
Introduction
During the past three decades, it has become clear that viruses may have important player in the development of various human malignancies [1, 2]. It has been estimated that viral infections contribute to approximately 18% of all human cancers [1, 2]. Human tumor-associated viruses belong to two main families, i.e., RNA viruses (Retroviridae and Flaviviridae) and DNA viruses (Hepadnaviridae, Herpesviridae, and Papillomaviridae) [3]. DNA viruses that have been found to be associated with human malignancies include EBV (Epstein-Barr virus, Herpesviridae), HBV (Hepatitis B virus, Hepadnoviridae), HPV (Human papilloma virus, Papillomaviridae), HHV-8 (Human Herpesvirus-8, also known as Kaposi’s sarcoma-associated herpesvirus, Herpesviridae), MCV (Merkel cell polyomavirus, Polyomaviridae) [4], and EVB (Erythrovirus B19, Parvovirus). RNA viruses that have been found to be correlated with human malignancies include HTLV-1 (Human T-cell leukemia virus, Retroviridae), HCV (Hepatitis C virus, Flaviviridae). HTLV-1 has been found to be associated with adult T-cell leukemia [5], HHV-8 with primary effusion lymphoma and Castleman’s disease [6], MCV with Merkel cell carcinoma [4], EBV with Burkitt’s lymphoma, nasopharyngeal, colorectal, gastric cancers [7-9], post-transplant lymphomas and Hodgkin’s disease; HBV and HCV with hepatocellular carcinoma [10], EVB with thyroid cancer [11], HPV with head and neck squamous cell carcinoma (HNSCC), cervical, anal, penile and vulvar cancers [12]. Viruses with possible roles in human malignancies include (i) Polyomaviridae (DNA viruses), i.e., Simian virus 40 (SV40) in mesothelioma, brain and bone cancers, BK virus (BKV) in prostate and bladder cancers [13]; John Cunningham virus (JCV) in brain cancer [14], (ii) Circoviridae (DNA viruses), i.e., Tonque teno virus (TTV) in multiple myeloma, gastrointestinal, lung and breast cancers [15-17], and (iii) Retroviridae (RNA viruses), i.e., Human endogenous retroviruses (HERVs) in melanoma, germ cell, prostate, breast and ovarian cancers [18-24], and human mammary tumor virus (HMTV) in breast and endometrial cancers [25, 26].
Advances in the next-generation sequencing (NGS) technologies, including whole genome sequencing (WGS), whole exome sequencing (WES), RNA sequencing (RNA-seq), miRNA sequencing (miRNA-seq), whole genome bisulfite sequencing (WGBS), its corresponding analytical tools and ‘omics’ techniques, have provided comprehensive novel genomic and epigenomic information on HPV-associated cancers. NGS approaches have e.g. successfully been used for the identification and characterization of viral integration sites into the human genome, the identification of disrupted genes including oncogenes, tumor suppressor genes, and DNA repair genes, the detection of HPV-prone genomic instability and altered cellular pathways. In this review, we will focus on HPV-associated cancers, HPV integration sites within the human genome, and its concomitant disruption at the genomic, transcriptomic and epigenomic levels.
HPV-associated cancers
Almost ~610,000 human cancers are the consequence of HPV infection every year in worldwide [27]. HPV infections are the primary cause of almost all cervical cancers (i.e., squamous cell cervical carcinoma and adenocarcinomas of the cervix, together 96.6%) [13, 28], a subset of HNSCC (12.9-33%) in which HPV has been found mainly in oropharyngeal tumors (50-64%), and vulvar (40-51%), vaginal (40-64%), anal (90-93%), penile (36-40%) (Table 1) [13, 29-39]. Currently, more than 200 HPV subtypes have been identified, and more than 40 of them are transmitted by sexual contact. Depending on their malignant potential, they can be classified into two groups: low-risk (LR) and high-risk (HR) HPVs [40, 41]. Twelve HPV types have been identified as belonging to the high-risk group, i.e., HPV16, HPV18, HPV31, HPV33, HPV35, HPV39, HPV45, HPV51, HPV52, HPV56, HPV58, and HPV59. HPV16 and HPV18 are predominantly (65.5% and 13.1% of the positive cases, respectively) found in cervical cancer [13, 42], whereas HPV16 and HPV33 are predominantly found in HNSCC (83.7% and 14.0% of positive cases, respectively) [13]. The occurrence of HPV in other human cancers is also rarer, such as HPV6b in bladder urothelial cancer, HPV16 in lung squamous cell carcinoma (LSCC) and uterine endometrial carcinomas (UEC) [13], and HPV16 and HPV18 in colorectal cancer (31% of colon and 0.4% in rectal cancer) [8, 43, 44], and HPV18 in gastrointestinal adenocarcinomas (19.8%) (Table 1) [8]. In the following sections, further information on the different HPV types and their respective effects on HPV-related cancers will be discussed.
