
INTRODUCTION
In the last years the modulation of immune checkpoint network is becoming an important therapeutic strategy for anti-cancer treatments. The activation of immune system able to kill the tumor represents the goal of cancer immunotherapy [1].
In anti-tumor immune response T lymphocytes represent the major components. The optimal recognition of the antigen induces a specific activation of T cells, followed by the acquisition of the effector function. It is particularly significant the differentiation of a specific subset of T cells, the cytotoxic T lymphocytes (CTLs), that are able to lyse target cells. In cancer, such as in chronic viral infection, the long exposure to the antigen leads to a dysfunction of T cells; in particular these cells lose their proliferation ability and progressively the capability to release cytokines, to eliminate pathogens and to kill target cells. This condition represents the state of “exhaustion”. Recent findings have defined the function of some receptors that negatively regulate T cell activity and promote exhaustion[2].
The intuition that targeting these receptors could dramatically influence T cell activity was originally of James P. Allison in his pioneer studies on cytotoxic T-lymphocyte antigen 4 (CTLA-4) inhibitory receptors. The idea was that if the negative regulation of T cells could be blocked, T cell responses would be expanded and sustained long enough to eliminate cancer [3][4]. The other important insight was translating the target from the cancer cell and its antigenic/genomic repertoire to the immune system unregarding the type of tumor and the antigens expressed. Several antibodies to different immune checkpoint inhibitors (ICIs) where then generated and tested in preclinical setting. They demonstrate capacity to unleash existing T cells in a unspecific mode, thus breaking the tolerance against self and non self neoantigens associated with the tumor and permitting the expansion of effector T cells able not only to recognize but also to destroy the tumor. Initial clinical trial results were exciting, ICI therapy led to tumor regression and improved survival in a subgroup of metastatic melanoma, lung carcinoma, renal cell carcinoma and lymphoma patients. Clinical trials are currently exploring combination therapies. The first ICIs approved by FDA are directed against the CTLA-4, programmed death receptor-1 (PD-1) and programmed death receptor-1 ligand (PD-L1) [5] .
A distinguishing feature that was observed since the first report of ICI anti-CTLA-4 treatment in metastatic melanoma patients is the observation that the responding patients showed durable complete responses. The response is maintained for a long time after the end of the treatment and long time survivors up to ten years and cured patients are now a reality [6] .
Several factors appear to governate the efficacy of these treatments. Pre existing endogenous natural or induced anti tumor immunity is one of the variables that has been associated with increased response. Interference with inhibitory pathways in the effector T cells and concomitant removal of immune-suppressive cells such as Treg cells are also dominant mechanisms of enhanced anti-tumor activity [7]. Oncologists have now tested the powerful potential of ICI treatment in cancer. Activated T cells unleashed from negative brakes are able to rapidly find target tumor cells, kill also significant tumor burden and maintain memory and control of recurrences. In order to proceed with novel combination of ICI and integration of these novel treatments with chemo/radiotherapy and target therapies, oncologists are now focalizing attention and research efforts on the management of novel array of immune related toxicities. The new side effects described for ICI treatments are in fact mainly immune related and autoimmunity classified, distinct from chemo and molecular targeted therapy and they have challenged greatly medical oncologists [8] . The most common toxicity observed included: diarrohea, colitis, thyroid disfunction, hypofisitis, liver disorder, dermatologic event and lung disorder. Altough these immune related side effects have become maneageable to some extent by the use of corticosteroid therapy, new predictive indicators of response and toxicity are necessary to improve the management and the compliance to immunotherapy.
In this setting among all the fields that are being explored, the study of the microbiome is showing interesting results mainly for two reasons, one comes from recent studies that have addressed the critical role that microbiome appears to have in the development of inflammation, cancer and in the integrity of mucosal immunity and the protection against pathogens. Second the high frequency of the severe diarrhea and colitis affecting ICI treated patients confirming a role of gut microbiome and suggesting possible microbiota influence on the therapeutic activity/toxicity of ICI immunotherapy.
In this review we discuss the new perspectives on the involvement of PD-1 and PDL-1 in the cross talk between gut microbiota and immune fitness and how gut microbiota impacts on the efficacy of anti-PD-1 and anti-PD-L1 treatments in cancer.
