molecules [43].
Intracellular antigen processing for MHC class II presentation by autophagy
Autophagy can deliver cytoplasmic constituents for lysosomal hydrolysis, which contributes to the processing of intracellular antigens for presentation by MHC-II molecules. Some previous studies revealed that equal to 20% of natural MHC class II ligands are derived from cytosolic and nuclear proteins. Subsequent studies demonstrated that antigens including viral antigens, self-proteins and tumor antigens can be presented on MHC-II molecules [44, 45]. Biosynthesized, intracellular antigens presented by MHC-II also documented in B cells and fibroblasts. Brazil et al. demonstrated that presentation of endogenous C5 protein (a component of complement) by macrophage could be achieved when the macrophage were treated with low doses of the lysosome tropic agent ammonium choride, whereas in the presence of an inhibitor of autophagy presentation of biosynthesized C5 was inhibited [46]. In another in vitro experiment, the agents specifically blocking autophagy (3-MA and wortmannin) had been shown to reduce the capacity of DCs to present MHC-II-restricted peptide derived from endogenously synthesized mucin 1 protein(MUC1) [47]. Because MUC1 is a heterodimeric protein that is aberrantly expressed in various cancer cells including acute myeloid leukemia (AML) blasts and AML stem cells [48, 49], it is likely that autophagy induced by chemotherapic drugs and small molecular inhibitors in AML enhances the loading of MUC1 onto MHC molecules. Presentation by MHC molecules of peptides that suffer post-translational modifications and may form neo-antigens is a key mechanism for the activation of T cells. Autophagy in antigen presenting cells (APCs) has been demonstrated to result in presentation of citrullinated peptides to CD4+ T cells, which can be reduced by either 3-MA or ATG5 siRNA [50].
In addition to self-proteins, pathogen derived antigens including some viral and bacterial antigens that escape after endocytosis or release into the cytosol could also get processed via autophagy for MHC class II presentation. For example, Epstein Barr virus nuclear antigen 1(EBNA1) was found in autophagosomes, which could be presented to CD4+ T cells by EBV transformed B cells via MHC class II pathway, and Atg12 (an essential autophagy-inducing gene) siRNA inhibited recognition by EBNA-1-specific CD4+T cells [51]. However, there is a limited CD4 epitope display from endogenously expressed EBNA1 because autophagy is predominantly a cytoplasmic process [52].
Together, these reports suggest the important role of autophagy in intracellular antigen processing for MHC class II presentation to CD4+ T specific cells (Figure 3A).
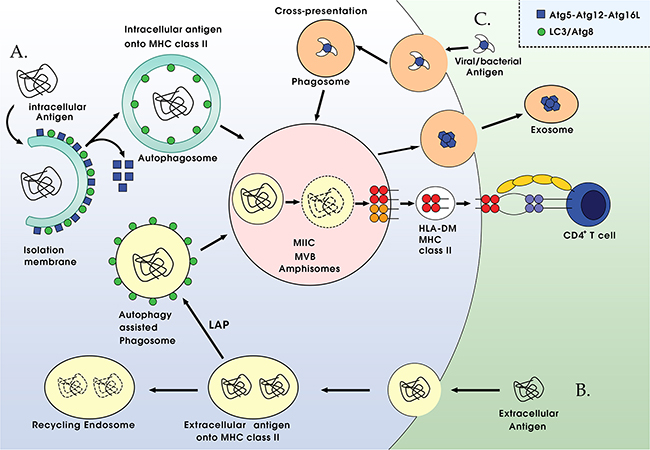
Figure 3: Autophagy-associated antigen presentation pathways. A. Autophagy is a novel pathway for intracellular antigen presentation. Autophagosomes, which recruit cytosolic antigens to MHC class II containing compartments (MIICs) for lysosomal degradation and presentation to CD4+ T cells with the assistance of the peptide-loading chaperone HLA-DM. B. Extracelluar antigens, including phagosomes or apoptotic cells, get decorated with Atg8/LC3 (a process termed LAP), which enhances fusion with lysosomes, and might also increase fusion with MIICs for antigen loading onto MHC class II molecules. C. Autophagy machinery seem to be beneficial for cross-presentation on MHC class I molecules in the donor cell. Both viral and tumor antigens benefit from the cross-presentation via autophagy in the infected or transformed cells. The process of multivesicular bodies (MVBs) releases exosomes may be the key molecular machinery for cross-presentation.
Extracellular antigen processing for MHC class II presentation by autophagy
Autophagy also plays an important role in facilitating the recognition of extracellular antigens phagocytesed by APCs in which antigens are delivered to autophagosomes [45, 53, 54]. For example, Atg5 and other proteins required for autophagy were demonstrated to be essential for optimal processing and presentation of a variety of forms of phagocytesed antigens containing Toll-like receptor (TLR) agonists [55]. It was reported that targeting of the Influenza Matrix Protein 1 (MP1) to autophagosomes via fusion to the Atg8/LC3 resulted in an enhanced MHC-II presentation to CD4+ T cells [44]. However, a more recent study showed that autophagy induced by influenza A virus failed to contribute to MHC-II-restricted presentation [56]. In addition to viral and bacterial antigen delivery for MHC II presentation after autophagy, another role for this catabolic process in tumor antigen delivery was recently suggested. Autophagic cargo that can be extruded into the extracellular matrix from cancer cells should be superior sources from which DCs can intake antigen for T cell priming [57].
