
INTRODUCTION
Glioblastoma is one of the most aggressive cancers and is often resistant to conventional anti-cancer treatments [1, 2]. The current standard treatment involves maximal surgical resection and concomitant chemo-radiotherapy followed by adjuvant chemotherapy with temozolomide [3]. However, the prognosis is still poor due to the refractoriness of glioblastoma to the various secondary treatments as well as the standard management [2, 4]. Therefore, the development of new therapeutic targets is urgently needed.
Cancer development is mediated by both aberrant genetic and epigenetic alterations [5]. Histone deacetylases (HDACs) are epigenetic regulators of gene expression that remove the acetyl group from histones, which is often associated with gene repression. Overexpression of HDACs in various types of cancer provides a rationale for targeting these enzymes in cancer [6–10]. Recently, many studies reported increased expression of HDACs in brain malignancies, including glioblastoma, among which HDAC4 (class IIA) is the most important target [8]. Previous publications confirmed the efficacy of HDAC inhibitors (HDACIs) as anti-cancer drugs or radiosensitizers in pre-clinical studies for glioblastoma, and several clinical trials have already been launched [8, 11–15]. For the anti-cancer effect against glioblastomas, cell cycle regulation is believed to be a key mechanism of HDACIs [16]. With the increasing focus on HDACIs, SAHA and TSA have been assessed in the treatment of many different types of cancer [17–19]. However, low doses of SAHA and TSA were not effective anti-cancer agents, and high doses had strong neurotoxic effects [20–23].
In the present study, we evaluated the anti-cancer effects of a novel pan-HDACI, 7-ureido-N-hydroxyheptanamide derivative (CKD5), on glioblastoma cell lines and patient-derived cells. CKD5 showed more potent cytotoxic effects than suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA) in vitro and superior therapeutic efficacy compared to SA in vivo.
Our study suggests that CKD5 might be a potent therapeutic agent for the treatment of glioblastoma.
RESULTS
CKD5 is a potent cytotoxic and cytostatic agent in glioblastoma cells
To compare the anti-cancer effects of the HDACIs (CKD5, SAHA and TSA) in glioblastoma cells, cell viability and proliferation assays were performed. All HDACIs suppressed the cell growth in a dose-dependent manner at 72 h (Figure 1A). The IC50 values of the HDACIs were calculated. Importantly, patient-derived glioblastoma cells were more sensitive to CKD5 than SAHA or TSA (IC50 of CKD5 vs. SAHA vs. TSA: 0.3 ± 0.41 μM vs. 9.3 ± 0.69 μM vs. 7.4 ± 0.86 μM, respectively, for SNU.GBM-2, and 0.4 ± 0.37 μM vs. 7.7 ± 0.40 μM vs. 52.1 ± 14.6 μM, respectively, for SNU.GBM-4, Supplementary Table S1). In cell proliferation assays, only CKD5 showed consistent anti-proliferative effects in all tested cell lines, even at the lowest concentration of 0.5 μM (Figure 1B). Next, we evaluated whether the anti-cancer effects of CKD5 were associated with apoptosis. With an exposure time of 48 h of CKD5, early apoptosis was significantly increased by approximately 1.6- to 4-fold in all glioblastoma cells (Figure 1C, Supplementary Table S2). We did not observe any increase in apoptosis in SNU.GBM-4 cells treated with TSA and in all cells treated with SAHA. Our results indicate that CKD5 was the most effective anti-cancer drug for glioblastoma cells among the tested pan-HDACIs.
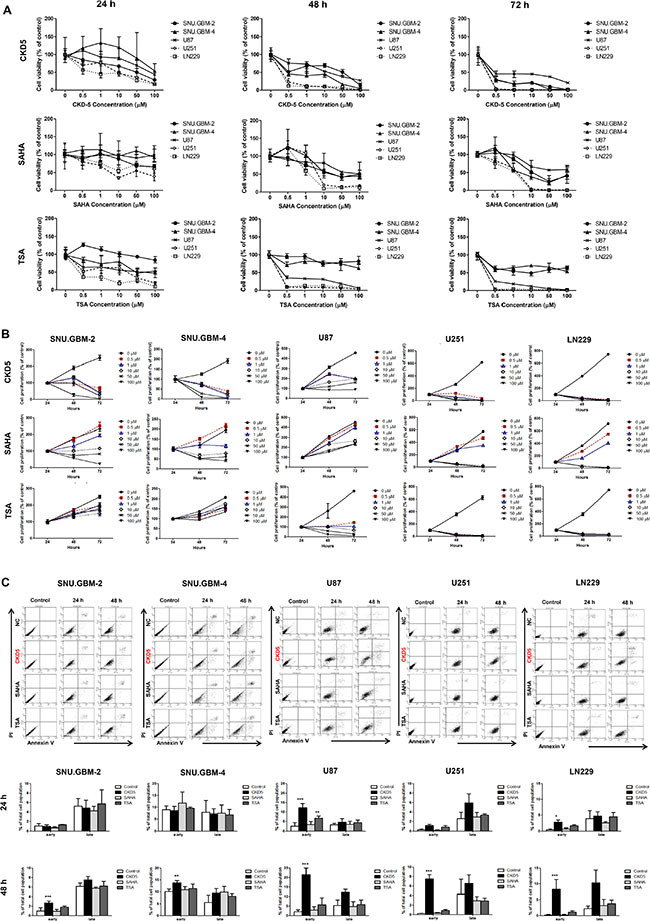
Figure 1: Cell viability, proliferation and apoptosis analyses. (A) The cell viability and (B) proliferation of glioblastoma cells treated with different concentrations of histone deacetylase inhibitors (HDACIs) at 24, 48 and 72 h. CKD5, SAHA and TSA inhibited the cell growth of glioblastoma. Following HDACIs treatment, the cell viability and proliferation were decreased. CKD5 is the most effective at reducing tumor cell growth. (C) Induction of apoptosis in glioblastoma cells. Early apoptosis was significantly increased by CKD5 compared to SAHA and TSA. *p < 0.05, **p < 0.01, ***p < 0.005.