PD-1/PD-L1 axis: immunological and clinical implications
PD-1 receptor, also called CD279, represents one of the most important target for immunological therapy. It is an inhibitory receptor expressed by activated T lymphocytes, B cells, natural killer T cells (NKT) and Treg cells [9]. PD-1 is a member of the CD28 co-receptor family [10] and has a key role in the modulation of T cell function in peripheral tissue, recognizing PD-L1 and PD-L2. Both these ligands are expressed on antigen presenting cells (APCs); in addition PD-L1 is present also on the surface of several cells of lymphoid and non lymphoid tissue and it is expressed by tumor cells [11]. The function of PD-1 is mainly regulated by its cytoplasmatic domain, containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM). When PD-1 recognizes its ligand, this interaction induces the phosphorylation of the tyrosine residue in ITSM, recruiting the tyrosine phosphatase SHP2 that induces the dephosphorylation and inactivation of Zap70 in T cells, down regulating TCR signaling activation. Therefore PD-1 down regulating T cell activity, affects negatively immune response. When in tumor microenvironment PD-1 binds PD-L1, T cell function is attenuated, so that T lymphocytes become unable to target tumor cells. Hence anti-tumor response results strongly restrained and tumor evasion favored. Initially the role of PD-1 in modulating T cell activity was described in chronic viral infection. It was shown that during chronic infection of LCMV all specific CD8+ T cells expressed PD-1, instead during acute infection this receptor has not been detected on LCMV-specific memory CD8+ T cells. [12]. Since the interaction between PD-1/PD-L1 can be blocked by monoclonal antibodies, these are now considered novel therapeutic approaches to unleash the anti-tumor immune response. In fact it has been strongly suggested that immune evasion of cancer can be favored by the expression of PD-1 by tumor infiltrating lymphocytes (TILs) along with the expression of PD-L1 by tumor cells [13]. Many studies have shown that blockade of PD-1 or PD-L1 restores T cell function, induce an increase of IFNγ [14] and a decrease of immune suppressive cell subsets, such as MDSCs [15]. In fact PD-1 and PD-L1 blocking represents an extremely efficient approach in controlling tumor growth by changing the dynamic of the tumor microenvironment. Currently different monoclonal anti PD-1 and anti-PD-L1 antibodies are in development for the treatment of advanced disease; they include Nivolumab (OPDIVO, anti-PD 1) [16-23], Pembrolizumab (Keytruda, anti-PD-1) [24-34], Atezolizumab (anti PD-L1)[35-36] , Durvalumab (anti-PDL-1)[37-38] and many others. These agents while are revolutionizing cancer patients care[39] , have a precise pattern of toxicity, that can be classified as immune related. It is important today to understand better the variability observed in patient outcomes together with strategies to improve efficacy and identify parameters to select responsive patients. Microbiota could represent one physiological mechanism that can influence and modulate response to ICI treatments. The involvement of gut microbiota in the outcome of anti cancer therapy and the role of immune response create new questions from a preclinical and clinical standpoint in the cancer field [40] .
MICROBIOME AND CANCER
Gut microbiota complexity and behaviour deserve the definition of tissue organ, as introduced and thoroughly discussed by Burcelin and collaborators [41], a major immunological organ which means metabolic organ, that influences different pathways of whole metabolism.Therefore the intricacy of microbiota components, metabolic functions and signaling control of the host leads to revise the concept of gut-host relationship in term of gut-microbiota-host network. In particular there is a close relationship between the acquisition of microbiome and the maturation of immune system during ontogeny. Intestinal homeostasis is then maintained through an efficient and interacting immune network that permits tolerance to the microbiota while allowing responsiveness to invading pathogens. Different members of the microbiota and their components have been demonstrated to interact with specific immune components influencing the synthesis of regulatory cytokines.
The final decision towards tolerance vs reactivity is the result of integrated signals from the microbiota and immune/non immune cells in the local microenvironment [42] . The perturbation of gut microbiota, called intestinal dysbiosis, is involved in many pathological mechanisms. Recent studies demonstrated the associations between microbiota profiles and the development of adiposity, diabetes, dyslipidemia and other inflammatory conditions.[43-48]
The close association between cancer susceptibility [49-61], responsiveness to cancer therapy and microbiome has just been investigated. Infact it was shown that the production of IL-17 in response to change of microbiota composition is associated to rapid progression of colo-rectal cancer. Furthermore enteric bacterial genes metabolizing estrogens could modify the risk to develop hormone positive breast cancer in postmenopausal women. [62]
Recentely it was demonstrated that cyclophosphamide changes composition of microbiota and induces traslocation of bacteria (Lactobacillus jonsoniii and Enterococco hirae) in secondary lymphoids organ, like spleen and mesenteric lymph nodes, stimulating the production of Th17 and Th1 cells [63], demonstrating that bacteria modulate chemotherapeutic drug efficacy. Furthermore in tumor bearing mice the perturbation of intestinal microbiota caused by antibiotics treatment is associated with the reduction of synthesis of cytokines and the decreasing effect of both CPG- oligonucleotides immunotherapy and chemotherapy. It was demonstrated that microbiome is also with inflammation modyfing the expression of gene involved.[64]. In this study the authors show that different microbiota profiles are associated with the TNF response. In particular the presence of Ruminococcus ( Gram negative), and Alistipes ( Gram-positive) is involved in TNF production , while an enriched Lactobacillus microbiota correlates with the fail of response.