Table 1: HPV-types infection is associated with related cancers
Cancer type |
HPV type |
Ref |
Cervical squamous cell cancer |
16, 18, 59, 58, 33, 11, 31, 52, 45, 68, 51, 56, 39, 70, 69, 68b |
|
Cervical adenocarcinoma |
18, 16, 45, 33, 31 |
|
HNSCC |
16, 33, 18, 35, 56 |
|
Oropharyngeal cancer |
16, 32, 35, 45, 53, 33 |
|
Vaginal carcinoma |
16, 18, 31, 33, 45, 52, 58, 6, 11 |
|
Vulvar carcinoma |
16, 18, 56, 31, 33, 35, 45, 51, 52, 59, 68, 58, 6, 11 |
|
Anal carcinoma |
16, 18, 6, 11, 31, 33,45 |
|
Penile carcinoma |
16, 18, 11, 45, 69 |
[38] |
Bladder urothelial carcinoma |
16, 6b |
[13] |
Lung squamous cell carcinoma |
16 |
[13] |
Uterine endometrioid carcinoma |
16 |
[13] |
Colon adenocarcinoma |
16, 18 |
|
Rectal adenocarcinoma |
18 |
|
Gastrointestinal adenocarcinoma |
18 |
[8] |
Bold represents the most frequent type of HPV infection; italic represents low-risk HPV’s, and the remaining HPV types are high-risk.
HPVS and cancer development
The first human papillomavirus (HPV) DNA was isolated from genital warts (HPV6) and subsequently cloned by Gissmann and de Villiers in the late 1970’s [45]. Shortly thereafter, the isolation of HPV16 and HPV18 DNA was reported [46]. Infection by HPVs can precipitate the development of cervical, vulvar, penile and anal intraepithelial cancers and HNSCC [47]. The HPV genome is composed of a double-stranded circular DNA that is ~8000 bp long, and contains a non-coding upstream regulatory regions (LCR), an early (E) region encompassing six open reading frames (ORFs) (E1, E2, E4, E5, E6, and E7) and late (L) region encompassing two ORFs (L1 and L2) (Figure 1) [48]. The transcription of the ORFs is complex, due to the existence of multiple promoters and the formation of multiple alternative splice isoforms. Infection of the host cell by HPV activates the early promoter, which leads to expression of a polycistronic primary RNA encompassing all six early ORFs [49]. The E1 and E2 proteins are required for replication and for conserving the viral DNA as a circular episome. The viral oncogenes E6 and E7 provide the primary transforming activities on the HR HPVs. E5 contributes to tumorigenesis by augmenting the role of E6 and E7, while E2 is negatively regulates their expression [50]. HR HPVs are required, but not sufficient, for the pathogenesis of anogenital and other epithelial carcinomas [51]. In early dysplastic low-grade lesions, the HR HPV genomes replicate as circular episomes (extrachromosomal circular DNA) in the normal life cycle and they retain an episomal state, while in some advanced HPV-associated cervical lesions and in most HR HPV-associated cancers the viral genome or fragments of that, integrates into the chromosomal DNA of the host cells [52, 53]. The integrated viral DNA transcripts exhibit an increased tumorigenic capacity compared to those of episomes [54, 55]. The integration rates of HPVs into the host genome are variable, depending on the tissue and HPV types involved. The HPV integration rate into the host genome is 86.5% in squamous cell carcinoma of cervix, and 53.3% in adenocarcinoma of cervix [56] and 71.2% in HPV-driven HNSCCs [57, 58]. In addition, the HPV integration rate into the host genome is 53.8% in cervical intraepithelial neoplasia and increases to 81.7% in cervical carcinomas, and by disease progression [56, 59]. Consistent with this variability, it has been found that the viral integration frequency of HPV18 is 100% and that of HPV16 is 58.5% in different HPV-related cancers [13, 60]. During the integration of HPV into the host genome, its DNA becomes fragmented. The breakpoints may occur throughout the entire HPV genome, but are mainly found in E1 in cervical cancers [56] and HNSCC [58], and in E2 in HNSCCs (Figure 1) [58, 61]. As a result, E2 is either disrupted or lost. Of note, E2 suppresses the expression of E6 and E7 in the episomal state. Thus, functional E2 loss prevents suppression of the E6 and E7 oncogenes, consequently an increased expression of E6 and E7 as viral-host fusion transcripts in HPV-positive tumors which, in turn, results in increased cellular proliferation and viral immortalization [54, 55].