CKD5 strongly induces cell cycle arrest mediated by p21, CDK4 and CCND1
The effect of CKD5 on the cell cycle profile was analyzed by flow cytometry. CKD5 induced significant accumulation of the cells in G2 phase and led to a concomitant decrease in the population of cells in G1 and S phase in all glioblastoma cells after 24 h compared with SAHA and TSA (Figure 2A and Supplementary Table S3). We found that CKD5 can induce apoptosis-specific DNA fragmentation following induction of G2 arrest in glioblastoma cells. The percentage of cells in G2 phase increased by 1.5- to 42.1-fold following CKD5 treatment at 48 h.
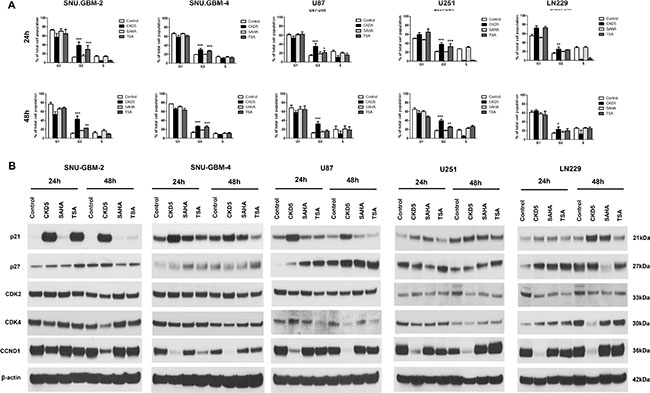
Figure 2: Cell cycle analysis and expression of cell cycle regulators. Cell cycle distributions in glioblastoma cells with HDACI treatment. The analysis of cell cycle arrest in glioblastoma cells showed that the percentage of cells in G2-M phase is induced by 1.5- to 42.1-fold by CKD5 at 48 h. *p < 0.05, **p < 0.01, ***p < 0.005.
Next, we investigated the molecular mechanism of cell cycle arrest by CKD5 by analyzing cell cycle related-proteins, such as p21, p27, CDK2, CDK4 and CCND1, with western blot analysis. There was a significant increase in expression of p21, and this was tightly linked to the reduction in CDK4 and CCND1 in all glioblastoma cells after CKD5 treatment (Figure 2B). This phenomenon was not found in cells after treatment of SAHA and TSA. To further explore the molecular mechanisms associated with the cell cycle arrest, we monitored expression of p27 and CDK2. However, there was no consistent pattern of changes in the levels of p27 and CDK2 in the glioblastoma cells. Overall, it is noteworthy that CKD5 was the most powerful regulator of the cell cycle, and its possible mediators are p21, CDK4 and CCND1.
CKD5 is a more effective HDACI than SAHA and TSA
To determine whether CKD5 efficiently inhibits HDAC enzyme activities, total HDAC enzyme activities were analyzed in different glioblastoma cells after treatment with CKD5, SAHA, and TSA at IC50 doses. CKD5 more significantly decreased the enzyme activities by approximately 6- to 8-fold at 24 h compared to SAHA and TSA, and it showed sustained inhibition at 48 h in all glioblastoma cells (Figure 3A). Additionally, we examined the acetylation status of histone H3 (Ac-H3) after 24 and 48 h. CKD5 more effectively induced histone H3 acetylation in all glioblastoma cells (Figure 3B)
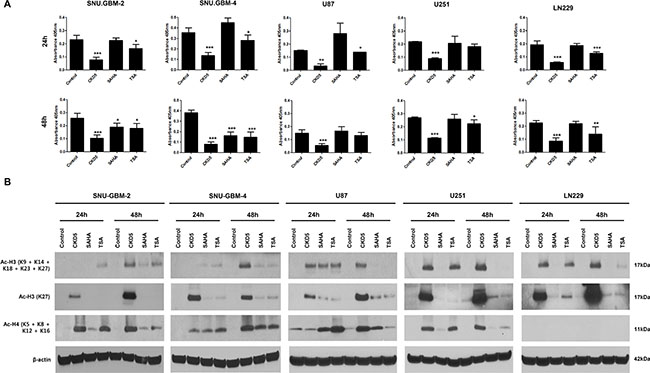
Figure 3: Histone deacetylase (HDAC) enzyme activity, histone H3 and H4 acetylation by HDACIs. (A) CKD5 strongly decreases the enzyme activities by approximately 6- to 8-fold at 24 h, which was stable at 48 h in all glioblastoma cells. (B) CKD5 induces the acetylation status of histone H3 (Ac-H3) at 24 and 48 h.