Autophagy machinery contributes to deliver phagosomes to lysosomes for extracellular antigen processing [58]. However, the exact mechanisms of this process remain elusive. There are two hypotheses that have been put forward to explain enhanced phagosome processing with the help of autophagy: One is Atg8/LC3-associated phagocytosis (LAP), which recruited to phagosome membrane for strengthened fusion with lysosomes. And the other one is amphisomes formation, the procedure prior to lysosome fusion. Phagocytosis, a prominent endocytic pathway, has been found to be regulated by Atg proteins. During this LAP, LC3 seemed to be transiently recruited to a subset of phagosome membrane, which surrounded by pathogen-associated molecular pattern (PAMP) receptors, including the TLR family, primarily TLR2, or the C-type lectin Dectin-1, the T-cell immunoglobulin mucin protein 4 (TIM4) or Fc receptors for immunoglobulins, thus enhances phagosome fusing with lysosomes [58–60]. The generation of ROS produced by NADPH oxidases (NOX2) at the phagosome was proposed to be needed to maintain the conjugation of LC3 to phagosomes in LAP [61]. The fate of these phagosomes depends on cellular background. In some cell types, primarily mouse macrophages, the contents of LAP phagosomes seem to be degraded more rapidly than LC3 negative phagosomes, possibly because of more efficient transport along microtubules through LC3 binding to FYVE and coiled-coil domain containing 1 (FYCO1) protein, which accelerates LAP phagosomes fusing with lysosomes [62, 63]. Whereas, in plasmacytoid dendritic cells (pDCs) and human macrophages, LAP vesicles seem to be stabilized for fusion with TLR-containing endosomes and postponed the presentation of extracellular antigens for MHC class II [61]. Thus, the autophagy machinery that mediates LAP can affect the fate of phagosomes and facilitate the presentation of exogenous antigens by MHC class II (Figure 3B).
Amphisomes have been characterized and defined as an intermediate organelles, formed during autophagy through the fusion of endosomes and autophagosomes. Complex multi-vesicular vacuoles, an amphisome-like structure, have been observed in various cell types [64, 65]. But, whether amphisomeis an alternative for phagosomes fusion with autophagosomes for more efficient delivery of endocytosed cargo, or just a tentative structure with no biological effects is still enigmatic.
Antigen packaging for cross-presentation via autophagy
Limited evidence displays that autophagy plays a role in the conventional MHC class I presentation, however, autophagy machinery has been implicated in the presentation of extracellular, endocytosed antigens by MHC class I molecules, a pathway termed cross-presentation that plays a critical role in cytotoxic T cell immunity against viruses and tumors. The autophagic exocytosis of antigen donor cell might benefit antigen processing and had been shown to facilitate the cross-presentation of tumor and viral antigens [66, 67]. Li, et al. demonstrated that autophagy in melanoma cells or ovalbumin antigen-expressing human HEK 293T cells is essential for cross-presentation by DCs both in vitro and in vivo [66]. Inhibition of autophagy by the RNA-interference (RNAi)-mediated depletion of Atg12 or Beclin-1abolished cross-presentation almost completely, whereas induction of autophagy by rapamycin or starvation dramatically enhanced cross-presentation of tumor antigens [66]. Recently, a working model has been established to explain antigen accumulation inside autophagosomes: when both proteasomes and lysosomes are inhibited, short-lived proteins, defective ribosomal products, likely component of the antigen pool in tumor cells, and misfolded proteins accumulate and form protein aggregates. This process then induces autophagy via p62 and Atg8/LC3 interaction [68]. Of note, further studies on mechanism of cross-presentation and therapeutic efficacy showed potent anti-tumor efficacy of the autophagosome-based DRibble (DRiPs-containing blebs) vaccine [69, 70].
But how do autophagosomes and their contents leave cells for cross-presentation is difficult to envision. One most possibility might be dependent on unconventional secretion of proteins, briefly, autophagosomes fuse with late endosomes to generate multi-vesicular bodies (MVBs), which could release exosomes, an immunogenic vesicles [69, 70]. Therefore, it is tempting to speculate that autophagy could facilitate exosomes release, and then antigens can be secreted by an unconventional pathway from MVBs that receive input from autophagosomes. Thus, autophagy might facilitate the packaging of antigens efficiently for cross-presentation on MHC I molecules (Figure 3C).
AUTOPHAGY AND T CELL HOMEOSTASIS
In addition to antigen processing, autophagy modifies adaptive immunity via its contribution to the development, repertoire selection, maturation, homeostasis and effector functions of CD4+ / CD8+ T cells. Basal autophagy maintains homeostasis in T cells and can be up-regulated following T cell receptor (TCR) stimulation, and the absence of autophagy causes T cells differentiation abnormalities and function deficiency [71, 72]. Autophagy-associated presentation plays a crucial role in positive and negative selection of naïve T cells. When autophagy is abrogated, the number of naïve T cells drastically decreased [73–75]. Similarly, autophagy is also essential for mature T cell homeostasis and maintaining periphery T cell survival during proliferation, which entails clearance of damaged mitochondria and sustains proper Ca2+ homeostasis by trimming the ER [72–76]. The lack of autophagy results in increasing CD4+ T cell death, because of increased ROS production, elevated amounts of p38 (a mitogen-activated protein kinase), and imbalance of pro- and pre-apoptotic protein [77]. Moreover, deficiency in autophagy causes impaired survival of memory CD8+ T cells during infection with virus [78]. Furthermore, another role for autophagy in inducible natural killer T (iNKT) cells development has been displayed. The early stage of iNKT development is retarded in mice with deficiency of vps34/Beclin-1 [79]. Interestingly, autophagy has recently been found to modulate energy metabolism in T cells. With autophagy inhibitors treatment, ATP production might not be normally increased accompanying with the T cells activation, and ultimately, exhibit some defective in T cells, which could be reversed by exogenous energy source [80]. Thus, autophagy might be required for T-cell homeostasis and function, though much remains to be explored (Figure 4A).
