
INTRODUCTION
Genome-wide association studies (GWAS) premised on the “common disease, common variants” hypothesis have made great strides in identifying common genetic variants associated with a variety of phenotypes [1]. Typically, the GWAS-identified variants for any particular phenotype cumulatively explain only a small portion of the phenotypic variation [2]. One explanation for the so-called missing heritability problem is that the variants identified by GWAS are often only proxies of the causal variants that still remain to be discovered [3, 4]. The association signal attenuates as the linkage disequilibrium (LD) between the causal variant(s) and the GWAS-discovered variant decreases, particularly when the causal variant(s) are rare in the population. Until recently, the majority of GWAS have focused on populations of European descent, where the causal variants can be far from the associated marker due to strong LD, making it difficult to localize the causal variants [5, 6]. Thus, trans-ethnic follow-up studies in populations with lower LD are becoming more common [7–13]. Studies in populations with lower average LD often yield shorter distances between causal variants and the associated marker, helping to narrow down the causal variants underlying the disease associations [5, 14].
We previously developed the novel preferential LD approach to identify causal variants that drive the GWAS signal of interest from a comprehensive genome-wide variant catalog [15]. This approach is premised on the notion that the LD between the causal variant(s) and the GWAS-discovered variant is stronger than the LD between the causal variant(s) and most other variants interrogated in the GWAS, even if the causal variants are rare and only weakly linked to the GWAS-discovered variant. The increasing number of publications where causal variants are in only weak LD with the GWAS hits emphasizes the need for approaches beyond the absolute magnitude of the LD [16–29]. Our approach selects candidate causal variants at the locus of the GWAS-discovered variant from the variant catalog and prioritizes them based on the tagging specificity of the candidate variants by the GWAS-discovered variant, as well as functional importance of the candidate variants. We showed that the approach could successfully pinpoint the known causal variants of diverse traits when the discovery population and follow-up population were from the same ethnic group [15]. We anticipated that the preferential LD approach would also perform well in a trans-ethnic setting, which leverages the benefit of shorter LD structure. As the preferential LD approach does not rely on any phenotypic information, another advantage of the approach is its ability to follow up GWAS of various phenotypes using the same comprehensive variant catalog, such as the 1000 Genomes project [30, 31] or other large-scale sequencing efforts. This feature allows the approach to make the most use of the rapidly accumulating variant data from next-generation sequencing.
In this study, we evaluated the performance of the preferential LD approach in an African American (AA) population, following up breast cancer GWAS hits. We carried out the study in the African American Breast Cancer Epidemiology and Risk (AMBER) consortium, which provides rich genetic and epidemiological resources for investigating breast cancer risk in AA women [32–36]. To date, a large number of GWAS and ensuing meta-analyses have been performed on breast cancer susceptibility, with more than 90 loci meeting the stringent criteria of genome-wide significance level identified [37–39]. However these GWAS were predominantly performed in European populations, with only two conducted among individuals of African ancestry [40, 41]. Even in the well-studied European populations, collectively, the GWAS-discovered variants only account for an estimated 16% of breast cancer heritability [39]. The large missing heritability emphasizes the need to pinpoint causal variants or better markers of breast cancer.
We first sought to evaluate the 74 GWAS-identified breast cancer risk variants (Supplementary Table S1) in 8,315 AA women (3,648 cases, including 1,977 ER+ cases and 1,092 ER- cases, and 4,667 controls) from the AMBER consortium [34–36]. We then followed up those GWAS signals that were replicated in AAs by utilizing the preferential LD approach and the comprehensive variant catalog of African population from 1000 Genomes Project, to search for candidate causal variants or better breast cancer markers in an AA population. The variants selected by the preferential LD approach were genotyped in the AMBER consortium and then compared with all neighboring 1000 Genomes variants in evaluation of the approach’s performance in a trans-ethnic setting.
RESULTS
Replication of GWAS-discovered variants in AA women from the AMBER consortium
Out of the 74 GWAS-discovered variants, only 18 were associated with overall breast cancer risk in AMBER with nominal p-value < 0.05 (Table 1). All of them had association directions consistent with previous reports. The replication rate was higher for the variants discovered initially in populations of African ancestry [53] (4/7 or 57.1%) than those discovered in populations of non-African ancestry (14/67 or 20.9%).
Table 1: Replicated GWAS-discovered variants in AMBER imputation data
GWAS-discovered Variants |
Region |
Neighboring Genes |
GWAS Population |
Risk allele |
RAFa |
Imputation Qualityb |
OR |
p-valuec |
|---|---|---|---|---|---|---|---|---|
Overall breast cancer risk |
||||||||
rs4849887 |
2q14.2 |
LOC84931, GLI2 |
European ancestry |
C |
0.7124 |
0.9967 |
1.1086 |
6.85E-03 |
rs13000023 |
2q35 |
TNP1, DIRC3 |
African American |
G |
0.8480 |
1.0079 |
1.1787 |
6.13E-04 |
rs16857609 |
2q35 |
DIRC3 |
European ancestry |
T |
0.2483 |
1.0099 |
1.1196 |
4.16E-03 |
rs13387042 |
2q35 |
TNP1, DIRC3 |
European ancestry |
A |
0.7291 |
1.0121 |
1.0841 |
0.0365 |
rs10069690 |
5p15.33 |
TERT |
European ancestry |
T |
0.5955 |
1.0237 |
1.1107 |
2.40E-03 |
rs1432679 |
5q33.3 |
EBF1 |
European ancestry |
C |
0.7989 |
1.0163 |
1.1373 |
2.58E-03 |
rs9693444 |
8p12 |
DUSP4, LINC00589 |
European ancestry |
A |
0.3833 |
0.9835 |
1.0781 |
0.0339 |
rs1011970 |
9p21.3 |
CDKN2B-AS1 |
European ancestry |
T |
0.3312 |
1.0244 |
1.0740 |
0.0473 |
rs2981578 |
10q26 |
FGFR2 |
African American |
C |
0.8542 |
1.001 |
1.2520 |
4.99E-06 |
rs2981579 |
10q26.13 |
FGFR2 |
European ancestry |
A |
0.6069 |
1.0172 |
1.1187 |
1.30E-03 |
rs1219648 |
10q26.13 |
FGFR2 |
European ancestry |
G |
0.4286 |
1.0068 |
1.0766 |
0.0324 |
rs2981582 |
10q26.13 |
FGFR2 |
European ancestry |
A |
0.4802 |
1.0005 |
1.0716 |
0.0438 |
rs3817198 |
11p15.5 |
LSP1 |
European ancestry |
C |
0.1651 |
0.9933 |
1.0962 |
0.0472 |
rs609275 |
11q13 |
MYEOV, CCND1 |
African American |
C |
0.5751 |
1.0104 |
1.1513 |
1.22E-04 |
rs6504950 |
17q22 |
STXBP4 |
European ancestry |
G |
0.6551 |
1.0166 |
1.0743 |
0.0452 |
rs3745185 |
19p13 |
BABAM1 |
African American |
G |
0.7775 |
1.0119 |
1.1853 |
3.85E-05 |
rs2363956 |
19p13.11 |
ANKLE1 |
European ancestry |
T |
0.5136 |
1.0042 |
1.1365 |
1.92E-04 |
rs8170 |
19p13.11 |
BABAM1 |
European ancestry |
A |
0.1993 |
1.0066 |
1.1465 |
1.36E-03 |
ER+ breast cancer risk |
||||||||
rs13387042 |
2q35 |
TNP1, DIRC3 |
European ancestry |
A |
0.7272 |
1.0028 |
1.1077 |
0.0287 |
rs2981579 |
10q26.13 |
FGFR2 |
European ancestry |
A |
0.6021 |
1.0196 |
1.1004 |
0.0224 |
rs3112572 |
16q12 |
LOC643714 |
African American |
A |
0.2151 |
0.9970 |
1.1447 |
6.45E-03 |
rs3745185 |
19p13 |
BABAM1 |
African American |
G |
0.7705 |
1.0119 |
1.1375 |
8.84E-03 |
ER– breast cancer risk |
||||||||
rs8170 |
19p13.11 |
BABAM1 |
European ancestry |
A |
0.1943 |
0.9951 |
1.1866 |
8.38E-03 |
rs2363956 |
19p13.11 |
ANKLE1 |
European ancestry |
T |
0.5069 |
1.0014 |
1.1823 |
1.44E-03 |
rs4245739 |
1q32.1 |
MDM4 |
European ancestry |
C |
0.2405 |
1.0135 |
1.1490 |
0.0198 |
rs10069690 |
5p15.33 |
TERT |
European ancestry |
T |
0.5972 |
1.0313 |
1.3217 |
2.47E-07 |
rs1432679 |
5q33.3 |
EBF1 |
European ancestry |
C |
0.7956 |
1.0240 |
1.2758 |
2.92E-04 |
a: risk allele frequency in AMBER imputation data.
b: the information metric from IMPUTE2.
c: the p-values were based on logistic regression between variant genotypes and breast cancer status while controlling for other covariates (see Methods).