CKD5 effectively reduces the tumor volume in an orthotopic xenograft glioblastoma mouse model
We confirmed the superior anti-cancer effects of CKD5 by in vivo experiments using an orthotopic xenograft glioblastoma mouse model. The overall design of the study, treatment groups, route of injection, and short-term/long-term treatment schedule are described in Figure 4A. We performed a pilot study to determine the optimal dosage of CKD5 (Supplementary Figure S1 and Supplementary Table S4). We found that two mice died after 0.8 mg/kg of CKD5 treatment. At high doses (1 and 2 mg/kg), CKD5 reduced the tumor volume by 70%, but toxic effects were observed. However, TSA had no therapeutic effect at any dose.
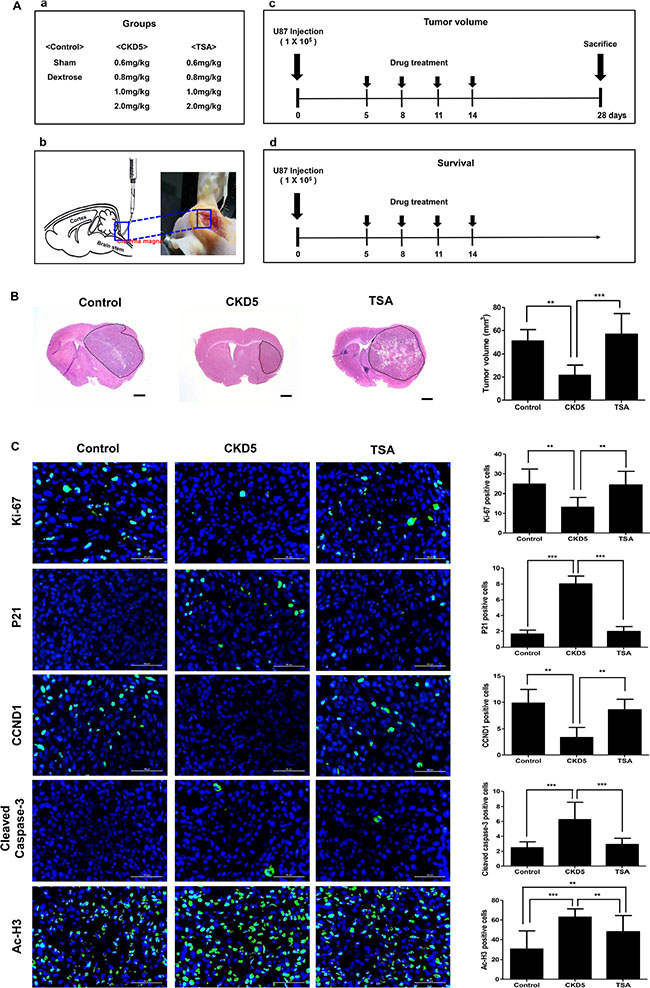
Figure 4: Short-term therapeutic efficacy of CKD5. CKD5 reduces tumor growth and prolongs survival rate in an orthotopic xenograft glioblastoma mice model. (A) Schematic plot of the study design and route of injection for short-term and long-term therapeutic efficacy. (B) Representative histological images show a 57% reduction in tumor volume by CKD5 (21.5 ± 8.9 mm3) compared with the control (50.9 ± 9.9 mm3, p < 0.01) or TSA (60.9 ± 9.2 mm3, p < 0.001). Hematoxylin and eosin (H&E) staining. Magnification, ×1.25. (C) Representative immunofluorescence images show Ki-67, p21, CCND1, cleaved caspase-3 and Ac-H3. Positive cells are shown in green. The graph indicates the number of positive cells compared with the control. Scale bar, 50 μm. Cells were counterstained with DAPI (blue). *p < 0.05, **p < 0.01, ***p < 0.005.