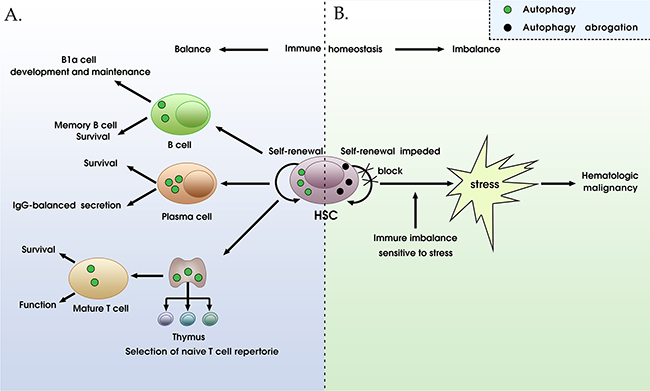
Figure 4: The roles of autophagy in immune homeostasis and tumorigenesis. A. The normal state of autophagy regulation mechanism is crucial for immune homeostasis. Autophagy plays a crucial role in positive and negative selection of naïve T cells and is essential for mature T cell homeostasis and maintaining periphery T cell survival during proliferation. Autophagy also affects self-renewal of hematopoietic stem cells (HSCs), B1a cell development, memory B cell maintenance, plasma cell survival, and IgG-balanced secretion. B. The abrogation of autophagy leads to immune homeostasis imbalance, HSCs self-renewal impeded, cells are more sensitive to various stresses, eventually resulting in the initiation and progression of blood cancers including leukemia, myelodysplastic syndrome, and lymphoproliferative disorder.
AUTOPHAGY IN THE DEVELOPMENT AND FUNCTION OF B CELLS
Current data from multiple studies have showed that autophagy plays a complex role in the development and function of B cells. Studies of Atg5 deficient mice revealed a key role for autophagy in B cell development and maintenance [81]. The quantity of B cells compromised when genes encoding essential Atg proteins are deficient, the decrease seems to originate from autophagy-deficient B-cell progenitors’ failure of transition between pro- and pre-B-cell stages in the bone marrow, suggesting autophagy is required for B cells development (Figure 4A).
The terminal step of the humoral immune response is attributed to plasma cells generate antibodies continuously. Besides, memory B cells can also be reactivated for antibody production upon encountering cognate antigen. Autophagy gene is essential for plasma cell homeostasis and the survival of memory B cells [82]. With the use of mice with B-cell-specific deletion of Atg5, Miller’s group testified that plasma cell differentiation initiated by T cell-dependent/-independent antibody responses required autophagy [81]. Moreover, the long-term survival of plasma cells in the bone marrow are diminished in these autophagy deficient mice [83]. The absence of autophagy in plasma cells had a larger ER and more ER stress signaling than did their wild-type counterparts, which led to higher expression of the transcriptional repressor Blimp-1 and triggered unfolded protein responses during the production of immunoglobulins. The uncontrolled stress response was associated with less intracellular ATP and induced plasma cell death [84]. In addition to the maintenance of plasma cells, autophagy is also required for the survival of memory B cells. Mice with autophagy deficient B cells have deadly impaired specific secondary antibody responses to influenza A virus due to compromised maintenance of memory B cells [85].
Taken together, autophagy functions as an important role in B cells development, memory B cells survival and plasma cells homeostasis.
THE ROLE OF AUTOPHAGY, AUTOPHAGY-ASSOCIATED ADAPTIVE IMMUNITY IN BLOOD CANCERS
Autophagy activity has been shown to be constitutively high in hematopoietic stem cells (HSC), and is required for the differentiation and self-renewal of HSCs [86–88]. It is well known that mechanisms protecting HSCs from cellular damage are essential to prevent hematopoietic malignancies. Autophagy is activated in response to various cellular stresses including DNA damage and genomic instability, removing unnecessary or harmful substances via lysosomal degradation mechinery [89, 90]. Several lines of evidence have shown that lack of autophagy in HSCs is involved in the pathogenesis of blood cancers [91, 92]. Beclin-1 haploinsufficiency contributes to the development of lymphoma and lymphoproliferative disease [91], whereas impairment of autophagy caused by Atg7 deletion leads to the expansion of progenitor cells in the bone marrow giving rise to a severe, invasive myeloproliferation [92]. Collectively, autophagy deficiencies impair HSCs, and cause oxidative stress, activation of the DNA damage response and genome instability, a known cause of blood cancer initiation and progression, which facilitates or even triggers tumorigenesis (Figure 4B).