Of the 74 GWAS-discovered variants, eight and fifteen have been reported to be associated with ER+ and ER- breast cancer, respectively [53–58] (Supplementary Table S2). We then tested the association of these particular variants with the corresponding breast cancer subtypes. Four of the eight GWAS-discovered ER+ breast cancer variants were found to affect ER+ breast cancer risk in the AMBER Consortium (nominal p-value < 0.05), including rs3112572 at chromosome 16q12, which was not replicated when tested for overall cancer risk (Table 1). For ER- breast cancer, five of the fifteen GWAS-discovered ER- breast cancer variants were associated, including rs4245739 at chromosome 1q32, which was not associated with overall breast cancer risk (Table 1). Taking into account these additional replicated loci by ER status, the replication rate increased to 27.0% (20/74) for all variants, and 71.4% (5/7) and 22.4% (15/67) for the variants discovered in populations of African and non-African ancestry, respectively. These observations are consistent with the notion that it is increasingly difficult to replicate GWAS findings across populations as the replication population becomes more genetically distant from the GWAS population.
Dissecting the replicated GWAS signals
We next used the preferential LD approach to search for nearby (±500 kb) candidate causal variants or better markers with lower association p-values for the 20 GWAS-discovered variants replicated in the AMBER consortium. A total of 5,451 candidates were identified by our approach from 127,697 variants in the 1000 Genomes variant catalog that lie within 500 kb of the GWAS-discovered variants (0.29−0.85 candidates per 1 kb for each GWAS-discovered variant). Among these, 77,608 of the 1000 Genomes variants including 4,932 of the preferential LD selected candidates were successfully genotyped or imputed in the AMBER consortium and were tested for association with breast cancer risk. After accounting for the pairwise correlations among these variants, the effective number of independent tests was 19,617 [59]. Using a Bonferroni correction for the effective number of independent tests, we required a significance level of 2.55 x 10-6 to reach study-wide significance. We compared the candidate variants selected by our approach with all 1000 Genomes variants to evaluate the performance of the preferential LD approach for association with breast cancer risk.
We first focused on the 18 loci where the GWAS-discovered variants were associated with overall breast cancer risk in the AMBER consortium (Table 2). In general, we observed that markers with lower p-values are more enriched among the candidate variants selected by the preferential LD approach than among all neighboring 1000 Genomes variants (Figure 1). Four variants passed the study-wide significance cutoff: rs73731716 (p=1.33×10-6) in TERT locus and three variants in ANKLE1-BABAM1 locus, rs11668840, rs8100241, and rs12982058 (p=1.51×10-6, 2.29×10-6, and 2.33×10-6 respectively) (Table 3 and Figure 2). All four variants except rs8100241 were selected by the preferential LD approach. In the ANKLE1-BABAM1 locus, variants rs8100241 and rs12982058 were no longer significant after conditioning on rs11668840 (p=0.7897 and 0.7954 respectively), suggesting the three variants represent the same signal. Previously, Chen et al found rs11668840 to be the strongest signal in this locus only for ER- breast cancer in AAs. For overall breast cancer risk in AAs, rs3745185, instead, was the most significant variant in this locus [53]. In the AMBER consortium, we found rs11668840 to be the most significant variant associated with both overall breast cancer and ER- breast cancer. For 14 of the 18 loci, the preferential LD approach was able to identify a better marker with a more significant p-value than the GWAS-discovered variant. Furthermore, for seven loci, the preferential LD approach was able to select the best marker with the lowest association p-value among all neighboring 1000 Genomes variants. In contrast, the best marker can only be found in one locus if selecting fine-mapping variants based on high LD with the GWAS signals (r2>0.6). For an additional six loci, the best candidate identified by the preferential LD approach was among the top 10 best markers in the corresponding locus. It is worth noting that for 11 of the above 13 loci, the best candidates identified by this approach were only in weak LD (r2<0.6) with the GWAS-discovered variants, which demonstrates the ability of this approach to pinpoint candidates even when they were not strongly linked with the GWAS-discovered variants. For example, rs73731716, the best marker identified by preferential LD approach by following up the GWAS signal rs10069690, is also the best marker among the 6,912 tested 1000 Genomes variants at the surrounding TERT locus. It is significantly associated with overall breast cancer risk with p-value 1.33×10-6 but only has weak LD with the GWAS signal (r2=0.015).
Table 2: The performance of the preferential LD approach in identifying the best markers in the GWAS locia
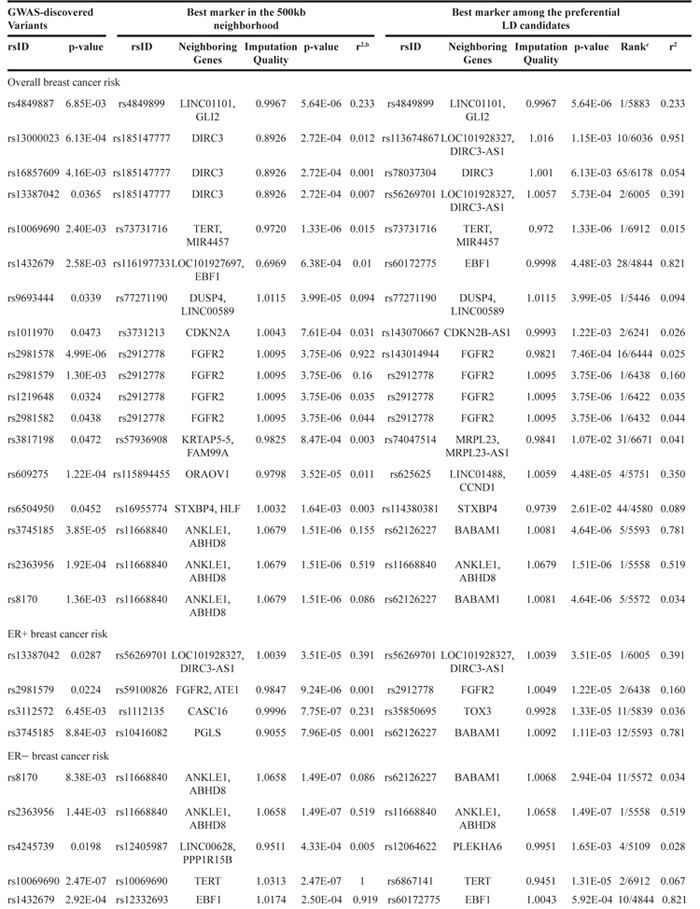
a: the p-values were based on logistic regression between variant genotypes and breast cancer status while controlling for other covariates (see Methods).
b: calculated from the 1000 Genomes African population using Haploview.
c: the rank of the best marker identified by the preferential LD approach among all variants from the 1000 Genomes African population in the 500kb neighborhood of the GWAS-discovered variant.