Thus microbiota may have a crucial role in influencing cancer treatment efficacy and considering the close interaction with immune system it’s reasonable to supposed its influence in response to ICIs or other immunotherapies.
In fact recently Vetizou et al. [65] demonstrated that germ free or antiobiotics treated mice had poor benefit from anti-CTLA-4 therapy and showed also that anti-CTLA-4 therapy can modify the composition of microbiota. Moreover a recent study established that microbiota composition enriched in Bacteroides phlilym can prevent the onset of immune colitis in patients treated with anti-CTLA-4 (Ipilimumab) [66]. This data support the idea that microbiota modifying immune response could influence the response of both chemotherapy and immunotherapy (Table 1).[67-72] Furthermore the microbiota profiles already studied in IBD and liver diseases could be useful to stratify cancer patients treated with ICIs [73-80].
Table 1: The immunological effects of gut microbiota
Bacteria |
Model |
Effects on immune system |
|
Lactobacillus johnsonii |
mouse |
Stimulates the differentiation of TH17 cells and Th1 cells |
Viaud 2013 |
Enterococcus hirae |
mouse |
Stimulates the differentiation of TH17 cells and Th1 cells |
Viaud 2013 |
Ruminococcus |
mouse |
TNF production, promotes response to immunotherapy |
Iida 2013 |
Alistipes shahii |
mouse |
TNF production, promotes response to immunotherapy |
Iida 2013 |
Lactobacillus fermentuum |
mouse |
TNF production , impairs response to immunotherapy |
Iida 2013 |
Bacteroides fragilis |
mouse |
Induces TH 1 in tumor draining lymph nodes. Promotes the maturation of intratumoral dendritic cells Increases the activity of anti-CTLA4 in vivo Reduces the inflammatory response Reduced histopathology signs of colitis induced by CTLA4 blockade |
Vetizou 2015 |
Bacteroides thetaiotamicron |
mouse |
Increseas the activity of anti-CTLA4 in vivo Reduced the inflammatory response |
Vetizou 2015 |
Bacteroidales |
mouse |
Decreased after CTLA4 blockade |
Vetizou 2015 |
Burkholderiales |
mouse |
Decreased after CTLA4 blockade |
Vetizou 2015 |
Clostridiales |
mouse |
Increased after CTLA4 blockade |
Vetizou 2015 |
Bifidobacterium breve, Bifidobacterium longum, Bifidobacterium adolescentis |
mouse |
Enhanced dendritic cells activation Increased CD8 +T cell accumulation, |
Sivan 2015 |
Bifidobacterium breve Bifidobacterium longu, |
mouse |
Improved the response to PDL-1 Improved IFNy levels |
Sivan 2015 |
Bacteroidetes |
human |
Enriched in colitis-resistant patients treated with ipilimumab |
Dubin 2015 |
Clostridium species |
mouse |
Stimulates the induction of suppressive FOXp3+ Treg |
Geuking 2011 |
Bacteroides fragilis |
mouse |
Stimulates the induction of suppressive FOXp3+ Treg |
Geuking 2011 |
Staphylococcus aureus |
mouse |
Converts CD4+ T cells into Foxp3+ Treg cell |
Hardis rabe 2013 |
Bacteroidaceae |
mouse |
Decreases in mice PD-1-/- |
Kawamoto 2012 |
Bifidobacterium |
mouse |
Decreases in mice PD-1-/- |
Kawamoto 2012 |
Enterobacteriaceae |
mouse |
Increases in mice PD1-/- |
Kawamoto 2012 |
Erysipelotrichaceae Prevvotellaceae Alcaligenacee TM7 incerte saedis |
mouse |
Increase in mice PD1-/- |
Kawamoto 2012 |
MICROBIOME and PD1-PD-L1 axis
It’s known that PD1-PDL1 axis plays a key role in the regulation of immune system and that immunotherapy is more efficient in T cell inflamed tumors rather than in T cell deficient tumors. Recent data support the hypothesis that microbiota shapes innate and adaptive immune system influencing PD-1-PD-L1 axis. In particular Sivan et al compared melanoma growth in mice derived from two different mouse facilities (JAX and TAC) harboring different intestinal microbiota but genetically similar [81]. They observed an higher rate of melanoma growth in TAC mice and a better response to PD-L1 treatment in JAX mice. Moreover the investigated the relationship between microbiota and immune cells demonstrating that Bifidobaterium seems to positively influence the number of activated antingen-presenting cells. Moreover the administration of Bifidobacterium to TAC mice improves tumor control and IFNγ production. Surprisingly the authors demonstrated that the combination of modulation of microbiota with anti-PD-L1 antibody improved tumor control. These data are very exciting because strongly suggest that different species can activate or conversely inhibit immune response.