On the other hand, when tumor is established, autophagy is able function as a pro-survival pathway. Indeed, cancer cells utilize autophagy as a means to adapt to the hypoxic, nutrient and growth factor deprivation, and metabolically stressful tumor microenvironment and therapeutically induced cell stress or damage [93]. Pharmacologic such as chloroquine and hydroxychloroquine approved by the U.S. Food and Drug administration for clinical use [94] or genetic inhibition of autophagy restores chemosensitivity and enhances tumor cell apoptosis in many types of hematopoietic malignancies [95]. However, autophagy also represents a distinct mechanism of cell death (autophagic cell death, ACD) in well-defined circumstances. For example, arsenic trioxide was showed to induce ACD in leukemic cell lines and AML progenitors, which could be reversed by knockdown of Beclin-1 or Atg7 [96]. We have reported that overexpression of Beclin-1 delivered by an oncolytic virus could induce significant ACD in a variety of leukemic cell lines and primary leukemic blasts [97]. ACD induced by anticancer agents in blood cancers has been summarized in an excellent review [95].
Recently, a deeper understanding of the process of immunogenic cell death (ICD) that can elicit a protective immune response against dead-tumor cell antigens induced by ICD inducers such as anticancer cytotoxic drugs and small molecular inhibitors has highlighted the importance of cancer immunotherapies and proposed novel antitumor strategies [98, 99]. There is increasing evidence to suggest some chemotherapeutic agents are intrinsically endowed with ability to trigger ICD (Table 1). These cytotoxic anticancer drugs are employed in the clinic for the treatment of hematologic malignancies, including various anthracyclines (such as doxorubicin, epirubicin, andidarubicin), mitoxantrone, oxaliplatin, cyclophosphamide, the histone deacetylase inhibitor (vorinostat) and bortezomib, a proteasomal inhibitor [99–101]. In AML cells, cytarabine, daunorubicin, all-trans retinoic acid (ATRA) and valproic acid were also found to induce increased calreticulin exposure (ecto-CRT) and release of HSP70 and HSP90, which indicating an induction of immunogenic apoptosis, although the level of CRT exposure/HSP release seems to depend on individual patients characteristics rather than the apoptosis-inducing drug [102]. Bortezomib had been showed to induce immunogenic death of human multiple myeloma, including primary tumor cells, which is dependent on cell-cell contact and linked to the expression of Hsp90 on the surface of dying cells [103]. More recently, diverse pro-apoptotic drugs, including topoisomerase II inhibitors, kinase inhibitors, and proteosome inhibitors have been shown to activate pannexin-1 channels and ATP release in Jurkat T cell acute lymphocytic leukemia model, which mediate immunogenic anti-tumor responses [104]. It is important to note that these anticancer agents could induce autophagy in both solid tumors and blood cancers (Table 1), thereby the relationship between autophagy in response to the cytotoxic agents and ICD in blood cancers deserve further attention.
Table 1: ICD-induced therapeutic compounds modulate autophagy in hematologic malignancies.
Compounds |
Bona fide ICD inducer |
Molecular basis for ICD |
Types of tumors (Ref.) |
Effect on autophagy |
Types of cancers |
|---|---|---|---|---|---|
Bortezomib |
Yes |
CALR exposure |
inducer |
B-ALL |
|
Bleomycin |
Yes |
CALR exposure |
Melanoma |
inducer |
HL |
Cyclophosphamide |
Yes |
CALR exposure |
inducer |
B-cell lymphoma |
|
Doxorubicin |
Yes |
CALR exposure |
inducer |
||
Gemcitabine |
No |
ATP secretion |
Pancreatic ductal adenocarcinoma |
inducer |
HL |
Idarubicin |
Yes |
CALR exposure |
inducer |
Leukemia |
|
Melphalan |
n.d |
HMGB1 release |
Lymphoma |
inducer |
MM |
Epirubicin |
No |
CALR exposure |
inducer |
||
Mitoxantrone |
Yes |
CALR exposure |
n.a. |
n.a. |
|
Temozolomide |
n.d. |
ATP secretion |
inducer |
NHM |
|
Cisplatin |
No |
ATP secretion |
inducer |
NHM |
|
Oxaliplatin |
Yes |
CALR exposure |
inducer |
NHM |
|
Vorinostat |
n.d. |
CALR exposure |
n.d. |
inducer |
Abbreviations: ICD, immunogenic cell death; CALR, calreticulin; HMGB1, high-mobility group box 1; ICD, IFN, interferon; MM, multiple myeloma; B-All, B cell acute lymphoblastic leukemia; HL, Hodgkin’s lymphoma; AML, Acute myeloid leukemia; MCL, Mantle cell lymphoma; n.a., not applicable; n.d., not determined; NHM, non-hematologic malignancies.