We compared the therapeutic effect of 0.8 mg/kg CKD5 with 0.8 mg/kg TSA and found that CKD5 showed stronger anti-cancer effects than SAHA in vitro. Cisternal injection of CKD5 significantly suppressed the tumor growth compared to the control (21.5 ± 8.9 mm3 vs. 50.9 ± 9.9 mm3, respectively, p < 0.01) or TSA (50.9 ± 9.9 mm3, p < 0.001, Figure 4B). There was no statistically significant difference between the control group and TSA-treated group in tumor volume (p > 0.05). Body weight monitoring of mice revealed that there were initial reductions in weight associated with HDACI administration. The reduction in weight continued until day 11 of treatment, remained stable through the treatment period, and was rapidly increased after completion of the treatment (Supplementary Figure S1). In the tumor samples, we found significantly increased p21, cleaved caspase-3 and Ac-H3 levels, with decreased numbers of cells that were positive for Ki-67 and CCND1 (Figure 4C).
CKD5 effectively prolongs the survival in the orthotopic xenograft glioblastoma mouse model
For the survival analysis, we employed a separate set of experimental mice with orthotopic xenografts. A Kaplan-Meier survival curve showed a significant increase in survival in CKD5-treated mice compared with the control (median survival 34 days vs 30 days, respectively, p = 0.0024) and TSA (median survival 29 days, p = 0.0009, Figure 5). There was no statistically significant difference between survival of the control group and the TSA-treated group (p = 0.68). Taken together, our results suggest that CKD5 is a promising novel therapeutic agent for glioblastoma treatment.
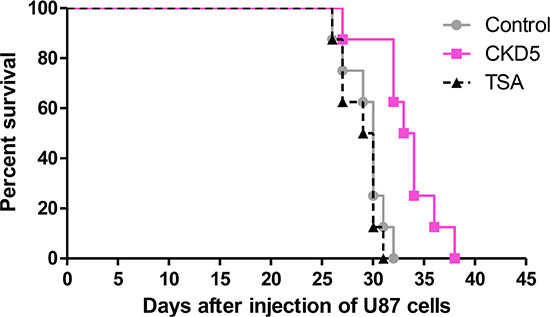
Figure 5: Long-term therapeutic efficacy of CKD5. Kaplan-Meier curves show that CKD5 prolongs the median survival of mice (34 days) compared with the control (30 days, p = 0.0024) or TSA (29 days, p = 0.0009) (control vs. CKD5 vs. TSA: 30 vs. 34 vs. 29 days, respectively).
DISCUSSION
Among the variety of HDACIs being developed as therapeutics, HDACI hydroxamate derivatives targeting multiple Zn2+-dependent HDACs, so-called pan-HDACIs, have advanced the furthest in clinical application as an anti-cancer drug. SAHA was approved in 2006 by the US Food and Drug Administration for the treatment of cutaneous T-cell lymphoma [24]. SAHA is known to inhibit HDACs 1, 2, 3, and 6 [8]. TSA is another pan-HDACI similar to SAHA that inhibits HDACs 1, 2, 3, 4, 6, 7, and 10 and showed potent anti-cancer effects in pre-clinical experiments [8, 25–27]. There are pre-clinical studies reporting encouraging results for both SAHA and TSA in the treatment of glioblastomas [18, 19, 28–31]. However, clinical trials showed that the effect was modest, and it is recommended to use HDACIs with other cytotoxic drugs for treating glioblastomas [32, 33]. There were several limitations in the use of SAHA for glioblastoma therapy. The activity of SAHA in the brain is weak due to its short half-life and low blood-brain barrier (BBB) permeability; the BBB permeability-surface area product to free drug of SAHA is less than 2 orders of magnitude of that predicted by passive diffusion [8, 34]. The solubility of SAHA is very low in water (0.0716 mg/ml). In terms of safety issues, SAHA has cardiac toxicity, such as QTc prolongation [35], and is positive in the Ames test [36], indicating that it induces DNA point mutations and genotoxicity. Compared to SAHA, CKD5 has improved drug-like properties. The basic tertiary amine structure of CKD5 allows the formation of salts, which improves its water solubility (> 1,000 mg/ml). Furthermore, CKD5 is negative in the Ames test, and CKD5 showed no cardiac toxicity in pre-clinical and GLP studies. Taken together, these data suggests that CKD5 has a wider therapeutic window than SAHA.
In the present study, we demonstrated more potent anti-cancer effects of a novel pan-HDACI, CKD5, than SAHA and TSA at a lower dose in glioblastoma. The general molecular mechanisms of HDACIs include cell cycle arrest, induction of apoptosis, and downregulation of survival signals. The mechanism of action of CKD5 is similar to that of the other pan-HDACIs, such as SAHA and TSA. Thus, we focused on the evaluation of its effect on the cell cycle, apoptosis and survival-related protein expression. We found that CKD5 strongly increased p21 protein expression, which was correlated with growth inhibition, whereas TSA-induced growth inhibition was p21-independent. Regulation of p21 by SAHA was cell-type dependent. As the G2 population increased only after 24 h of CKD5 treatment in all cells, induction of apoptosis was accelerated with rapid cell cycle arrest at an earlier stage. This result was consistent with patient-derived glioblastoma cells, which showed the greatest sensitivity to CKD5; inhibition of cell growth was the highest in patient-derived glioblastoma cells. Additionally, p21 levels were the highest, and CDK4 and CCND1 levels were the lowest in these cells.