Moreover the microbiota influences the development of regulatory T cells in mice, in particular germ free mice showed a lower amount of suppressive Foxp3+ Treg cells in the gut and the colonization of Clostridium species or Bacteroides fragilis stimulates the induction of suppressive Foxp3+ Treg cells in the intestine of these mice [82]. Furthermore neonatal human CD4+ T cells can be converted into Foxp3+ Treg cells by Staphylococcus aureus.
In fact S.aureus increases the expression of PD-L1 on APCs, and this is linked to the APCs ability to induce Foxp3+ Tregs. The interaction between PD-L1 and PD-1, expressed on T cells, prevents the TCR signaling within T cells, which leads to differentiation into Foxp3+ Tregs.
These data demonstrate a significant role of specific gut bacteria in influencing immune system and response to cancer therapies. But it’s equally true that the gut microbiota is itself modulated by immune response. In fact intestinal microbiota plays a crucial role in the development of gut immune system representing one of the first barrier against pathogens. Germ free mice presented reduced Pejer’s patches, levels of immunoglobulin A, intraepithelial lymphocytes and production of antimicrobial peptide. It was also demonstrated that recolonization with healthy mouse commensal microbiota can correct the immune deficiency.
Fargarsan showed that PD-1-/- mice have a significant alteration in microbiota composition (reduction of anaerobic bacteria, of Bifidobacterium and Bacteroidaceae, increase in Enterobacteriaceae and at the general level, increase in members of the Erysipelotrichaceae, Prevotellaceae, Alcaligenaceae and TM7 genera incertae sedis) and it’s supposed to be caused by a decreased capacity of IgA of binding bacteria[83]. Thus PD-1 is strongly associated with the maturation of antibody to maintain the integrity of intestinal mucosal barrier [84] .
One accredited hypothesis, proposed by Rescigno speculates that the immune system can be manipulated to alter gut microbiota composition. In this way microbiota could be induced to be less pro-inflammatory (i.e. more diverse and with a reduced level of innate immune activators), thus reducing susceptibility to inflammation or minimizing the progression of the damage [85-90].
FUTURE DIRECTION OF IMMUNOTHERAPEUTIC APPROACH
The main challenge today for the oncologists is to fully utilize the potential of ICI treatment in order to treat and cure the majority of patients, to limit the immune related events and toxicity and to better understand the dynamics of response to treatment. It is conceivable that in a very short time ICI treatment will be proposed for all tumors and in earlier setting in the different protocols. In this review we outline several recent findings that could help to draw a roadmap of clinical and laboratory criteria to help the oncologist in designing more efficient protocols of ICIs treatment (Figure 1). We hypothesize that the identification of different microbiome profiles (for example Bifidus enriched or Bacterioides enriched) could help us to establish classes of patients responders or at major risk to develop high grade toxicities. To better define the profile of our patients we could also consider the nutritional status and immune repertoire. The possibility of intervention is attractive. In fact diet, use of probiotics, prebiotics and antibiotics or stool transfer that can change microbiota profile, drugs that can modulate mucosal permeability and homeostasis as well as pretreatment immunotherapy/chemotherapy to increase the specific anti tumor T cell compartment are some of the strategies. We are today dealing with oncology treatments that have moved the attention from the tumor to the patients immune system and the multiple intersecting immunity regulatory networks. The further understanding of these mechanisms and the relation with clinical outcome will be the key for the development of protocols and guidelines for ICI treatment with maximized curative potential.