A detailed discussion of the molecular and cellular mechanisms involved in chemotherapy induced ICD can be found in Ref [100, 105]. Briefly, ICD is preceded or accompanied by the emission by dying cancer cells of immunostimulatory molecules called damage-associated molecular patterns (DAMPs). DAMPs that are crucial for ICD consist of ATP, high-mobility group protein B1 (HMGB1), and exposed molecules on the outer membrane of dying cells such as calreticulin, heat-shock proteins (Hsp90 and Hsp70), and ER sessile proteins. Mounting evidence indicates that autophagy plays a critical role in the induction of ICD. Michaud et al [106]. reported that the process of autophagy is necessary for the anti-tumor immune response evoked by apoptotic tumor cells in response to chemotherapy by regulation of ATP release. HMGB1, a key DAMP factor, serve as powerful immunological adjuvants and mediates ICD in cancer therapy [107]. Autophagy regulates passive HMGB1 release from dying cells and active HMGB1 secretion [105, 108]. Additionally, ACD in cancer cells exhibits an ICD property, especially in the ICD induced by some cytotoxic agents (anthracyclines, mitoxantrone, and oxaliplatin), vorinostat and bortezomib [99]. However, there is a few studies suggest that autophagic response of melanoma cells to ER stress suppresses basal ecto-CRT and restrains ICD induced by photodynamic therapy, suggesting that the role of autophagy in ICD needs to be assessed in context with the cancer model and the type of ICD inducer [109, 110].
CONCLUSIONS AND PERSPECTIVES
Autophagy, as an evolutionarily conserved catabolic process, has been implicated in regulation of various aspects of biological process, including cell survival/death, proliferation, differentiation, senescence, and carcinogenesis. Current evidence suggests an important role for autophagy in both innate and adaptive immunity. In fact, autophagy is widely involved in antigen processing, and presentation, or function of antigen donor cells, APCs and T cells. Furthermore, premortem autophagy has been to determine the immunogenicity of chemotherapy-induced cancer cell death via promoting release of ATP. Like apoptotic cell death, autophagic cell death also entails immunogenicity after anticancer treatments. As autophagy plays multiple roles in antitumor immune, it is a potential target for therapies in cancer including hematologic malignancy.
Dysregulation of autophagy in HSCs is linked to the initiation and progression of blood cancers including leukemia [111], myelodysplastic syndrome [112], and lymphoproliferative disorder [113]. In blood cancer cells, autophagy is also a double-edged sword. Recently, many clinical trials using autophagy inhibitor, such as chloroquine and its analogue hydroxychloroquine, are being applied to multiple blood cancer types, including acute myeloid leukemia, chronic myeloid leukemia and multiple myeloma, in combination with chemotherapies or target therapies (ClinicalTrials.gov ID: NCT02631252; NCT01227135; NCT01689987 and NCT00568880). However, an interesting unresolved is whether systemic autophagy inactivation will be sufficiently selective to kill cancer cells while sparing immune cells from the deleterious consequences. Although autophagy-triggered modulation of anticancer immune response is a very complicated process, we need to elucidate some aspects of antitumor immunity in the patients with hematologic malignancy when autophagy inhibitor is administered.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (Grant No. 81670178, 81500110 and 81370645), Funds of Science Technology Department of Zhejiang Province (Grant No. 2014C33235), and Special Scientific Construction Research Funds of National Chinese Medicine Clinical Research Center, SATCM (Grant No. JDZX2015113).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Ogier-Denis E and Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta.2003;1603:113-128.
2. AH and A Ciechanover. The ubiquitin system. Annual Review of Biochemistry.1998;67:425.
3. Noboru Mizushima. Methods for monitoring autophagy. The International Journal of Biochemistry & Cell Biology.2004;36:2491-2502.
4. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis.2013;4:e838.
5. Mizushima N, Levine B, Cuervo AM and Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature.2008;451:1069-1075.
6. Levine B and Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell.2004;6:463-677.
7. Klionsky DJ and Emr SD. Autophagy as a regulated pathway of cellular degradation. Science.2000;290:1717-1721.
8. Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy.2005;1:1-10.
9. Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, et al. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. BiochemBiophys Res Commun.1998;246:222-227.
10. Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci.2006;119:3888-3900.
11. Xiao G. Autophagy and NF-kappaB: fight for fate. Cytokine Growth Factor Rev.2007;18:233-243.
12. Mizushima N1, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol.2001;152:657-668.
13. Nakatogawa H, Suzuki K, Kamada Y and Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol.2009;10:458-467.
14. Maiuri MC, Zalckvar E, Kimchi A and Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol.2007;9:741-752.
15. Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. MolBiol Cell.2008;19:4762-4775.
16. Bernard A and Klionsky DJ. Autophagosome formation: tracing the source. Developmental cell.2013;25:116-117.
17. Pan H, Chen L, Xu Y, Han W, Lou F, Fei W, Liu S, Jing Z, Sui X. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget.2016;7:21235-21246. doi: 10.18632/oncotarget.6908.
18. Ning Chen and VassilikiKarantza-Wadsworth. Role and regulation of autophagy in cancer. BiochimBiophysActa.2009;1793:1516-1523.
19. Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science.2011;332:966-970.
20. Joungmok Kim, MondiraKundu, Benoit Viollet and Kun-Liang Guan. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol.2011;13:132-141
21. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem.2001;276:35243-35246.
22. Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H and Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem.1997;243:240-246.
23. Shintani T and Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science.2004;306:990-995.
24. Nicholson KM and Anderson NG. The protein kinase B/Akt signaling pathway in human malignancy. Cell Signal.2002;14:381-395.
25. Hanada M, Feng J and Hemmings BA. Structure, regulation and function of PKB/AKT--a major therapeutic target. BiochimBiophysActa.2004;1697:3-16.
26. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet.2004;36:585-595.
27. Yu X, Long YC and Shen HM. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy.2015;11:1711-1728.
28. Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagytriggers apoptosis. Mol Cell Biol.2005;25:1025-1040.