For therapeutic use of CKD5 in glioblastoma, delivery methods and dose of CKD5 should be titrated. CKD5 was administered intracisternally because it does not penetrate the BBB. We determined the effective concentration of CKD5 (Supplementary Figure S1 and Supplementary Table S4). Although CKD5 had powerful therapeutic effects at more than 0.8 mg/kg, this dosage resulted in serious toxicity. Therefore, we used the 0.8 mg/kg concentration and confirmed the reduction in tumor volume and the increase in survival in an orthotopic xenograft glioblastoma mouse model. Fine tuning of the optimal biological and chemotherapy scheduling is needed to maximize therapeutic efficacy and safety for clinical application of CKD5. The severe toxicity of the other pan-HDACIs, such as thrombocytopenia, and severe fatigue significantly limited their clinical use [37, 38]. CKD5 has a better safety profile and a wider therapeutic index compared to SAHA. Therefore, CKD5 may be used at a higher dose to reach the clinically effective range in cancer patients. To minimize the toxicity, CKD5 may be used as a therapeutic agent in combination with other drugs such as temozolomide or radiation for further improvement of glioblastoma therapy [39]. Confirmation of the effect for monotherapy is a priority before testing the combined therapy.
In this study, we demonstrated for the first time that a novel HDACI, CKD5, has strong anti-cancer effects with more susceptible patient-derived glioblastoma cells in vitro. We also confirmed that CKD5 was more powerful than TSA in vivo. Taken together, our results suggest that CKD5 can be used for improvement of the therapeutic strategy for glioblastoma.
MATERIALS AND METHODS
Isolation of glioblastoma tumor cells and cell culture
Glioblastoma tissue samples (World Health Organization Grade IV) were obtained from two patients (a 64-year-old man: MGMT promoter methylation, SNU.GBM-2 and a 55-year-old woman: MGMT promoter unmethylation, SNU.GBM-4) who underwent surgical excision at the Seoul National University Hospital and provided written informed consent approved by the Institutional Review Board (IRB # 1410-033-616). The tumor cells were isolated immediately after surgical resection and cultured as reported previously [24]. Only the cells that were under 4 passages in primary culture were used in this study. Human glioblastoma cell lines (U87, U251 and LN229) were purchased from the American Tissue Culture Collection (ATCC, Manassas, VA). All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; WelGENE, Seoul, Korea) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) and 1% antibiotics/antimycotics and incubated at 37°C in a humidified atmosphere of 5% CO2.
HDAC inhibitors
CKD5 was obtained from Chong Kun Dang Research Institute, CKD Pharmaceuticals (Korea). It is a hydroxamic acid-based small molecule that chelates Zn in the active site of HDAC enzymes. The structure of HDACIs is composed of three parts (Zn binder, a linker and a CAP group). A novel pan-HDACI, CKD5 is structurally different from other pan-HDACIs, such as SAHA and TSA. All three compounds have the same Zn-chelating moiety, hydroxamate, but CKD5 has an aliphatic linker and urea for the CAP group. SAHA and TSA were purchased from Sigma-Aldrich (St. Louis, MO, USA). All inhibitors were dissolved in dimethyl sulfoxide (Sigma Aldrich) to concentrations of 10 mM and stored at −80°C.
Cell viability and proliferation assay
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8; Dojindo, Japan). Briefly, cells (4×103 cells/well, SNU.GBM-2 and SNU.GBM-4 or 2×103 cells/well, U87, U251 and LN229) were plated in each well of a 96-well plate and exposed to different concentrations of HDACIs for 24, 48 and 72 h. The relative cell viability (%) was calculated using the equation ODT/ODC×100% (where ODT represents the absorbance of the treatment group, and ODC represents the absorbance of the control group) as reported previously [24]. The median inhibitory concentration (IC50) was defined as the drug concentration required to inhibit 50% of the cells relative to the controls. IC50 values were estimated from the dose-response curve. For further studies, the IC50 values for CKD5, SAHA and TSA were used for 72 h (Supplementary Table S1).
Apoptosis assay
Apoptotic cells were determined by annexin V-FITC and propidium iodide (PI) staining with a FITC-Annexin V apoptosis detection kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. The stained cells were then analyzed by FACS (BD), and the results were analyzed by CellQuest software (BD).
Cell cycle analysis
Cells (2 × 106 cells/well, SNU.GBM-2 and SNU.GBM-4 or 1 × 106 cells/well, U87, U251 and LN229) were plated in 100 mm plates and treated with IC50 values of the chemicals. After HDACI treatment with the given concentrations, cells were fixed using ice-cold 70% ethanol, washed with 1 × PBS and then suspended in propidium iodide (10 μg/ml) and ribonuclease A (0.1%). Cells were incubated for 30 min in the dark at room temperature. Propidium fluorescence was quantified after laser excitation of the fluorescent dye by FACS (BD) with a cell count of 10,000 cells per sample. Finally, DNA content of the cells in different phases of the cell cycle was determined using CellQuest Software (BD). We used synchronized cell population to perform the cell cycle and western blot analysis.