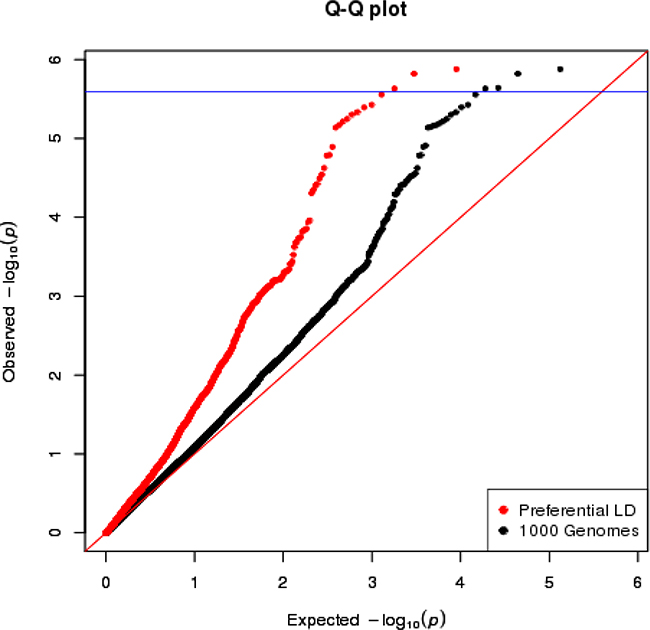
Figure 1: The QQ plot of overall breast cancer association p-values in AMBER consortium. The variants selected by the preferential LD approach in the 18 replicated loci are in red. The 1000 Genomes variants in the same 18 loci are in black. The blue horizontal line corresponds to the study-wide significance cutoff 2.55×10-6.
Table 3: The 1000 Genome variants that passed study-wide significance when tested for association with breast cancer risk
rsIDa |
Position |
Neighboring Genes |
Allele |
Frequency |
Imputation Quality |
OR |
p-valueb |
Conditional p-valuec |
|---|---|---|---|---|---|---|---|---|
Overall breast cancer risk |
||||||||
rs73731716 |
5:1298680 |
TERT, MIR4457 |
G |
0.1073 |
0.9720 |
1.3123 |
1.33E-06 |
- |
rs11668840 |
19:17399625 |
ANKLE1, ABHD8 |
C |
0.4081 |
1.0679 |
0.8493 |
1.51E-06 |
- |
rs8100241 |
19:17392894 |
ANKLE1 |
A |
0.3980 |
1.0114 |
0.8476 |
2.29E-06 |
0.7897 |
rs12982058 |
19:17409380 |
ABHD8 |
T |
0.3995 |
1.0130 |
0.8477 |
2.33E-06 |
0.7954 |
ER+ breast cancer risk |
||||||||
rs1112135 |
16:52639755 |
CASC16 |
T |
0.3250 |
0.9996 |
1.2420 |
7.75E-07 |
- |
rs4238750 |
16:52639236 |
CASC16 |
T |
0.3250 |
1.0003 |
1.2417 |
7.90E-07 |
- |
ER– breast cancer risk |
||||||||
rs11668840 |
19:17399625 |
ANKLE1, ABHD8 |
C |
0.4132 |
1.0658 |
0.7589 |
1.49E-07 |
- |
rs10069690 |
5:1279790 |
TERT |
T |
0.5972 |
1.0313 |
1.3217 |
2.47E-07 |
- |
rs61494113 |
19:17401859 |
ANKLE1, ABHD8 |
A |
0.4019 |
0.9992 |
1.3153 |
2.51E-07 |
0.1068 |
rs12974508 |
19:17401521 |
ANKLE1, ABHD8 |
T |
0.3983 |
1.0045 |
0.7594 |
4.10E-07 |
0.9825 |
rs12982058 |
19:17409380 |
ABHD8 |
T |
0.4044 |
1.0129 |
0.7614 |
4.23E-07 |
0.7954 |
rs8100241 |
19:17392894 |
ANKLE1 |
A |
0.4032 |
1.0099 |
0.7610 |
4.28E-07 |
0.7897 |
rs28473003 |
19:17406167 |
ABHD8 |
T |
0.3443 |
0.9850 |
1.3092 |
9.79E-07 |
0.1241 |
a: the variants selected by the preferential LD approach are in bold.
b: the p-values were based on logistic regression between variant genotypes and breast cancer status while controlling for other covariates (see Methods).
c: p-value after conditioning on rs11668840.
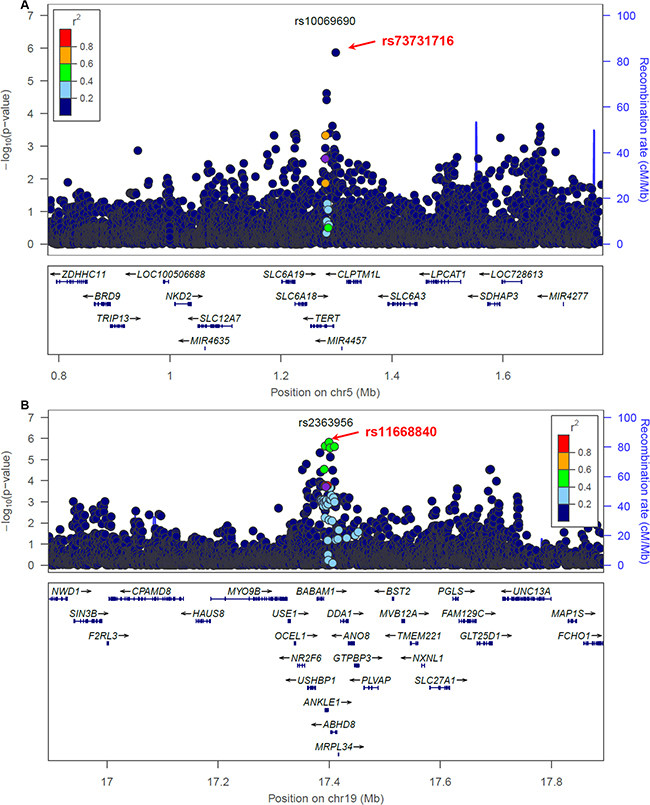
Figure 2: Breast cancer association of variants within 500 kb of rs10069690 A. and rs2363956 B. in the AMBER cohort. The GWAS-discovered variants were denoted by the purple circles.
We next investigated the 9 loci where the GWAS-discovered variants were associated with breast cancer by ER status in the AMBER consortium (Table 2). The association between ER+ breast cancer and two variants in the neighborhood of GWAS-discovered variant rs3112572, rs1112135 and rs4238750, passed study-wide significance (Table 3 and Figure 3). These two variants were not included in the preferential LD candidates because they are more common than the GWAS-discovered variant rs3112572 in the 1000 Genomes African population (see Method). We will loosen up this requirement in future trans-ethnic applications of the preferential LD approach, as the allele frequencies can change substantially when the follow-up population is not the same as the GWAS population. In two replicated ER+ breast cancer loci, the best candidates identified by the preferential LD approach were among the top 10 best markers (Table 2). These include rs2912778 (p=1.22×10-5, ranked no.2 among 6438 tested 1000 Genomes variants) for the locus surrounding GWAS signal rs2981579 (p=0.0224), and rs56269701 (p=3.51×10-5, ranked no.1 among 6005 tested 1000 Genomes variants) for the locus surrounding GWAS signal rs13387042 (p=0.0287). The association between ER- breast cancer and seven 1000 Genomes variants passed study-wide significance, including the GWAS-discovered variant rs10069690 and six variants in ANKLE1-BABAM1 locus (Table 3). All six variants except rs8100241 were selected by the preferential LD approach but none were significant after conditioning on rs11668840. In four of the five replicated ER- breast cancer loci, the best candidates identified by the preferential LD approach were among the top 10 best markers (Table 2).