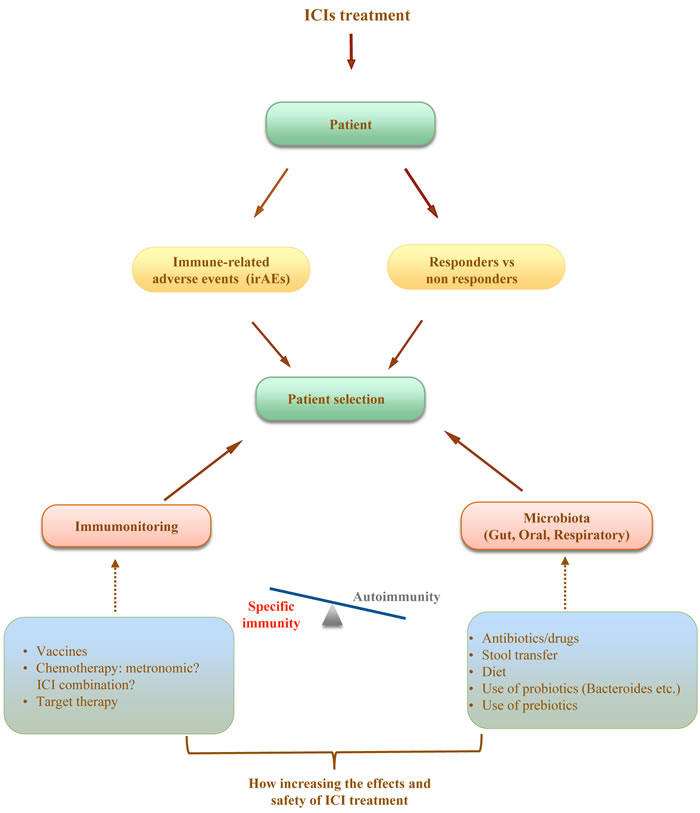
Figure 1: A new proprosed approach for the management of immunotherapy cancer treatment.
ACKNOWLEDGEMENTS
This work was supported by Associazione Italiana per la Ricerca sul Cancro (Marianna Nuti: AIRC 2015 cod.17432).
ConflictS of Interest
There is no conflict of interest.
REFERENCES
1. McNutt M. Cancer immunotherapy. Science [Internet]. 2013 [cited 2016 Oct 24]; 342: 1417. doi: 10.1126/science.1249481.
2. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat Rev Immunol. 2014; 15: 45–56. doi: 10.1038/nri3790.
3. Allison JP, Krummel MF. The Yin and Yang of T cell costimulation. Science [Internet]. 1995 [cited 2016 Oct 24]; 270: 932–3. Available from http://www.ncbi.nlm.nih.gov/pubmed/7481795.
4. Allison JP. Checkpoints. Cell [Internet]. 2015 [cited 2016 Oct 24]; 162: 1202–5. doi: 10.1016/j.cell.2015.08.047.
5. Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Letters. 2014. p. 368–76. doi: 10.1016/j.febslet.2013.10.015.
6. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen T-T, Berman DM, Wolchok JD. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015; 33: 1889–94. doi: 10.1200/JCO.2014.56.2736.
7. Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015; 15: 457–72. doi: 10.1038/nrc3973.
8. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, Massard C, Fuerea A, Ribrag V, et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. European Journal of Cancer. 2016. p. 139–48. doi: 10.1016/j.ejca.2015.11.016.
9. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013; 14: 1212–8. doi: 10.1038/ni.2762.
10. Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci U S A. 2008; 105: 10483–8. doi: 10.1073/pnas.0804453105.
11. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002; 169: 5538–45. doi: 10.4049/jimmunol.169.10.5538.
12. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439: 682–7. doi: 10.1038/nature04444.
13. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008; 8: 467–77. doi: 10.1038/nri2326.
14. Zang X, Allison JP. The B7 family and cancer therapy: Costimulation and coinhibition. Clinical Cancer Research. 2007. p. 5271–9. doi: 10.1158/1078-0432.CCR-07-1030.
15. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013; 19: 5636–46. doi: 10.1158/1078-0432.CCR-13-0458.
16. Sui X, Ma J, Han W, Wang X, Fang Y, Li D, Pan H, Zhang L. The anticancer immune response of anti-PD-1/PD-L1 and the genetic determinants of response to anti-PD-1/PD-L1 antibodies in cancer patients. Oncotarget. 2015; 6: 19393–404. doi: 10.18632/oncotarget.5107.
17. Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, Gerber DE, Shepherd FA, Antonia S, Goldman JW, Juergens RA, Laurie SA, Nathan FE, et al. Nivolumab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016; . doi: 10.1200/JCO.2016.66.9929.
18. Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jäger D, Pietanza MC, Le DT, de Braud F, Morse MA, Ascierto PA, Horn L, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. The Lancet Oncology. 2016; . doi: 10.1016/S1470-2045(16)30098-5.
19. Rizvi NA, Hellmann MD, Brahmer JR, Juergens RA, Borghaei H, Gettinger S, Chow LQ, Gerber DE, Laurie SA, Goldman JW, Shepherd FA, Chen AC, Shen Y, et al. Nivolumab in Combination With Platinum-Based Doublet Chemotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J Clin Oncol [Internet]. 2016 [cited 2016 Oct 24]; 34: 2969–79. doi: 10.1200/JCO.2016.66.9861.
20. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015; : 1803–13. doi: 10.1056/NEJMoa1510665.
21. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015; 373: 123–35. doi: 10.1056/NEJMoa1504627.
22. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015; 373: 23–34. doi: 10.1056/NEJMoa1504030.
23. McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I, Hodi FS, Kluger HM, Topalian SL, et al. Survival, Durable Response, and Long-Term Safety in Patients With Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab. J Clin Oncol. 2015; 33: 2013–20. doi: 10.1200/JCO.2014.58.1041.
24. Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, Tsiouris AJ, Cohen J, Vortmeyer A, Jilaveanu L, Yu J, Hegde U, Speaker S, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol [Internet]. 2016 [cited 2016 Oct 24]; 17: 976–83. doi: 10.1016/S1470-2045(16)30053-5.
25. Armand P, Shipp MA, Ribrag V, Michot J-M, Zinzani PL, Kuruvilla J, Snyder ES, Ricart AD, Balakumaran A, Rose S, Moskowitz CH. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J Clin Oncol [Internet]. American Society of Clinical Oncology; 2016 [cited 2016 Oct 24]; : JCO673467. doi: 10.1200/JCO.2016.67.3467.
26. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, Cheng JD, Chow LQ. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016; 17: 956–65. doi: 10.1016/S1470-2045(16)30066-3.
27. Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, Eder JP, Golan T, Le DT, Burtness B, McRee AJ, Lin CC, Pathiraja K, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016; 17: 717–26. doi: 10.1016/S1470-2045(16)00175-3.
28. Chatterjee M, Turner DC, Felip E, Lena H, Cappuzzo F, Horn L, Garon EB, Hui R, Arkenau H-TT, Gubens MA, Hellmann MD, Dong D, Li C, et al. Systematic evaluation of pembrolizumab dosing in patients with advanced non-small-cell lung cancer. TL - 27. Ann Oncol. 2016; 27 VN-r: 1291–8. doi: 10.1093/annonc/mdw174.
29. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu W-J, Weber JS, Gangadhar TC, Hersey P, Dronca R, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA [Internet]. 2016 [cited 2016 Oct 24]; 315: 1600–9. doi: 10.1001/jama.2016.4059.
30. Sul J, Blumenthal GM, Jiang X, He K, Keegan P, Pazdur R. U.S. Food and Drug Administration Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non-Small Cell Lung Cancer Whose Tumors Express Programmed Death-Ligand 1. Oncologist. 2016; : 1–8. doi: 10.1634/theoncologist.2015-0498.
31. Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, De Castro G, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet. 2016; 387: 1540–50. doi: 10.1016/S0140-6736(15)01281-7.
32. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, Cranmer LD, Blank CU, O’Day SJ, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015; 16: 908–18. doi: 10.1016/S1470-2045(15)00083-2.
33. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn M, Felip E, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372: 2018–28. doi: 10.1056/NEJMoa1501824.
34. Robert C, Schachter J, Long G V., Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015; 372: 2521–32. doi: 10.1056/NEJMoa1503093.
35. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016; 387: 1837–46. doi: 10.1016/S0140-6736(16)00587-0.
36. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar A V, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016; . doi: 10.1016/S0140-6736(16)00561-4.
37. Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, Curiel TJ, Colon-Otero G, Hamid O, Sanborn RE, ODonnell PH, Drakaki A, Tan W, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016; 34: 3119–25. doi: 10.1200/JCO.2016.67.9761.
38. Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, Narwal R, Steele K, Gu Y, Karakunnel JJ, Rizvi NA. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: A multicentre, phase 1b study. Lancet Oncol. 2016; 17: 299–308. doi: 10.1016/S1470-2045(15)00544-6.
39. Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Molecular Immunology. 2015. p. 4–17. doi: 10.1016/j.molimm.2015.02.009.
40. Karin M, Jobin C, Balkwill F. Chemotherapy, immunity and microbiota--a new triumvirate? Nat Med [Internet]. NIH Public Access; 2014 [cited 2016 Oct 24]; 20: 126–7. doi: 10.1038/nm.3473.
41. Burcelin R, Serino M, Chabo C, Garidou L, Pomié C, Courtney M, Amar J, Bouloumié A. Metagenome and metabolism: The tissue microbiota hypothesis. Diabetes, Obesity and Metabolism. 2013. p. 61–70. doi: 10.1111/dom.12157.
42. Flannigan KL, Geem D, Harusato A, Denning TL. Intestinal Antigen-Presenting Cells: Key Regulators of Immune Homeostasis and Inflammation. Am J Pathol. 2015; 185: 1809–19. doi: 10.1016/j.ajpath.2015.02.024.