Western blot analysis
After treatment with HDACIs, the cells were lysed with lysis buffer (Cell Signaling, Danvers, MA) including a protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Western blot analysis was performed as previously reported [40]. Antibodies used for immunodetection were anti-p21 (1:1,000, Abcam, Cambridge, MA), anti-p27 (1:500, Abcam), anti-CDK2 (1:500, Thermo Scientific, Rockford, IL), anti-CDK4 (1:200, Abcam), anti-acetyl H3 (K9 + K14 + K18 + K23 + K27, 1:1,000, Abcam), anti-acetyl H3 (K27,1:2,000, Abcam), anti-acetyl H4 (K5 + K8 + K12 + K16,1:2,000, Abcam) and anti-β-actin (1:10,000, Sigma-Aldrich).
After the blot was washed, it was incubated with a horseradish peroxidase-conjugated species-specific secondary antibody (1:5,000, Jackson Laboratory, West Grove, PA) for 1 h at room temperature. The blots were developed using a chemiluminescence detection system (Invitrogen, Carlsbad, CA). Band density was analyzed using NIH ImageJ software. Densitometric measurements were performed on individual immunoblot for each antibody tested, and the values indicate the protein level normalized to the corresponding β-actin levels.
HDAC enzyme activity assay
Total HDAC enzyme activity was determined by the HDAC activity colorimetric assay kit (Biovision, Mountain View, CA) following the manufacturer’s protocol. The cells were treated with HDACIs and collected using lysis buffer (Cell Signaling, Danvers, MA). The protein concentration was determined with the BCA assay (Pierce, Rockford, IL). Equal amounts of proteins (30 μg) were analyzed at 405 nm.
In vivo short-term and long-term therapeutic efficacy
Animal experiments were carried out using a protocol approved by the animal facility of the Seoul National Institutional Animal Care and Use Committee (IACUC # 13-0363-C1A0) in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health. Seven-week-old female BALB/c nude mice were purchased from OrientBio (Seongnam, Korea). The mice were anesthetized by intraperitoneal injection of a solution of 30 mg/kg zoletil (Virbac) and 10 mg/kg xylazine (Bayer Korea). U87 cells (1 × 105) were injected stereotactically into the brains using a stereotactic device (1 mm anterior and 2 mm lateral to the bregma, 3 mm depth from the dura).
Five days after U87 tumor implantation, intracisternal injection of HDACIs was applied [41, 42]. Mice were randomly divided into three groups: the control, CKD5 or TSA treatment group (N = 8–11 per group). The groups were administered various concentrations (0.6, 0.8, 1.0 or 2.0 mg/kg) of CKD5 or TSA dissolved in 0.5% dextrose buffer every three days for two weeks. The mouse intrathecal catheter (32G, ALZET Osmotic Pumps, Cupertino, CA) was prepared as described previously [41] and inserted into a microsyringe (10 μl; Hamilton Company, NV). The infusion pump (Harvard Apparatus) was connected to the microsyringe. After anesthesia, the mice were secured in a stereotaxic frame, and the cisterna magna was punctured with a 30G needle to drain the cerebrospinal fluid. Then, the prepared catheter was inserted into the cisterna magna. HDACIs were slowly delivered in 5 μl volume at a rate of 1 μl/min. In control mice (N = 8), the cisterna magna was punctured and injected with only 0.5% dextrose.
To confirm short-term therapeutic efficacy, the mice were sacrificed after 28 days, and tumor volumes were measured as described previously [40]. To assess long-term therapeutic efficacy, mouse survival was measured as described previously [40]. Discomfort or distress was assessed by animal care personnel with no knowledge of the protocol design. All euthanized animals were verified as bearing tumors by necropsy.
Immunofluorescence staining
After sacrifice, the mice were perfused, and the whole brain was fixed in 4% paraformaldehyde and embedded in OCT compound with 10 μm sections. Hematoxylin-eosin staining and immunofluorescence with antibodies against anti-Ki67 (1:200, Abcam), anti-p21 (1:400, Cell Signaling), anti-CCND1 (1:100, Thermo Scientific), anti-cleaved caspase-3 (1:200, Abcam), and anti-acetyl (Ac)-H3 (1:200, Abcam) were performed. The number of positive cells was counted in randomly taken pictures (5 pictures from each tumor, 8 tumors in each experimental group) by a confocal microscope and graphed.
Statistical analysis
Statistical analysis of the data was performed using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA). All the experiments were performed more than three times to confirm the significance. To compare the mean variables among the groups and procedures, one-way or two-way ANOVA was used. Group means were compared by a Bonferroni’s post -test. A Kaplan-Meier curve was used for survival analysis. p < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (1420020).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest except Chang Sik Lee and Ju-Hee Lee. Chang Sik Lee and Ju-Hee Lee are employees of CKD Pharm, which owns patents related to CKD5.