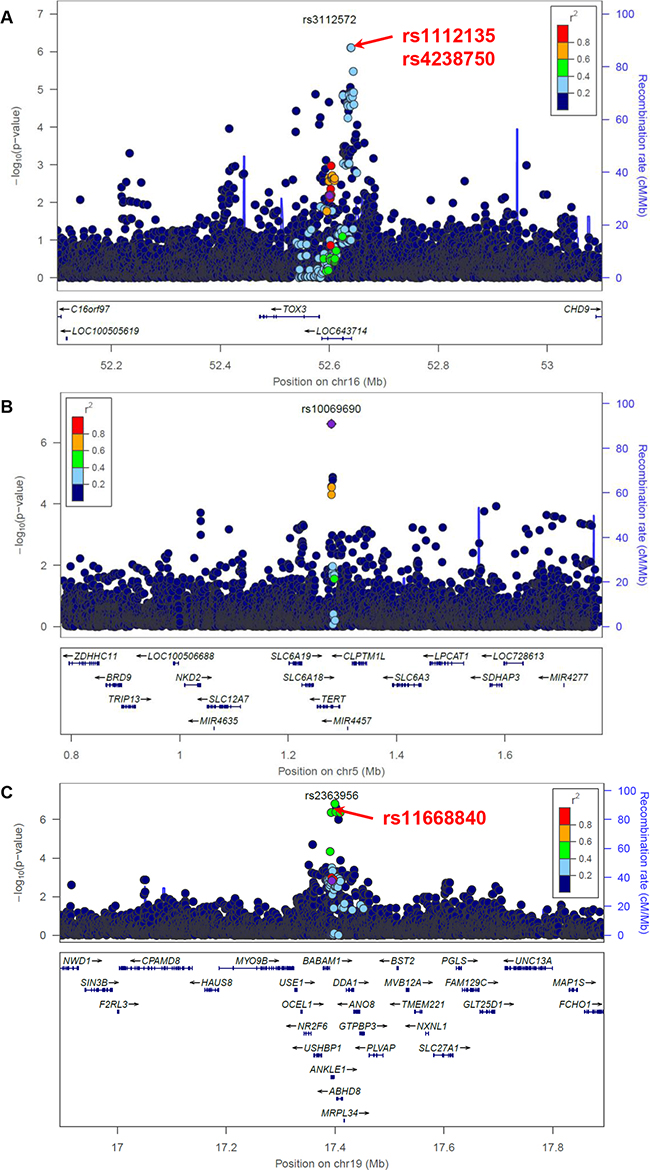
Figure 3: Association between variants within 500 kb of rs3112572 and ER+ breast cancer A., between variants within 500 kb of rs10069690 B. and rs2363956 C. and ER- breast cancer in the AMBER cohort. The GWAS-discovered variants were denoted by the purple circles.
An example: dissecting a GWAS signal with known causal variant within FGFR2 locus in AA
FGFR2 locus was one of the first breast cancer loci identified by GWAS. The most strongly associated GWAS-variant is rs2981582, a non-coding variant in intron 2 of FGFR2, which encodes the fibroblast growth factor receptor 2 [7]. Further fine-mapping studies led to the identification of rs2981578 as the most likely causal variant in this locus [8, 60, 61]. The variant rs2981578 resides in a FoxA1 binding site in an enhancer of FGFR2 gene. The cancer risk allele (C) triggers stronger FoxA1 and PolII binding, enhanced transcription activity, and increased FGFR2 expression level [8, 60, 61]. The GWAS-discovered variant rs2981582, a tag of the causal variant, correlates with rs2981578 in European and Asian populations (r2=0.64 and 0.31 respectively), but the correlation is much lower in AAs (r2=0.05). Consistent with the LD, the association between rs2981582 and breast cancer risk is very strong in European individuals, more modest in Asian individuals, and mostly attenuated in AAs [8, 61]. In the AMBER Consortium, we also found rs2981582 was only nominally associated with overall breast cancer risk (p=0.0438). Nevertheless, rs2981578 was still the most likely causal variant of this locus in African-descent populations (p=4.99×10-6). After conditioning on the causal variant rs2981578, the major association signal in this locus was completely eliminated (Figure 4). The MAF for rs2981578 is much lower than the GWAS-discovered variant rs2981582 in the 1000 Genomes AFR population (7.9% and 48.8% respectively). When using the preferential LD approach to follow up rs2981582, we assumed HapMap II variants from YRI population (~2.9 M markers) as the genotyped variants in the original GWAS because the variants in the GWAS genotyping platform, an early SNP array at Perlegen Sciences with 266,732 variants, were not available (see Method). Although this assumption resulted in the removal of causal variant rs2981578 from the preferential LD candidates as it is a HapMAP variant in YRI population, our approach did identify rs2912778, which was most strongly associated with breast cancer in this locus in the AMBER consortium (p=3.75×10-6), as a candidate causal variant. Variant rs2912778 is also a non-coding variant in intron 2 of FGFR2 and it is highly correlated with the causal variant rs2981578 (r2=0.92, MAF= 8.5% in 1000 Genomes AFR population). This finding further confirms the ability of the preferential LD approach to identify rarer and weakly tagged causal variants in a trans-ethnic setting.
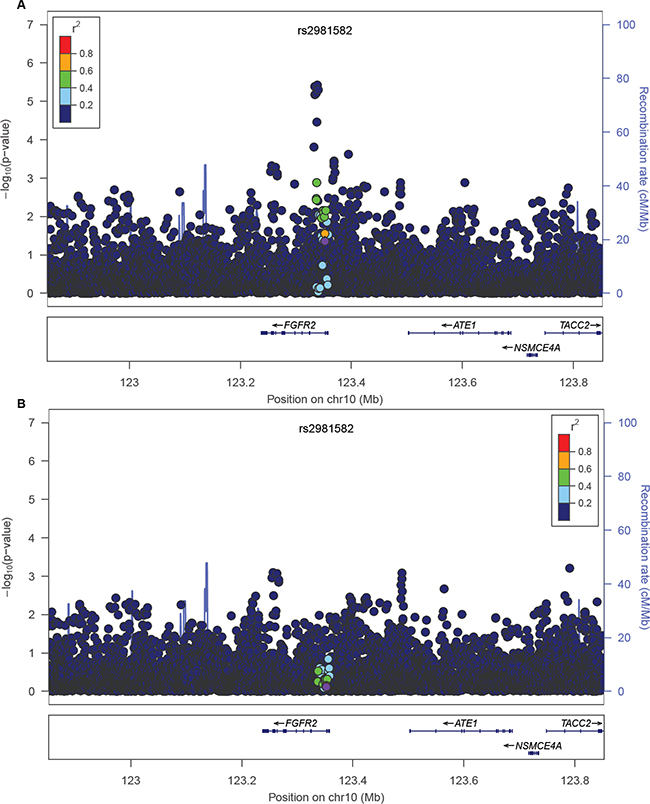
Figure 4: Breast cancer association of variants within 500 kb of rs2981582 in the AMBER cohort. The GWAS-discovered variant rs2981582 is denoted by the purple circle. The -logP values before A. and after B. conditioning on the causal variant rs2981578 were shown.
DISCUSSION
In this study, we investigated the associations of 74 breast cancer risk variants previously discovered by GWAS in 8,315 AA women from the AMBER consortium. We found that the majority of the GWAS-discovered variants identified from non-African populations could not be replicated in AA women in the AMBER consortium, which is consistent with the literature. Previously, Long et al. investigated 67 GWAS-discovered variants in 1,231 AA cases and 2,069 controls, and found that only 10 of them (14.9%) were significantly associated (p<0.05) with overall breast cancer risk and by subtype. Chen et al. examined 19 risk variants identified by GWAS and Feng et al. further tested an additional 54 GWAS-discovered variants in AA women [53, 62]. Together, they showed that 12 of the 73 GWAS-discovered variants (16.4%) could be replicated in their study population of 5,761 AA women at p<0.05. A recent large multi-consortium (including AMBER) fine-mapping study of breast cancer risk variants revealed that 10 of 73 (13.7%) tested GWAS hits were associated with breast cancer in 6,522 AA cases and 7,643 controls (under review, Haiman et al.). The consistently low replication rate from studies conducted by us and others [63, 64] reinforce the conclusion that it is challenging to extrapolate the GWAS variants identified from non-African populations to African ancestry populations, and highlight the challenge of trans-ethnic follow-up studies of GWAS hits.