43. Mai V. Dietary modification of the intestinal microbiota. Nutr Rev. 2004; 62: 235–42. doi: 10.1111/j.1753-4887.2004.tb00045.x.
44. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology [Internet]. 2009 [cited 2016 Oct 24]; 136: 65–80. doi: 10.1053/j.gastro.2008.10.080.
45. Tlaskalová-Hogenová H, Štěpánková R, Hudcovic T, Tučková L, Cukrowska B, Lodinová-Žádníková R, Kozáková H, Rossmann P, Bártová J, Sokol D, Funda DP, Borovská D, Řeháková Z, et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunology Letters. 2004. p. 97–108. doi: 10.1016/j.imlet.2004.02.005.
46. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky A V. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008; 455: 1109–13. doi: 10.1038/nature07336.
47. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012; 13: 991–9. doi: 10.1038/ni.2416; 10.1038/ni.2416.
48. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013; 500: 541–6. doi: 10.1038/nature12506.
49. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012; 22: 292–8. doi: 10.1101/gr.126573.111.
50. Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des. 2009; 15: 1524–7. doi: 10.2174/138161209788168191.
51. Zhu Q, Gao R, Wu W, Qin H. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumor Biology. 2013. p. 1285–300. doi: 10.1007/s13277-013-0684-4.
52. Chen H-M, Yu Y-N, Wang J-L, Lin Y-W, Kong X, Yang C-Q, Yang L, Liu Z-J, Yuan Y-Z, Liu F, Wu J-X, Zhong L, Fang D-C, et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am J Clin Nutr. 2013; 97: 1044–52. doi: 10.3945/ajcn.112.046607.
53. Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012; 6: 320–9. doi: 10.1038/ismej.2011.109.
54. Gold JS, Bayar S, Salem RR. Association of Streptococcus bovis bacteremia with colonic neoplasia and extracolonic malignancy: Commentary. Diseases of the Colon and Rectum. 2005. p. 180. doi: 10.1007/s10350-004-0732-7.
55. Peek Jr. RM, Blaser MJ, Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002; 2: 28–37. doi: 10.1038/nrc703.
56. Yang Y, Wang X, Huycke T, Moore DR, Lightfoot S a, Huycke MM. Colon Macrophages Polarized by Commensal Bacteria Cause Colitis and Cancer through the Bystander Effect. Transl Oncol. 2013; 6: 596–606. doi: 10.1593/tlo.13412.
57. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012; 22: 299–306. doi: 10.1101/gr.126516.111.
58. McIntosh GH, Royle PJ, Playne MJ. A probiotic strain of L. acidophilus reduces DMH-induced large intestinal tumors in male Sprague-Dawley rats. Nutr Cancer. 1999; 35: 153–9. doi: 10.1207/S15327914NC352_9.
59. Sonnenberg GF, Artis D. Innate Lymphoid Cell Interactions with Microbiota: Implications for Intestinal Health and Disease. Immunity. 2012. p. 601–10. doi: 10.1016/j.immuni.2012.10.003.
60. Wu S, Shin J, Zhang G, Cohen M, Franco A, Sears CL. The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect Immun. 2006; 74: 5382–90. doi: 10.1128/IAI.00060-06.
61. Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med [Internet]. NIH Public Access; 2009 [cited 2016 Oct 24]; 15: 1016–22. doi: 10.1038/nm.2015.
62. Kwa M, Plottel CS, Blaser MJ, Adams S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J Natl Cancer Inst. 2016; 108: djw029. doi: 10.1093/jnci/djw029.
63. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science (80- ). 2013; 342: 971–6. doi: 10.1126/science.1240537.
64. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten R a, Molina D a, Salcedo R, Back T, Cramer S, Dai R-M, Kiu H, Cardone M, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013; 342: 967–70. doi: 10.1126/science.1240527.
65. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM, Poirier-Colame V, Roux A, Becharef S, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (80- ). 2015; 350: 1079–84. doi: 10.1126/science.aad1329.
66. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, No D, Gobourne A, Littmann E, Huttenhower C, Pamer EG, Wolchok JD. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016; 7: 10391. doi: 10.1038/ncomms10391.
67. Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D’Urso MT, Belardelli F, Gabriele L, Proietti E, Bracci L. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res [Internet]. 2011 [cited 2016 Oct 24]; 71: 768–78. doi: 10.1158/0008-5472.CAN-10-2788.
68. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004; 34: 336–44. doi: 10.1002/eji.200324181.