Authors’ contributions
SKK supervised the development of the work, obtained funding, gave precious advices concerning data interpretation and finally approved the article. SAC performed the experiments, interpreted data and wrote the manuscript. PAK performed animal experiments. CKP reviewed and developed the basic concept of the project. KCW, JHP and JYL critically reviewed the manuscript. CSL and JHL contributed the CKD5 and reviewed the manuscript. All authors read and approved the final manuscript.
REFERENCES
1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008; 359:492–507. doi: 10.1056/NEJMra0708126.
2. Haar CP, Hebbar P, Wallace GCt, Das A, Vandergrift WA, 3rd, Smith JA, Giglio P, Patel SJ, Ray SK, Banik NL. Drug resistance in glioblastoma: a mini review. Neurochem Res. 2012; 37:1192–200. doi: 10.1007/s11064-011-0701-1.
3. Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007; 110:13–24. doi: 10.1002/cncr.22741.
4. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10:459–66. doi: 10.1016/S1470-2045(09)70025-7.
5. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006; 6:38–51. doi: 10.1038/nrc1779.
6. Adamopoulou E, Naumann U. HDAC inhibitors and their potential applications to glioblastoma therapy. Oncoimmunology. 2013; 2:e25219. doi: 10.4161/onci.25219.
7. Jin H, Liang L, Liu L, Deng W, Liu J. HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J Cell Biochem. 2013; 114:1498–509. doi: 10.1002/jcb.24491.
8. Lee P, Murphy B, Miller R, Menon V, Banik NL, Giglio P, Lindhorst SM, Varma AK, Vandergrift WA, 3rd, Patel SJ, Das A. Mechanisms and clinical significance of histone deacetylase inhibitors: epigenetic glioblastoma therapy. Anticancer Res. 2015; 35:615–25.
9. Singh MM, Johnson B, Venkatarayan A, Flores ER, Zhang J, Su X, Barton M, Lang F, Chandra J. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro Oncol. 2015; 17:1463–73. doi: 10.1093/neuonc/nov041.
10. Singh MM, Manton CA, Bhat KP, Tsai WW, Aldape K, Barton MC, Chandra J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011; 13:894–903. doi: 10.1093/neuonc/nor049.
11. Cuperlovic-Culf M, Touaibia M, St-Coeur PD, Poitras J, Morin P, Culf AS. Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites. 2014; 4:807–30. doi: 10.3390/metabo4030807.
12. Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol. 2014; 31:985. doi: 10.1007/s12032-014-0985-5.
13. Egler V, Korur S, Failly M, Boulay JL, Imber R, Lino MM, Merlo A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin Cancer Res. 2008; 14:3132–40. doi: 10.1158/1078-0432.CCR-07-4182.
14. Entin-Meer M, Yang X, VandenBerg SR, Lamborn KR, Nudelman A, Rephaeli A, Haas-Kogan DA. In vivo efficacy of a novel histone deacetylase inhibitor in combination with radiation for the treatment of gliomas. Neuro Oncol. 2007; 9:82–8. doi: 10.1215/15228517-2006-032.
15. Bojang P, Jr., Ramos KS. The promise and failures of epigenetic therapies for cancer treatment. Cancer Treat Rev. 2014; 40:153–69. doi: 10.1016/j.ctrv.2013.05.009.
16. Cornago M, Garcia-Alberich C, Blasco-Angulo N, Vall-Llaura N, Nager M, Herreros J, Comella JX, Sanchis D, Llovera M. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 2014; 5:e1435. doi: 10.1038/cddis.2014.412.
17. Rahman R, Osteso-Ibanez T, Hirst RA, Levesley J, Kilday JP, Quinn S, Peet A, O’Callaghan C, Coyle B, Grundy RG. Histone deacetylase inhibition attenuates cell growth with associated telomerase inhibition in high-grade childhood brain tumor cells. Mol Cancer Ther. 2010; 9:2568–81. doi: 10.1158/1535-7163.MCT-10-0272.
18. Horing E, Podlech O, Silkenstedt B, Rota IA, Adamopoulou E, Naumann U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. 2013; 33:1351–60.
19. Baek SY, Kim SR, Bae MK, Hwang JW, Kim JS, Choi YH, Wee HJ, Kim BS, Kim JB, Yoon S, Bae SK. Trichostatin A increases the thermosensitivity of human glioblastoma A172 cells. Neurosci Lett. 2006; 396:230–4. doi: 10.1016/j.neulet.2005.11.041.
20. Huang WJ, Lin CW, Lee CY, Chi LL, Chao YC, Wang HN, Chiou BL, Chen TJ, Huang CY, Chen CN. NBM-HD-3, a novel histone deacetylase inhibitor with anticancer activity through modulation of PTEN and AKT in brain cancer cells. J Ethnopharmacol. 2011; 136:156–67. doi: 10.1016/j.jep.2011.04.034.