Besides sample size and statistical power, a number of additional factors could contribute to the low replication rate and therefore have direct influence on the trans-ethnic application of the preferential LD approach. First, the causal variants can be population specific [4, 14]. For example, the causal variants of nondiabetic end-stage renal disease in APOL1 gene are common in AAs but absent in European Americans [65]; the cardiomyopathy causal variant at MYBPC3 is common in individuals from South Asia but not observed elsewhere [66]; and the ABCA1 variant that reduces cholesterol efflux is Native-American ancestry specific [67]. As the GWAS-discovered variants, in general, are not the disease causing mutations but are linked to the causal variants, GWAS association signals resulting from a population-specific causal variant may disappear in other populations. Evidence is emerging that rare variants significantly contribute to cancer susceptibility including bladder cancer [23], ovarian cancer [19], glioma [27], prostate cancer [28, 29], and colorectal cancer [68–70]. As rare variants tend to be population specific, the cross-population replication rate of GWAS findings in cancer is expected to be low. Second, even if the causal variants are identical across populations, they may have different effect size and allele frequencies [14]. For a replication population where the effect size or allele frequency of the causal variant is lower than in the discovery population, a much larger sample size is required to identify the same signal. Third, the tagging efficiency of the variants in the genotyping platform differs in populations [4, 5]. As has been seen in the FGFR2 locus, the GWAS-discovered variant in European population, rs2981582, is no longer a good proxy of the causal variant in AAs. With all of these considerations, well-powered breast cancer GWAS in African populations will not only improve the replication rate of the GWAS findings from other populations, but will also lead to identification of novel breast cancer loci that are specific to African ancestry. To date, there are only two moderately sized (~3,000 cases and ~3,000 controls) GWAS of breast cancer focused on women of African ancestry [40, 41], indicating the under representation of this population. In addition, as most commercially existing genotyping arrays were designed with a focus on populations of European descent, the overall genomic coverage based on LD is reduced in other populations, especially in populations of African ancestry, which are known for increased genome diversity and decreased levels of LD [5]. Therefore, the causal variants in African populations may not be well-captured using the existing genotyping platforms. Illumina has released its newly designed Infinium Multi-Ethnic Genotyping Array, which empowers GWAS studies in understudied populations including African-ancestry populations. Knowing that the GWAS signals exist in the populations of interest is essential to the success of follow-up studies using the preferential LD approach.
For the 20 loci where the GWAS-discovered variants were replicated in AA women in the AMBER consortium, we applied the preferential LD approach to search for causal variant candidates or better markers for AA breast cancer risk. The preferential LD approach could identify better markers in 16 of the 20 loci, and in 13 of them, the approach could identify markers that were top 10 among all genotyped and imputed 1000 genomes variants in the corresponding locus, which indicates the ability of this approach to follow up GWAS hits trans-ethnically. The preferential LD approach was developed to identify causal variants by following the GWAS-discovered variants even if the causal variants are much rarer. It is important to note that the goal of the preferential LD is to follow up a particular GWAS signal to search causal variants driving the corresponding association instead of fine mapping a GWAS locus (eg. selecting variants based on LD pruning) where additional risk variants independent of the GWAS signal can exist. The success of this approach relies on two factors: the presence of the causal variant in the variant catalog used by the approach, and the GWAS-discovered variant being the best tag for the causal variant. However, there were limitations for both of these two assumptions in the current trans-ethnic study. First, the currently best publicly available variant resource for African Population is from the 1000 Genomes Project, which includes 246 individuals of African-descent. As the causal variants of breast cancer are likely rare in the populations without breast cancer, they are expected to be depleted in this variant catalog, which was compiled from a relatively small number of healthy individuals. In our previous intra-ethnic application of the preferential LD approach, we utilized a comprehensive genome-wide variant catalog from 479 deeply sequenced individuals of European ancestry [15]. Being an understudied population, a comprehensive variant catalog is not readily available for populations of African descent. The recent availability of the extensive variant catalog from the UK10K project [71] and the Haplotype Reference Consortium [72] will significantly boost the genetic studies in European populations. The African Genome Variation Project (AGVP) [73] released recently and the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA) [74] are filling the gap for African populations. In contrast to CAAPA, the variants in AGVP were generated from genotyping and low-coverage sequencing and therefore rare variants may still be underrepresented in this dataset. Second, as described above, most of the GWAS-discovered variants in breast cancer were discovered in non-African population. Thus, the assumption of the GWAS-discovered variants being the best tags for the causal variants in African population is likely violated. Despite these limitations, we found that the preferential LD approach performed reasonably well in dissecting the GWAS signals in the AMBER consortium. Future studies with a more comprehensive variant catalog of African ancestry and the completion of well-powered breast cancer GWAS in African populations will be needed to better reveal the causal variants of breast cancer risk in AA women. In that case, the preferential LD approach can be directly carried out using the variant catalogs of AAs to follow up the AA GWAS-discovered variants, which is expected to be more powerful than the trans-ethnic application of this approach. On the other hand, given the promising trans-ethic performance of the preferential LD approach observed in this study, we anticipate more and more trans-ethic application of this approach in other human traits to be carried out in the future, especially when GWAS in the population of interest is not available.
MATERIALS AND METHODS
The AMBER consortium
The AMBER Consortium [32, 33] was formed in 2011 by combining data and biospecimens from four of the largest epidemiological studies of breast cancer in AA women: the Carolina Breast Cancer Study (CBCS) [42], the Women’s Circle of Health Study (WCHS) [43, 44], the Black Women’s Health Study (BWHS) [45] and the Multiethnic Cohort Study (MEC) [46].
The CBCS is a North Carolina population-based case control study of breast cancer. Breast cancer cases were identified using Rapid Case Ascertainment in cooperation with the NC Central Cancer Registry. Controls were identified using Division of Motor Vehicles lists for women under age 65 and Health Care Financing Administration lists for women 65 and older. The WCHS is a multi-site case control study in New York City (NYC) and New Jersey (NJ) aimed at evaluating risk factors for early and aggressive breast cancer in AA and European American (EA) women. Recruitment in NYC involved hospital-based ascertainment of cases, while controls were identified through random digit dialing (RDD). Cases in NJ were identified by the NJ State Cancer Registry using rapid case ascertainment. Controls were recruited though RDD and community-based efforts [47]. The BWHS is an ongoing prospective cohort study of health and illness among AA women, with a focus on cancer. Women diagnosed with breast cancer are identified by self-report in follow-up questionnaires, and confirmed by medical records, state cancer registries and the National Death Index. The MEC is a prospective cohort study that was designed to provide prospective data on cancer and other chronic diseases. Identification of incident breast cancer in study participants is by regular linkage with the Los Angeles County Cancer Surveillance Program and the State of California Cancer Registry. Controls in BWHS and MEC were chosen from among participants without breast cancer, and were frequency matched to cases on geographical region, sex, race, and 5-year age group.
All study participants provided consent for using their data and specimens for research purposes and the study was approved by Institutional Review Boards at participating institutions. ER status for cases was determined using pathology data from hospital records or cancer registry records.
Preferential LD approach
Our approach identifies candidate causal variants from a comprehensive variant catalog with four major steps [15]. First, we select variants that are: 1) in a 1 Mb interval centered on a GWAS-discovered variant, 2) have not been evaluated in the GWAS of interest, and 3) are rarer than the GWAS-discovered variant. Second, we identify the candidate variants that are preferentially tagged by the GWAS-discovered variant by calculating the preferential LD statistic, which estimates the percentage of all GWAS investigated variants that can tag the candidate variant better than or as well as the GWAS-discovered variant. Third, we perform permutation tests and keep the candidate variants that have non-random LD with the GWAS-discovered variant. Finally, we prioritize the candidate variants that are preferentially tagged by the GWAS-discovered variant and are functionally important on the basis of a sorting score that incorporates both the preferential LD statistic and evolutionary conservation. Candidate variants with statistically significant sorting scores are considered to be the candidates for causal variants driving the association between the GWAS-discovered variant and the phenotype of interest.
We used the preferential LD approach to follow up the GWAS-discovered breast cancer risk variants that were replicated in the AMBER consortium by utilizing the variant catalog from the 1000 Genomes African population (phase I release 3). As the preferential LD approach excludes variants that were already interrogated in the corresponding GWAS from the search of candidate causal variants, we used the variants in the HapMap phase II YRI (Yoruba in Ibadan, Nigeria) samples as the variants analyzed for meta-GWAS, where multiple genotyping platforms and imputation were used, or when the GWAS platform content was unavailable (Supplementary Table S3). When assessing the performance of the preferential LD approach, we compared the candidate causal variants selected by our approach to all 1000 Genome variants that were in the 1Mb interval centered on the GWAS-discovered variants and were available in the AMBER dataset.