69. Viaud S, Flament C, Zoubir M, Pautier P, LeCesne A, Ribrag V, Soria JC, Marty V, Vielh P, Robert C, Chaput N, Zitvogel L. Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res. 2011; 71: 661–5. doi: 10.1158/0008-5472.CAN-10-1259.
70. Yang J, Liu K, Qu J, Wang X. The changes induced by cyclophosphamide in intestinal barrier and microflora in mice. Eur J Pharmacol [Internet]. 2013 [cited 2016 Oct 24]; 714: 120–4. doi: 10.1016/j.ejphar.2013.06.006.
71. Zwielehner J, Lassl C, Hippe B, Pointner A, Switzeny OJ, Remely M, Kitzweger E, Ruckser R, Haslberger AG. Changes in Human Fecal Microbiota Due to Chemotherapy Analyzed by TaqMan-PCR, 454 Sequencing and PCR-DGGE Fingerprinting. Heimesaat MM, editor. PLoS One [Internet]. Public Library of Science; 2011 [cited 2016 Oct 24]; 6: e28654. doi: 10.1371/journal.pone.0028654.
72. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007; 13: 1050–9. doi: 10.1038/nm1622.
73. Strauss J, Kaplan GG, Beck PL, Rioux K, Panaccione R, Devinney R, Lynch T, Allen-Vercoe E. Invasive potential of gut mucosa-derived fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011; 17: 1971–8. doi: 10.1002/ibd.21606.
74. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host and Microbe. 2014. p. 317–28. doi: 10.1016/j.chom.2014.02.007.
75. Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: Inflammatory bowel disease and colitis-associated colon cancer. Frontiers in Immunology. 2012. doi: 10.3389/fimmu.2012.00107.
76. Imray CH, Radley S, Davis a, Barker G, Hendrickse CW, Donovan I a, Lawson a M, Baker PR, Neoptolemos JP. Faecal unconjugated bile acids in patients with colorectal cancer or polyps. Gut. 1992; 33: 1239–45. doi: 10.1136/gut.33.9.1239.
77. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007; 7: 41–51. doi: 10.1038/nri1995.
78. Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004; 127: 80–93. doi: 10.1053/j.gastro.2004.03.054.
79. Bonfrate L, Tack J, Grattagliano I, Cuomo R, Portincasa P. Microbiota in health and irritable bowel syndrome: current knowledge, perspectives and therapeutic options. Scand J Gastroenterol. 2013; 48: 995–1009. doi: 10.3109/00365521.2013.799220.
80. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009; 9: 313–24. doi: 10.1038/nri2515.
81. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Man Lei Y, Jabri B, Alegre M-L, Chang EB, Gajewski TF. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (80- ). 2015; 350: 1084–9. doi: 10.1126/science.aac4255.
82. Rabe H, Nordström I, Andersson K, Lundell AC, Rudin A. Staphylococcus aureus convert neonatal conventional CD4+ T cells into FOXP3+ CD25+ CD127low T cells via the PD-1/PD-L1 axis. Immunology. 2014; 141: 467–81. doi: 10.1111/imm.12209.
83. Maruya M, Kawamoto S, Kato LM, Fagarasan S. Impaired selection of IgA and intestinal dysbiosis associated with PD-1-deficiency. Gut Microbes [Internet]. Taylor & Francis; 2013 [cited 2016 Oct 24]; 4: 165–71. doi: 10.4161/gmic.23595.
84. Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato LM, Fagarasan S. The Inhibitory Receptor PD-1 Regulates IgA Selection and Bacterial Composition in the Gut. Science (80- ). 2012; 336: 485–9. doi: 10.1126/science.1217718.
85. Rescigno M. A “fit” microbiota to potentiate cancer immunotherapy. Genome Med. 2015; 7: 1–2. doi: 10.1186/s13073-015-0256-x.
86. Zagato E, Mazzini E, Rescigno M. The variegated aspects of Immunoglobulin A. Immunol Lett [Internet]. 2016 [cited 2016 Oct 24]; 178: 45–9. doi: 10.1016/j.imlet.2016.04.009.
87. Rescigno M. Tfr cells and IgA join forces to diversify the microbiota. Immunity.2014 Jul 17;41(1):9-11. doi: 10.1016/j.immuni.2014.06.012.
88. FransenF, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S, Chiavelli A, D’Erchia AM, Sethi MK, PabstO, Marzano M, Moretti S, Romani L et al. BALB/c and C57BL/6 mice differ in generation of antigen-specific IgA and microbiota diversity.Immunity 2015:43:527–40. doi: 10.1016/j.immuni.2015.08.011.
89. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–5. doi:10.1073/pnas.1002601107
90. Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007 Nov 15;2(5):328 39.10.1016/j.chom.2007.09.013