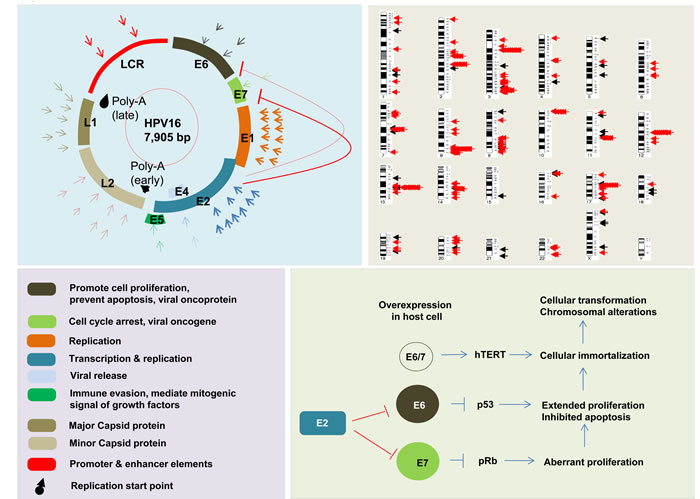
Figure 1: Representative figure of HPV genome, function of HPV genome components, and interaction of those components with each other in tumor development. Breakpoints are distributed through the HPV genome with the most frequent in E1 in HNSC and cervical cancers. Arrows represent distribution of breakpoints in HPV genome; first circle of arrows in cervical cancer, and second circle of arrows in HNSCC (on left panel). Black mark indicates the start location for late and early replication. Distribution of HPV integration breakpoints in across the human genome is shown in upper right panel. Black arrows indicate HPV integration breakpoint in HNSCC, and red arrows indicate HPV integration breakpoints in cervical cancers. Breakpoint distribution data are based on results from TCGA, Parfenov et al, Hu et al, and Ojesina et al [34, 56, 58, 71].
In most clinical samples E6 and E7 are found to be amplified as viral-host fusion genes. The E6 and E7 proteins inactivate the p53 and Rb tumor suppressor proteins, respectively [62, 63], thereby conferring a selective growth advantage to the cells [64-66]. E6 interacts with the host E6-associated protein and leads to p53 proteosomal degradation, which prevents apoptosis. E7 binds to the Rb protein, which releases E2F and leads to transactivation of its targets, thereby promoting cell cycle progression [65, 66]. This information indicates that integration of the HPV genome may play a crucial role in tumor development and the progression to invasive carcinoma. However, HPV integration does not always result in an increased expression of E6 and E7 oncoproteins. An increased expression of E6 and E7 oncoproteins may not sufficient for cancer development. Indeed, the expression of viral proteins may be regulated through its corresponding gene promoters as also post-transcriptionally through (host cell-derived) microRNAs such as miR-145 [67]. MiR-145 is the only microRNA known to directly regulate viral transcription and, thereby, its life cycle [68].