Genotyping, quality control, and imputation
The GWAS-discovered variants and their corresponding followed-up variants selected by the preferential LD approach were added as part of the custom content to the Illumina Human Exome Beadchip v1.1 [48–51], and genotyped in CBCS, WCHS, and BWHS by the Center for Inherited Disease Research (CIDR). Variants successfully genotyped were subjected to stringent quality control (QC) metrics. Variants that are monomorphic variants, failed CIDR technical filters, or had missing rate ≥ 2%, > 1 Mendelian error in 17 HapMap trios, or > 2 discordant calls between 192 duplicate samples were omitted. DNA from a total of 6,828 unique participants from CBCS, WCHS, and BWHS were successfully genotyped and passed sample-level QC, including removal of samples with missing rate ≥ 2% and samples with unresolved identity. Variants from the 1000 Genomes project (Phase I v3, 11/23/2010) were imputed using SHAPEIT2 and IMPUTE2. In addition, 1528 AA subjects from MEC were previously genotyped with Illumina 1M-Duo chip and imputed with the 1000 Genomes reference panel. The genotyped and imputed genotypes for the CBCS, BWHS, and WCHS were combined with the MEC data to generate a complete AMBER analytical dataset. Variants were excluded if the minor allele frequency (MAF) was less than 0.6%, the imputation info score (INFO) was less than 0.5 in either set, or if the allele frequencies between the two sets differed by > 0.15.
Statistical analysis
As principal component analysis (PCA) using variants genotyped in all four studies of the AMBER consortium revealed little heterogeneity across studies, we analyzed all studies jointly. PCA analysis identified 35 population outliers. These outliers and six additional samples with missing phenotype information were omitted from association analyses. We used PLINK [52] to test the association between allelic dosage and susceptibility to breast cancer, ER+ breast cancer, and ER- breast cancer using logistic regression. Study, age, geographic location, DNA source, and principal components from the PCA analysis that had p-value <0.1 in the covariate-only models for the corresponding phenotype were included as covariates in the association models.
ACKNOWLEDGMENTS
This research was funded by the National Institutes of Health: P01 CA151135 (CBA, JRP and AFO), R01 CA058420 (LR), UM1 CA164974 (JRP and LR), R01 CA098663 (JRP), R01 CA100598 (CBA), R01 CA185623 (EVB, CCH, KD), UM1 CA164973 (LNK), R01 CA54281 (LNK), P50 CA58223 (MAT, AO), U01 CA179715 (MAT, AO), the Komen for the Cure Foundation, the Breast Cancer Research Foundation (CBA); and the University Cancer Research Fund of North Carolina. The Biostatistics and Bioinformatics Shared Resource is Roswell Park Cancer Center Support Grant (CCSG) shared resource supported by P30CA016056 from the National Cancer Institute.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Research. 2014; 42: D1001-D6. doi: 10.1093/nar/gkt1229.
2. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, et al. Finding the missing heritability of complex diseases. Nature. 2009; 461: 747-53.
3. Lee JC, Parkes M. Genome-wide association studies and Crohn’s disease. Briefings in Functional Genomics. 2011; 10: 71-6. doi: 10.1093/bfgp/elr009.
4. Bustamante CD, De La Vega FM, Burchard EG. Genomics for the world. Nature. 2011; 475: 163-5.
5. Teo Y-Y, Small KS, Kwiatkowski DP. Methodological challenges of genome-wide association analysis in Africa. Nat Rev Genet. 2010; 11: 149-60.
6. Li Y, Keating B. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Medicine. 2014; 6: 91.
7. Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007; 447: 1087-93. doi: http://www.nature.com/nature/journal/v447/n7148/suppinfo/nature05887_S1.html.
8. Udler MS, Meyer KB, Pooley KA, Karlins E, Struewing JP, Zhang J, Doody DR, MacArthur S, Tyrer J, Pharoah PD, Luben R, Collaborators S, Bernstein L, et al. FGFR2 variants and breast cancer risk: fine-scale mapping using African American studies and analysis of chromatin conformation. Human Molecular Genetics. 2009; 18: 1692-703. doi: 10.1093/hmg/ddp078.
9. Todd JA, Mijovic C, Fletcher J, Jenkins D, Bradwell AR, Barnett AH. Identification of susceptibility loci for insulin-dependent diabetes mellitus by trans-racial gene mapping. Nature. 1989; 338: 587-9.
10. Helgason A, Palsson S, Thorleifsson G, Grant SFA, Emilsson V, Gunnarsdottir S, Adeyemo A, Chen Y, Chen G, Reynisdottir I, Benediktsson R, Hinney A, Hansen T, et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet. 2007; 39: 218-25. doi: http://www.nature.com/ng/journal/v39/n2/suppinfo/ng1960_S1.html.
11. McKenzie CA, Abecasis GR, Keavney B, Forrester T, Ratcliffe PJ, Julier C, Connell JMC, Bennett F, McFarlane-Anderson N, Lathrop GM, Cardon LR. Trans-ethnic fine mapping of a quantitative trait locus for circulating angiotensin I-converting enzyme (ACE). Human Molecular Genetics. 2001; 10: 1077-84. doi: 10.1093/hmg/10.10.1077.
12. Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen W-M, Bonnycastle LL, Shen H, Timpson N, Lettre G, Usala G, Chines PS, Stringham HM, Scott LJ, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet. 2008; 40: 198-203. doi: http://www.nature.com/ng/journal/v40/n2/suppinfo/ng.74_S1.html.
13. Wu Y, Waite LL, Jackson AU, Sheu WHH, Buyske S, Absher D, Arnett DK, Boerwinkle E, Bonnycastle LL, Carty CL, Cheng I, Cochran B, Croteau-Chonka DC, et al. Trans-Ethnic Fine-Mapping of Lipid Loci Identifies Population-Specific Signals and Allelic Heterogeneity That Increases the Trait Variance Explained. PLoS Genet. 2013; 9: e1003379. doi: 10.1371/journal.pgen.1003379.
14. Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nature Reviews Genetics. 2010; 11: 356-66. doi: 10.1038/nrg2760.
15. Zhu Q, Ge D, Heinzen Erin L, Dickson Samuel P, Urban Thomas J, Zhu M, Maia Jessica M, He M, Zhao Q, Shianna Kevin V, Goldstein David B. Prioritizing Genetic Variants for Causality on the Basis of Preferential Linkage Disequilibrium. The American Journal of Human Genetics. 2012; 91: 422-34. doi: http://dx.doi.org/10.1016/j.ajhg.2012.07.010.
16. Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet. 2010; 86: 730-42. doi: S0002-9297(10)00204-1 [pii] 10.1016/j.ajhg.2010.04.003.
17. Fellay J, Thompson AJ, Ge D, Gumbs CE, Urban TJ, Shianna KV, Little LD, Qiu P, Bertelsen AH, Watson M, Warner A, Muir AJ, Brass C, et al. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature. 2010; 464: 405-8. doi: http://www.nature.com/nature/journal/v464/n7287/suppinfo/nature08825_S1.html.
18. Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C, Magnusson OT, Helgason A, Saemundsdottir J, Gylfason A, Stefansdottir H, Gretarsdottir S, Matthiasson SE, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011; 43: 316-20. doi: http://www.nature.com/ng/journal/v43/n4/abs/ng.781.html#supplementary-information.
19. Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, Besenbacher S, Lundin P, Stacey SN, Gudmundsson J, Magnusson OT, le Roux L, Orlygsdottir G, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011; 43: 1104-7. doi: http://www.nature.com/ng/journal/v43/n11/abs/ng.955.html#supplementary-information.
20. Sanna S, Li B, Mulas A, Sidore C, Kang HM, Jackson AU, Piras MG, Usala G, Maninchedda G, Sassu A, Serra F, Palmas MA, Wood WH, III, et al. Fine Mapping of Five Loci Associated with Low-Density Lipoprotein Cholesterol Detects Variants That Double the Explained Heritability. PLoS Genet. 2011; 7: e1002198.
21. Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, Whittaker P, Ranganath V, Kumanduri V, McLaren W, Holm L, Lindh J, Rane A, et al. A Genome-Wide Association Study Confirms VKORC1, CYP2C9, and CYP4F2 as Principal Genetic Determinants of Warfarin Dose. PLoS Genet. 2009; 5: e1000433. doi: 10.1371/journal.pgen.1000433.
22. Wadelius M, Chen L, Eriksson N, Bumpstead S, Ghori J, Wadelius C, Bentley D, McGinnis R, Deloukas P. Association of warfarin dose with genes involved in its action and metabolism. Human Genetics. 2007; 121: 23-34. doi: 10.1007/s00439-006-0260-8.
23. Tang W, Fu Y-P, Figueroa JD, Malats N, Garcia-Closas M, Chatterjee N, Kogevinas M, Baris D, Thun M, Hall JL, De Vivo I, Albanes D, Porter-Gill P, et al. Mapping of the UGT1A locus identifies an uncommon coding variant that affects mRNA expression and protects from bladder cancer. Human Molecular Genetics. 2012. doi: 10.1093/hmg/ddr619.
24. Thun GA, Imboden M, Ferrarotti I, Kumar A, Obeidat Me, Zorzetto M, Haun M, Curjuric I, Couto Alves A, Jackson VE, Albrecht E, Ried JS, Teumer A, et al. Causal and Synthetic Associations of Variants in the SERPINA Gene Cluster with Alpha1-antitrypsin Serum Levels. PLoS Genet. 2013; 9: e1003585. doi: 10.1371/journal.pgen.1003585.
25. Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, Quismorio FP, Reiff A, Myones BL, Kaufman KM, McCurdy D, Harley JB, Silverman E, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proceedings of the National Academy of Sciences. 2012; 109: E59–E67. doi: 10.1073/pnas.1113251108.
26. Croteau-Chonka DC, Wu Y, Li Y, Fogarty MP, Lange LA, Kuzawa CW, McDade TW, Borja JB, Luo J, AbdelBaky O, Combs TP, Adair LS, Lange EM, et al. Population-specific coding variant underlies genome-wide association with adiponectin level. Human Molecular Genetics. 2012; 21: 463-71. doi: 10.1093/hmg/ddr480.
27. Jenkins RB, Xiao Y, Sicotte H, Decker PA, Kollmeyer TM, Hansen HM, Kosel ML, Zheng S, Walsh KM, Rice T, Bracci P, McCoy LS, Smirnov I, et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet. 2012; 44: 1122-5. doi: http://www.nature.com/ng/journal/v44/n10/abs/ng.2388.html#supplementary-information.
28. Kote-Jarai Z, Amin Al Olama A, Leongamornlert D, Tymrakiewicz M, Saunders E, Guy M, Giles GG, Severi G, Southey M, Hopper JL, Sit KC, Harris JM, Batra J, et al. Identification of a novel prostate cancer susceptibility variant in the KLK3 gene transcript. Human Genetics. 2011; 129: 687-94. doi: 10.1007/s00439-011-0981-1.
29. Saunders EJ, Dadaev T, Leongamornlert DA, Jugurnauth-Little S, Tymrakiewicz M, Wiklund F, Al Olama AA, Benlloch S, Neal DE, Hamdy FC, Donovan JL, Giles GG, Severi G, et al. Fine-Mapping the HOXB Region Detects Common Variants Tagging a Rare Coding Allele: Evidence for Synthetic Association in Prostate Cancer. PLoS Genet. 2014; 10: e1004129. doi: 10.1371/journal.pgen.1004129.
30. The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010; 467: 1061-73. doi: http://www.nature.com/nature/journal/v467/n7319/abs/nature09534.html#supplementary-information.
31. The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491: 56-65. doi: http://www.nature.com/nature/journal/v491/n7422/abs/nature11632.html#supplementary-information.
32. Palmer JR, Viscidi E, Troester MA, Hong C-C, Schedin P, Bethea TN, Bandera EV, Borges V, McKinnon C, Haiman CA, Lunetta K, Kolonel LN, Rosenberg L, et al. Parity, Lactation, and Breast Cancer Subtypes in African American Women: Results from the AMBER Consortium. Journal of the National Cancer Institute. 2014; 106. doi: 10.1093/jnci/dju237.
33. Palmer J, Ambrosone C, Olshan A. A collaborative study of the etiology of breast cancer subtypes in African American women: the AMBER consortium. Cancer Causes & Control. 2014; 25: 309-19. doi: 10.1007/s10552-013-0332-8.
34. Yao S, Haddad SA, Hu Q, Liu S, Lunetta KL, Ruiz-Narvaez EA, Hong CC, Zhu Q, Sucheston-Campbell L, Cheng TY, Bensen JT, Johnson CS, Trump DL, et al. Genetic variations in vitamin D-related pathways and breast cancer risk in African American women in the AMBER consortium. Int J Cancer. 2016; 138: 2118-26. doi: 10.1002/ijc.29954.
35. Haddad SA, Lunetta KL, Ruiz-Narvaez EA, Bensen JT, Hong CC, Sucheston-Campbell LE, Yao S, Bandera EV, Rosenberg L, Haiman CA, Troester MA, Ambrosone CB, Palmer JR. Hormone-related pathways and risk of breast cancer subtypes in African American women. Breast Cancer Res Treat. 2015; 154: 145-54. doi: 10.1007/s10549-015-3594-x.
36. Cheng TY, Ambrosone CB, Hong CC, Lunetta KL, Liu S, Hu Q, Yao S, Sucheston-Campbell L, Bandera EV, Ruiz-Narvaez EA, Haddad S, Troester MA, Haiman CA, et al. Genetic variants in the mTOR pathway and breast cancer risk in African American women. Carcinogenesis. 2016; 37: 49-55. doi: 10.1093/carcin/bgv160.
37. Hindorff L, Junkins H, Hall P, JP M, TA M. A Catalog of Published Genome-Wide Association Studies. Available at: www.genome.gov/gwastudies. Accessed 9-15-2015.
38. Michailidou, K., et al., Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet, 2013. 45: p. 353-361. doi: http://www.nature.com/ng/journal/v45/n4/abs/ng.2563.html#supplementary-information.
39. Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Maranian MJ, Bolla MK, Wang Q, Shah M, Perkins BJ, Czene K, Eriksson M, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015; 47: 373-80.
40. Chen F, Chen G, Stram D, Millikan R, Ambrosone C, John E, Bernstein L, Zheng W, Palmer J, Hu J, Rebbeck T, Ziegler R, Nyante S, et al. A genome-wide association study of breast cancer in women of African ancestry. Human Genetics. 2013; 132: 39-48. doi: 10.1007/s00439-012-1214-y.
41. Song C, Chen GK, Millikan RC, Ambrosone CB, John EM, Bernstein L, Zheng W, Hu JJ, Ziegler RG, Nyante S, Bandera EV, Ingles SA, Press MF, et al. A Genome-Wide Scan for Breast Cancer Risk Haplotypes among African American Women. PLoS ONE. 2013; 8: e57298. doi: 10.1371/journal.pone.0057298.
42. Chen F, Chen GK, Millikan RC, John EM, Ambrosone CB, Bernstein L, Zheng W, Hu JJ, Ziegler RG, Deming SL, Bandera EV, Nyante S, Palmer JR, et al. Fine-mapping of breast cancer susceptibility loci characterizes genetic risk in African Americans. Hum Mol Genet. 2011; 20: 4491-503.
43. Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007; 39: 865-9.
44. Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K, Chatterjee N, Garcia-Closas M, Gonzalez-Bosquet J, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet. 2009; 41: 579-84.
45. Antoniou AC, Wang X, Fredericksen ZS, McGuffog L, Tarrell R, Sinilnikova OM, Healey S, Morrison J, Kartsonaki C, Lesnick T, Ghoussaini M, Barrowdale D, Peock S, et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet. 2010; 42: 885-92.
46. Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, Orr N, Rhie SK, Riboli E, Feigelson HS, Le Marchand L, Buring JE, Eccles D, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013; 45: 392-8.