29. Botti J, Djavaheri-Mergny M, Pilatte Y and Codogno P. Autophagy signaling and the cogwheels of cancer. Autophagy.2006;2:67-73.
30. Spasić MR, Callaerts P and Norga KK. AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist.2009;15:309-316.
31. Koren I and Kimchi A. Promoting tumorigenesis by suppressing autophagy. Science.2012;338:889-890.
32. Jiang Q, Li F, Shi K, Wu P, An J, Yang Y, et al. Involvement of p38 in signal switching from autophagy to apoptosis via the PERK/eIF2alpha/ATF4 axis in selenitetreated NB4 cells. Cell Death & Disease.2014;5:e1270.
33. Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer letters.2014;344:174-179.
34. Inbal B, Bialik S, Sabanay I, Shani G and Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. Cell Biol.2002;157:455-468.
35. Zeng X, Yan T, Schupp JE, Seo Y and Kinsella TJ. DNA mismatch repair initiates 6-thioguanine--induced autophagy through p53 activation in human tumor cells. Clin Cancer Res.2007;13:1315-1321.
36. Kim JH, Hong SK, Wu PK, Richards AL, Jackson WT and Park JI. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Experimental cell research.2014;327:340-352.
37. Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY).2011;3:192-222. doi: 10.18632/aging.100296.
38. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.2009;29:2570-2581.
39. Daniel J, Puleston, Anna Katharina and Simon. Autophagy in the immune system. Immunology. 2014;141:1-8.
40. Martins JD, Liberal J, Silva A, Ferreira I, Neves BM and Cruz MT. Autophagy and inflammasome interplay. DNA Cell Biol.2015;34:274-281.
41. Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ.2005;12:1178-1190.
42. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. ProcNatlAcadSci USA.2005;102:7922-7927.
43. Rammensee HG, Friede T and Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics.1995;41:178-228.
44. Christian Münz. Antigen processing via autophagy-not only for MHC class II presentation anymore? CurrOpinImmunol.2010;22:89-93.
45. Schmid D, Pypaert M and Münz C. MHC class II antigen loading compartments continuously receive input from autophagosomes. Immunity.2007;26:79-92.
46. Schmid D, Münz C. Immune surveillance of intracellular pathogens via autophagy. Cell Death Differ. 2005;12:1519-1527.
47. Brazil MI, Weiss S and Stockinger B. Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur J Immunol.1997;27:1506-1514.
48. Dorfel D, Appel S, Grunebach F, Weck MM, Muller MR, Heine A, et al. Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro transcribed MUC1 RNA. Blood.2005;105:3199-3205.
49. Liu S, Yin L, Stroopinsky D, Rajabi H, Puissant A, Stegmaier K, et al. MUC1-C oncoprotein promotes FLT3 receptor activation in acute myeloid leukemia cells. Blood.2014;123:734-742.
50. Lau SK, Weiss LM and Chu PG. Differential expression of MUC1, MUC2, and MUC5AC in carcinomas of various sites: an immunohistochemical study. Am J Clin Pathol.2004;122:61-69.
51. Ireland JM and Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4+ T cells. J Exp Med.2011;208:2625-2632.
52. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen aftr autophagy. Science.2005;307:593-596.
53. Leung CS, Haigh TA, Mackay LK, Rickinson AB and Taylor GS. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4+ epitope display. ProcNatlAcadSci USA.2010;107:2165-2170.
54. Y Ma, L Galluzzi, L Zitvogel and G Kroemer. Autophagy and Cellular Immune Responses. Immunity.2013;39:211-227.
55. Münz C. Enhancing immunity through autophagy. Annu Rev Immunol.2009;27:423-449.
56. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity.2010;32:227–239.
57. Comber JD, Robinson TM, Siciliano NA, Snook AE and Eisenlohr LC. Functional macroautophagy induction by influenza A virus without a contribution to major histocompatibility complex class II-restricted presentation. J Virol.2011;85:6453-6463.
58. Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res.2011;17:654-666.
59. Shibutani ST, Saitoh T, Nowag H, Münz C and Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol.2015;16:1014-1024.
60. Florey O, Kim SE, Sandoval CP, Haynes CM and Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol.2011;13:1335-1343.
61. Ma J, Becker C, Lowell CA and Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem.2012;287:34149-34156.
62. Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol.2013;203:757-766.
63. Ma J, Becker C, Reyes C and Underhill DM. Cutting edge: FYCO1 recruitment to dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J Immunol.2014;192:1356-1360.
64. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature.2007;450:1253-1257.
65. Tooze SA and Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol.2010;12:831-835.
66. Nakatogawa H, Ichimura Y and Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell.2007;130:165-178.
67. Li Y, Wang LX, Yang G, Hao F, Urba WJ and Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res.2008;68:6889-6895.
68. Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G and Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen crosspriming of virus-specific CD8+ T cells. Cell Death Differ.2009;16:991-1005.
69. Li Y, Wang LX, Pang P, Twitty C, Fox BA, Aung S, et al. Cross-presentation of tumor associated antigens through tumor-derived autophagosomes. Autophagy.2009;5:576-577.
70. Li Y, Wang LX, Pang P, Cui Z, Aung S, Haley D, et al. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res.2011;17:7047-7057.
71. Ren H, Zhao S, Li W, Dong H, Zhou M, Cao M, et al. Therapeutic antitumor efficacy of B cells loaded with tumor-derived autophagasomes vaccine (DRibbles). J Immunother.2014;37:383-793.