21. Kulp SK, Chen CS, Wang DS, Chen CY, Chen CS. Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin Cancer Res. 2006; 12:5199–206. doi: 10.1158/1078-0432.CCR-06-0429.
22. Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, Richon VM. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002; 99:11700–5. doi: 10.1073/pnas.182372299.
23. Kang MR, Kang JS, Han SB, Kim JH, Kim DM, Lee K, Lee CW, Lee KH, Lee CH, Han G, Kang JS, Kim HM, Park SK. A novel delta-lactam-based histone deacetylase inhibitor, KBH-A42, induces cell cycle arrest and apoptosis in colon cancer cells. Biochem Pharmacol. 2009; 78:486–94. doi: 10.1016/j.bcp.2009.05.010.
24. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011; 3:166–79.
25. Kim HR, Kim EJ, Yang SH, Jeong ET, Park C, Lee JH, Youn MJ, So HS, Park R. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp Mol Med. 2006; 38:616–24. doi: 10.1038/emm.2006.73.
26. Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. J Surg Res. 2007; 142:219–26. doi: 10.1016/j.jss.2006.12.555.
27. Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, Girard L, Martinez ED. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer. 2012; 106:116–25. doi: 10.1038/bjc.2011.532.
28. Sarcar B, Kahali S, Chinnaiyan P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J Neurooncol. 2010; 99:201–7. doi: 10.1007/s11060-010-0127-7.
29. Tamannai M, Farhangi S, Truss M, Sinn B, Wurm R, Bose P, Henze G, Riabowol K, von Deimling A, Tallen G. The inhibitor of growth 1 (ING1) is involved in trichostatin A-induced apoptosis and caspase 3 signaling in p53-deficient glioblastoma cells. Oncol Res. 2010; 18:469–80.
30. Wolf IM, Fan Z, Rauh M, Seufert S, Hore N, Buchfelder M, Savaskan NE, Eyupoglu IY. Histone deacetylases inhibition by SAHA/Vorinostat normalizes the glioma microenvironment via xCT equilibration. Sci Rep. 2014; 4:6226. doi: 10.1038/srep06226.
31. Xie L, Santhoshkumar P, Reneker LW, Sharma KK. Histone deacetylase inhibitors trichostatin A and vorinostat inhibit TGFbeta2-induced lens epithelial-to-mesenchymal cell transition. Invest Ophthalmol Vis Sci. 2014; 55:4731–40. doi: 10.1167/iovs.14-14109.
32. Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, Richon VM, Scheithauer B, Giannini C, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009; 27:2052–8. doi: 10.1200/JCO.2008.19.0694.
33. Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E, Jaeckle K, Galanis E. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012; 14:215–21. doi: 10.1093/neuonc/nor198.
34. Palmieri D, Lockman PR, Thomas FC, Hua E, Herring J, Hargrave E, Johnson M, Flores N, Qian Y, Vega-Valle E, Taskar KS, Rudraraju V, Mittapalli RK, et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin Cancer Res. 2009; 15:6148–57. doi: 10.1158/1078-0432.CCR-09-1039.
35. Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, Garcia-Vargas J, Sanz-Rodriguez C, Rizvi S. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009; 2:31. doi: 10.1186/1756-8722-2-31.
36. Kerr JS, Galloway S, Lagrutta A, Armstrong M, Miller T, Richon VM, Andrews PA. Nonclinical safety assessment of the histone deacetylase inhibitor vorinostat. Int J Toxicol. 2010; 29:3–19. doi: 10.1177/1091581809352111.
37. San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Gunther A, Nakorn TN, Siritanaratkul N, Corradini P, Chuncharunee S, Lee JJ, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014; 15:1195–206. doi: 10.1016/S1470-2045(14)70440-1.
38. Dimopoulos M, Siegel DS, Lonial S, Qi J, Hajek R, Facon T, Rosinol L, Williams C, Blacklock H, Goldschmidt H, Hungria V, Spencer A, Palumbo A, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 2013; 14:1129–40. doi: 10.1016/S1470-2045(13)70398-X.
39. Carraway HE, Gore SD. Addition of histone deacetylase inhibitors in combination therapy. J Clin Oncol. 2007; 25:1955–6. doi: 10.1200/JCO.2006.09.8293.
40. Choi SA, Choi JW, Wang KC, Phi JH, Lee JY, Park KD, Eum D, Park SH, Kim IH, Kim SK. Disulfiram modulates stemness and metabolism of brain tumor initiating cells in atypical teratoid/rhabdoid tumors. Neuro Oncol. 2015; 17:810–21. doi: 10.1093/neuonc/nou305.
41. Chen Y, Imai H, Ito A, Saito N. Novel modified method for injection into the cerebrospinal fluid via the cerebellomedullary cistern in mice. Acta Neurobiol Exp (Wars). 2013; 73:304–11.
42. Rangroo Thrane V, Thrane AS, Plog BA, Thiyagarajan M, Iliff JJ, Deane R, Nagelhus EA, Nedergaard M. Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci Rep. 2013; 3:2582. doi: 10.1038/srep02582.