Disruption of cellular genes through HPV integration
Integration of a viral genome leads to disruption the host genome. Several studies have shown that half of the HPV integration sites are within or in close proximity (5Mb) of fragile sites (e.g. FRA2K, FRAD3D) [69-71], and that the remaining integration sites are within or adjacent to loci exhibiting DNA copy number alterations, including focal amplifications or deletions (e.g. PROX1, FANCC, C9orf3, LINC00475, EGFL7, LOC100506190, RARA, KRT39, CEACAM5, TP63, ERBB2, and RAD51B) or to regions exhibiting intra- or inter-chromosomal translocations, genic and miRNA regions [56, 58, 71]. It has been noted that integration occurs in regions of micro-homology among the HPVs and host genome [56, 58]. These observations suggest that HPV integration goes along with variable structural alterations within the host genome [60]. Indeed, HPV integrations may occur either within UTRs (5’ UTR: 12.6%, or 3’ UTR: 15.3%), within genic regions (exons: 2.7%, introns: 39.6%) or within intergenic regions (29.7%) [70], indicating that HPV-based disruptions may lead to the introduction of aberrant gene promoters, aberrant enhancers and/or aberrant microRNA expression patterns, thereby inactivating tumor-suppressor genes, activating proto-oncogenes, inactivating DNA repair genes and promoting genomic instability. Interestingly, different types of HPV (HPV16, HPV18 and HPV52) have been found to integrate into the intronic regions of the RAD51B locus in different tumors [71]. This observation supports the idea that HPV integration is a nonrandom process. Integration of HPV into the exonic or promoter regions of genes may result in their over-expression, as has e.g. been shown for c-MYC, ERBB2 and TP63, while integration into intronic regions of genes may result in median-to-low expression, as has e.g. been shown for RAD51B and FANCC. HPV integration into the first introns of FHIT and LRP1B has been found to result in a decreased expression of these genes [56, 71]. In some HPV integrants, high expression levels were found to correlate with DNA copy number gains of loci encompassing e.g. the c-MYC or ERBB2 (HPV16) genes, but this was not noted for other over-expressed genes such as MAFA (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog A). Moreover, in some integrants miscellaneous gene expression was found to correlate with copy number changes, including those of TP63 and RAD51B in cervical cancers [71], suggesting that distinct mechanisms may underlie altered gene expression, such as gene amplification and viral promoter deregulation, in HPV-driven cancers.
Deregulation of key cellular genes by HPV integration, which may present a selective growth advantage to cells, are thought to occur through five distinctive mechanisms. A first mechanism represents integration into exon, results by truncated protein, and integration into introns, results by decreased protein [56, 58, 71]. For instance, integration of HPV into ETS2 has been found to result in deletion of exons 7 and 8 at the integration site, resulting in a truncated form of the ETS2 protein in HNSCC [58], and integration of HPV16 into introns of FHIT and LRP1B, resulting in decreased or aberrant protein expression in cervical intraepithelial neoplasia and cervical squamous cell carcinomas [56]. A second mechanism represents amplification results with loss-of-function by integration into genes. For example, integration of HPV into RAD51B has been found to result in a 28-fold amplification of a 42 kb segment of intron 8 along with fragment of the viral genome, and over-expression of exons located downstream of the integrated virus, but nonfunctional protein in HNSCC [58]. Also, integration of HPV16, HPV18 and HPV52 into the same gene has also been noted in cervical cancers [71]. A third mechanism represents aberrant promoter introduction into the host gene. It has e.g. been found that the introduction of the aberrant promoter to TP63 by HPV can cause for viral-host fusion transcript, and novel over-expressed but truncated form of p63 protein, which inhibits the action of pro-apoptotic protein [72]. A fourth mechanism represents integration of HPV within or in close proximity of a proto-oncogene or other genes, resulting in its amplification and/or over-expression. For example, HPV integration upstream of the NR4A2 proto-oncogene has been found to result in a 248 fold amplification of a 75 kb genomic region encompassing NR4A2 and a concomitant over-expression of this gene. Similarly, HPV18 has been found to be integrated downstream, upstream (at POU5F1B and OCT4) or within MYC, resulting in its amplification (20 fold) and over-expression (4.3-fold induced) in cervical cancers [56, 71, 73]. Also, integration of HPV into upstream, downstream or intronic region of HMGA2 has been found to be resulted with its increased expression [56]. A fifth mechanism represents integration of HPV resulting in the disruption of neighboring genes by complex rearrangement (intra- and/or inter-chromosomal rearrangements). For example, amplification for certain exons of DIAPH2, and deletion of >100 kb segment flanking the same gene have been noted in head and neck cancer-derived UD-SCC-2 cells [70], and amplification and over-expression for complex rearrangement including TPRG1, TP63 and KLF5 have been found in HNSCC [58]. Alterations in this gene have been found to lead to chromosomal instability by misalignment of sister chromatids during mitosis [74]. In cervical cancer many HPV-integration sites have been identified, including a few hot spots in POU5F1B (near c-MYC) (9.7%), FHIT (8.7%), KLF12 (7.8%), HMGA2 9 (7.8%), KLF5 (6.8%), LRP1B (5.8%), LEPREL1 (4.9%), DLG2 (4.9%) and SEMA3D (4.9%) [56], whereas no integration hot spots (including in or near c-MYC) have been reported in HNSCC [34]. This observation may be due to the small number of HPV-positive HNSCCs tested and, therefore, requires evaluation in a larger cohort.