72. Travassos LH, Carneiro LA, Girardin S and Philpott DJ. Nod proteins link bacterial sensing and autophagy. Autophagy.2010;6:409-411.
73. Pua HH, Dzhagalov I, Chuck M, Mizushima N and He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med.2007;204:25-31.
74. Pua HH, Guo J, Komatsu M and He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol.2009;182:4046-4055.
75. Anand PK, Tait SW, Lamkanfi M, Amer AO, Nunez G, Pages G, et al. TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenesvia extracellular signal-regulated kinase (ERK) activation. J Biol Chem.2011;286:42981-42991.
76. Klein L, Kyewski B, Allen PM and Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol.2014;14:377-391.
77. Willinger T and Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naïve T-cell homeostasis. Proc Natl Acad Sci USA.2012;109:8670-8675.
78. Sian M Henson, Alessio Lanna, Natalie E Riddell, Ornella Franzese, Richard Macaulay, Stephen J Griffiths, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest.2014;124:4004-4016.
79. Xiaojin Xu, Koichi Araki, Shuzhao Li, Jin-Hwan Han, Lilin Ye, Wendy G Tan, et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat Immunol.2014;15:1152-1161.
80. Sun JR, Desai MM, Soong L and Ou JHJ. IFN alpha/beta and autophagy Tug-of-war between HCV and the host. Autophagy.2011;7:1394-1396.
81. Crisan TO, Plantinga TS, van de Veerdonk FL, Farcas MF, Stoffels M, Kullberg BJ, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One.2012;6:e18666.
82. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy.2008;4:309-314.
83. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med.2014;20:503-510.
84. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol.2013;14:298-305.
85. Tan Y, Kusuma CM, St John LJ, Vu HA, Alibek K and Wu A. Induction of autophagy by anthrax lethal toxin. BiochemBiophys Res Commun.2009;379:293-297.
86. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med.2014;20:503-510.
87. Salemi S, Yousefi S, Constantinescu MA, Fey MF and Simon HU. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res.2012;22:432-435.
88. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med.2011;208:455-467.
89. Phadwal K, Watson AS and Simon AK. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci.2013;70:89-103.
90. Levine B and Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell.2004;6:463-477.
91. Liu D, Shaukat Z, Xu T, Denton D, Saint R and Gregory S. Autophagy regulates the survival of cells with chromosomal instability. Oncotarget.2016;doi:10.18632/oncotarget.11736.
92. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest.2003;112:1809-1820.
93. Mortensen M, Watson AS and Simon AK. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy.2011;7:1069-1070.
94. Dalby KN, Tekedereli I, Lopez-Berestein G and Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy.2010;6:322-329.
95. Evangelisti C, Evangelisti C, Chiarini F, Lonetti A, Buontempo F, Neri LM, et al. Autophagy in acute leukemias: a double-edged sword with important therapeutic implications. Biochim Biophys Acta.2015;1853:14-26.
96. Nencioni A, Cea M, Montecucco F, Longo VD, Patrone F, Carella AM, et al. Autophagy in blood cancers: biological role and therapeutic implications. Haematologica.2013;98:1335-1343.
97. Goussetis DJ, Altman JK, Glaser H, McNeer JL, Tallman MS and Platanias LC. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J Biol Chem.2010;285:29989-29997.
98. Tong Y, You L, Liu H, Li L, Meng H and Qian W. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget.2013;4:860-874. doi: 10.18632/oncotarget.1018.
99. Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buqué A, Senovilla L, Baracco EE, Bloy N, Castoldi F, Abastado JP, Agostinis P, Apte RN, Aranda F, Ayyoub M, et al. Classification of current anticancer immunotherapies. Oncotarget.2014;5:12472-12508. doi: 10.18632/oncotarget.2998.
100. Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ.2014;21:39-49.
101. Gebremeskel S and Johnston B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies. Oncotarget.2015;6:41600-41619. doi: 10.18632/oncotarget.6113.
102. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ.2014;21:79-91.
103. Fredly H, Ersvær E, Gjertsen BT and Bruserud O. Immunogenic apoptosis in human acute myeloid leukemia (AML): primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol Rep.2011;25:1549-1556.
104. Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S and Dhodapkar MV. Bortezomib enhances dendritic cell (DC)–mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood.2007;109:4839-4845.
105. Boyd-Tressler A, Penuela S, Laird DW and Dubyak GR. Chemotherapeutic drugs induce ATP release via caspase-gated pannexin-1 channels and a caspase/pannexin-1-independent mechanism. J Biol Chem.2014;289:27246-27263.
106. Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii HJ, Kang R, et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis.2013;4:e966.
107. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science.2011;334:1573-1577.
108. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P and Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer.2012;12:860-875.
109. Colangelo T, Polcaro G, Ziccardi P, Muccillo L, Galgani M, Pucci B, et al. The miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis.2016;7:e2108.
110. Garg AD, Dudek AM and Agostinis P. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology.2013;2:e26260.
111. Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy.2013;9:1292-1307.
112. Evangelisti C, Evangelisti C, Chiarini F, Lonetti A, Buontempo F, Neri LM, et al. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim Biophys Acta.2015;1853:14-26.