47. Purrington KS, Slager S, Eccles D, Yannoukakos D, Fasching PA, Miron P, Carpenter J, Chang-Claude J, Martin NG, Montgomery GW, Kristensen V, Anton-Culver H, Goodfellow P, et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis. 2014; 35: 1012-9.
48. Gao X, Starmer J, Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol. 2008; 32: 361-9.
49. Meyer KB, Maia A-T, O’Reilly M, Teschendorff AE, Chin S-F, Caldas C, Ponder BAJ. Allele-Specific Up-Regulation of FGFR2 Increases Susceptibility to Breast Cancer. PLoS Biol. 2008; 6: e108. doi: 10.1371/journal.pbio.0060108.
50. Meyer Kerstin B, O’Reilly M, Michailidou K, Carlebur S, Edwards Stacey L, French Juliet D, Prathalingham R, Dennis J, Bolla MK, Wang Q, de Santiago I, Hopper John L, Tsimiklis H, et al. Fine-Scale Mapping of the FGFR2 Breast Cancer Risk Locus: Putative Functional Variants Differentially Bind FOXA1 and E2F1. The American Journal of Human Genetics. 2013; 93: 1046-60. doi: http://dx.doi.org/10.1016/j.ajhg.2013.10.026.
51. Feng Y, Stram DO, Rhie SK, Millikan RC, Ambrosone CB, John EM, Bernstein L, Zheng W, Olshan AF, Hu JJ, Ziegler RG, Nyante S, Bandera EV, et al. A comprehensive examination of breast cancer risk loci in African American women. Human Molecular Genetics. 2014; 23: 5518-26. doi: 10.1093/hmg/ddu252.
52. Huo D, Zheng Y, Ogundiran TO, Adebamowo C, Nathanson KL, Domchek SM, Rebbeck TR, Simon MS, John EM, Hennis A, Nemesure B, Wu SY, Leske MC, et al. Evaluation of 19 susceptibility loci of breast cancer in women of African ancestry. Carcinogenesis. 2012; 33: 835-40.
53. Hutter CM, Young AM, Ochs-Balcom HM, Carty CL, Wang T, Chen CT, Rohan TE, Kooperberg C, Peters U. Replication of breast cancer GWAS susceptibility loci in the Women’s Health Initiative African American SHARe Study. Cancer Epidemiol Biomarkers Prev. 2011; 20: 1950-9.
54. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010; 329: 841-5. doi: science.1193032 [pii] 10.1126/science.1193032.
55. Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, Tharkan JM, Vaideeswar P, Rathinavel A, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009; 41: 187-91. doi: http://www.nature.com/ng/journal/v41/n2/suppinfo/ng.309_S1.html.
56. Acuña-Alonzo V, Flores-Dorantes T, Kruit JK, Villarreal-Molina T, Arellano-Campos O, Hünemeier T, Moreno-Estrada A, Ortiz-López MG, Villamil-Ramírez H, León-Mimila P, Villalobos-Comparan M, Jacobo-Albavera L, Ramírez-Jiménez S, et al. A functional ABCA1 gene variant is associated with low HDL-cholesterol levels and shows evidence of positive selection in Native Americans. Human Molecular Genetics. 2010; 19: 2877-85. doi: 10.1093/hmg/ddq173.
57. Frayling IM, Beck NE, Ilyas M, Dove-Edwin I, Goodman P, Pack K, Bell JA, Williams CB, Hodgson SV, Thomas HJW, Talbot IC, Bodmer WF, Tomlinson IPM. The APC variants I1307K and E1317Q are associated with colorectal tumors, but not always with a family history. Proceedings of the National Academy of Sciences. 1998; 95: 10722-7.
58. Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature genetics. 2008; 40: 695-701. doi: 10.1038/ng.f.136.
59. Fearnhead NS, Wilding JL, Winney B, Tonks S, Bartlett S, Bicknell DC, Tomlinson IPM, Mortensen NJM, Bodmer WF. Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101: 15992-7. doi: 10.1073/pnas.0407187101.
60. The UKKC. The UK10K project identifies rare variants in health and disease. Nature. 2015; advance online publication. doi: 10.1038/nature14962. http://www.nature.com/nature/journal/vaop/ncurrent/abs/nature14962.html#supplementary-information.
61. McCarthy S, Das S, Kretzschmar W, Durbin R, Abecasis G, Marchini J. A reference panel of 64,976 haplotypes for genotype imputation. bioRxiv. 2015.
62. Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, Karthikeyan S, Iles L, Pollard MO, Choudhury A, Ritchie GRS, Xue Y, Asimit J, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015; 517: 327-32. doi: 10.1038/nature13997. http://www.nature.com/nature/journal/v517/n7534/abs/nature13997.html#supplementary-information.
63. The Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA).
64. Newman B, Moorman PG, Millikan R, Qaqish BF, Geradts J, Aldrich TE, Liu ET. The Carolina Breast Cancer Study: integrating population-based epidemiology and molecular biology. Breast Cancer Res Treat. 1995; 35: 51-60.
65. Ambrosone CB, Ciupak GL, Bandera EV, Jandorf L, Bovbjerg DH, Zirpoli G, Pawlish K, Godbold J, Furberg H, Fatone A, Valdimarsdottir H, Yao S, Li Y, et al. Conducting Molecular Epidemiological Research in the Age of HIPAA: A Multi-Institutional Case-Control Study of Breast Cancer in African-American and European-American Women. Journal of Oncology. 2009; 2009: 15. doi: 10.1155/2009/871250.
66. Bandera EV, Chandran U, Zirpoli G, McCann SE, Ciupak G, Ambrosone CB. Rethinking sources of representative controls for the conduct of case-control studies in minority populations. BMC Med Res Methodol. 2013; 13: 1471-2288.
67. Rosenberg L, Adams-Campbell L, Palmer JR. The Black Women’s Health Study: a follow-up study for causes and preventions of illness. J Am Med Womens Assoc. 1995; 50: 56-8.
68. Kolonel LN, Henderson BE, Hankin JH, Nomura AM, Wilkens LR, Pike MC, Stram DO, Monroe KR, Earle ME, Nagamine FS. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. Am J Epidemiol. 2000; 151: 346-57.
69. Bandera EV, Chandran U, Zirpoli G, McCann SE, Ciupak G, Ambrosone CB. Rethinking sources of representative controls for the conduct of case-control studies in minority populations. BMC Med Res Methodol. 2013; 13: 71. doi: 10.1186/1471-2288-13-71.
70. Huyghe JR, Jackson AU, Fogarty MP, Buchkovich ML, Stancakova A, Stringham HM, Sim X, Yang L, Fuchsberger C, Cederberg H, Chines PS, Teslovich TM, Romm JM, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet. 2013; 45: 197-201. doi: http://www.nature.com/ng/journal/v45/n2/abs/ng.2507.html#supplementary-information.
71. Peloso Gina M, Auer Paul L, Bis Joshua C, Voorman A, Morrison Alanna C, Stitziel Nathan O, Brody Jennifer A, Khetarpal Sumeet A, Crosby Jacy R, Fornage M, Isaacs A, Jakobsdottir J, Feitosa Mary F, et al. Association of Low-Frequency and Rare Coding-Sequence Variants with Blood Lipids and Coronary Heart Disease in 56,000 Whites and Blacks. The American Journal of Human Genetics. 2014; 94: 223-32. doi: http://dx.doi.org/10.1016/j.ajhg.2014.01.009.
72. Auer PL, Teumer A, Schick U, O’Shaughnessy A, Lo KS, Chami N, Carlson C, de Denus S, Dube M-P, Haessler J, Jackson RD, Kooperberg C, Perreault L-PL, et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet. 2014; advance online publication. doi: 10.1038/ng.2962. http://www.nature.com/ng/journal/vaop/ncurrent/abs/ng.2962.html#supplementary-information.
73. Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W, Guo Y, Zhang J, Langhammer A, Lochen M-L, Ganesh SK, Vatten L, Skorpen F, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet. 2014; 46: 345-51. doi: 10.1038/ng.2926. http://www.nature.com/ng/journal/v46/n4/abs/ng.2926.html#supplementary-information.
74. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics. 2007; 81: 559-75. doi: 10.1086/519795.