113. Watson AS, Mortensen M and Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle.2011;10:1719-1725.
114. Pierdominici M, Barbati C, Vomero M, Locatelli SL, Carlo-Stella C, Ortona E, et al. Autophagy as a pathogenic mechanism and drug target in lymphoproliferative disorders. FASEB J.2014;28:524-535.
115. Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R, et al. Primary effusion lymphoma cell death induced by bortezomib and AG 490 activates dendritic cells through CD91. PLoS One.2012;7:e31732.
116. Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC, et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol.2012;189:3209-3220.
117. Wang Z, Zhu S, Zhang G and Liu S. Inhibition of autophagy enhances the anticancer activity of bortezomib in B-cell acute lymphoblastic leukemia cells. American journal of cancer research.2015;5:639-650
118. Bugaut H, Bruchard M, Berger H, Derangere V, Odoul L, Euvrard R, et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS One.2013;8:e65181.
119. Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E and Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol.2011;33:369-383.
120. Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res.2011;71:768-778.
121. Chen X, Yang Y, Zhou Q, Weiss JM, Howard OZ, McPherson JM, et al. Effective chemoimmunotherapy with anti-TGFbeta antibody and cyclophosphamide in a mouse model of breast cancer. PLoS One.2014;9:e85398.
122. Huang JJ, Zhu YJ, Lin TY, Jiang WQ, Huang HQ and Li ZM. Beclin 1 expression predicts favorable clinical outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Hum Pathol.2011;42:1459-1466.
123. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med.2005;202:1691-1701.
124. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med.2007;13:54-61.
125. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res.2011;71:4821-4833.
126. Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med.2011;208:491-503.
127. Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD, et al. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res.2010;70:9062-9072.
128. Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang Y, et al. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer Res.2011;17:3248-3258.
129. Zhao T, Ren H, Jia L, Chen J, Xin W, Yan F, Li J, Wang X, Gao S, Qian D, Huang C, Hao J. Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget.2015;6:2250-2262. doi: 10.18632/oncotarget.2948.
130. Amaylia Oehadian, Naoki Koide, Ferdaus Hassan, Shamima Islam, Isamu Mori, Tomoaki Yoshida, et al. Differential Expression of Autophagy in Hodgkin Lymphoma Cells Treated with Various Anti-Cancer Drugs. Acta Med Indones.2009;39:153-156.
131. Wemeau M, Kepp O, Tesniere A, Panaretakis T, Flament C, De Botton S, et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis.2010;1:e104.
132. Ristic B, Bosnjak M, Arsikin K, Mircic A, Suzin-Zivkovic V, Bogdanovic A, et al. Idarubicin induces mTOR-dependent cytotoxic autophagy in leukemic cells. Experimental cell research.2014;326:90-102.
133. Dudek-Peric AM, Ferreira GB, Muchowicz A, Wouters J, Prada N, Martin S, et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res.2015;75:1603-1614.
134. Bilir A, Altinoz MA, Erkan M, Ozmen V and Aydiner A. Autophagy and nuclear changes in FM3A breast tumor cells after epirubicin, medroxyprogesterone and tamoxifen treatment in vitro. Pathobiology.2001;69:120-126.
135. Sun WL1, Chen J, Wang YP and Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitatesepirubicin-resistance development. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J.2009;28:578-590.
136. Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ.2008;15:1499-1509.
137. Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther.2003;21:1212-1223.
138. Rubner Y, Muth C, Strnad A, Derer A, Sieber R, Buslei R, et al. Fractionated radiotherapy is the main stimulus for the induction of cell death and of Hsp70 release of p53 mutated glioblastoma cell lines. Radiat Oncol.2014;9:89.
139. lkka Liikanen, Laura Ahtiainen, Mari LM Hirvinen, Simona Bramante, Vincenzo Cerullo, Petri Nokisalmi1, et al. Oncolytic Adenovirus With Temozolomide Induces Autophagy and Antitumor Immune Responses in Cancer Patients. Mol Ther.2013;21:1212-1223.
140. Aranda F, Bloy N, Galluzzi L, Kroemer G and Senovilla L. Vitamin B6 improves the immunogenicity of cisplatin-induced cell death. Oncoimmunology.2014;3:e955685.
141. Aranda F, Bloy N, Pesquet J, Petit B, Chaba K, Sauvat A, et al. Immunedependent antineoplastic effects of cisplatin plus pyridoxine in non-small-cell lung cancer. Oncogene.2015;34:3053-3062.
142. Cirone M, Garufi A, Di Renzo L, Granato M, Faggioni A and D’orazi G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology.2013;2:e26198.
143. Shen M, Duan WM, Wu MY, Wang WJ, Liu L, Xu MD, et al. Participation of autophagy in the cytotoxicity against breast cancer cells by cisplatin. Oncol Rep.2015;34:359-367.
144. Gou HF, Huang J, Shi HS, Chen XC and Wang YS. Chemo-immunotherapy with oxaliplatin and interleukin-7 inhibits colon cancer metastasis in mice. PLoS One.2014;9:e85789.
145. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene.2010;29:482-491.
146. Ding ZB, Hui B, Shi YH, Zhou J, Peng YF, Gu CY, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res.2011;17:6229-6238.
147. Maria Lyngaas Torgersen, Nikolai Engedal, Stig-Ove Bøe, Peter Hokland, and Anne Simonsen. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood.2013;122:2467-2476.
148. Yazbeck VY1, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol.2008;36:443-450.