Similarly, a high viral integration rate has been found in HBV-positive tumors (76.5%), whereas none were found in HHV-positive tumors [13]. Interestingly, copy number alterations have been found near ~25% of the HBV-integration sites in hepatocellular carcinomas, with the hotspots in TERT locus. Overall, HBV integration has been found to be associated with copy number alterations and deregulation of gene expression in the host genome [75-77]. Also, MLL4 and FN1 contain hot spot integration sites for HBV in hepatocellular cancer [13], and such integration have been found to correlate with increased expression of MLL4, but not FN1 [13].
Table 2: Genomic alterations in cervical, HPV-positive and negative HNSC carcinomas.
Alterations |
HPV-positive |
HPV-negative |
Ref |
Mutation |
|||
Cervical (squamous cell) carcinoma |
PIK3CA, PTEN, TP53, STK11, KRAS, MAPK1, HLA-B, EP300, FBXW7, NFEL2, ERBB2 |
[71] |
|
Cervical (adenocarcinoma) |
ELF3, CBFB, PIK3CA, STK11, EP300, PTEN, TP53, ERBB2, KRAS |
||
HNSCC |
FBXW7, TRAF3, PIK3CA, FGFR3 |
TP53, CDKN2A, FAT1, AJUBA, CASP8, HRAS, NOTCH1, SMAD4 |
|
Amplification |
|||
Cervical* (squamous cell carcinoma) |
MYC, ERBB2, GLI2, TNIK, NRA2, PROX1, EIF2C2, FAM179B, SERPINB4, BIRC3, YAP1, NFE2L2, TP63 |
[71] |
|
Cervical (adenocarcinoma) |
MYC, ERBB2, MCL1, ELF3, PIK3CA, SOX2 |
||
HNSCC |
E2F1 |
CCND1, FADD, CTTN, BIRC2, YAP1, EGFR, ERBB2, FGFR1, MYC |
|
Deletion |
|||
Cervical (squamous cell carcinoma) |
IDH1, PAX3, CREB1, ATM, CBL, MLL, FLI1, CDK4 |
[71] |
|
Cervical (Adenocarcinoma) |
|||
HNSCC |
TRAF3, PTEN, CASP8 |
CDKN2A, CDKN2B, NSD1, FAT1, NOTCH1, SMAD4, Let-7c |
|
Amplification |
|||
Cervical (squamous cell carcinoma) |
1p, 1q, 3q, 5p, 8q, 11, 14q, 17q,19q, 20p, 20q |
||
Cervical (adenocarcinoma) |
1q, 3q, 17q, 19q, 20p |
[71] |
|
HNSCC |
1q25.1, 3q26.32, 3q28, 5p15.33, 11q13.3, 12p13.31, 14q24.1, 20p11.21, 20q11.22, 21q22.3 |
2q11.2, 2q31.2, 3q26.33, 5p12, 5p15.33, 6p12.1,6q12, 7q22.1, 8p11.23, 8q11.21, 8q24.21, 9p13.3, 9p23, 9p24.1, 11p13, 12p13.33, 12q15, 13q22.1, 17q12, 18p11.31, 18q11.2 |
|
Loss |
|||
Cervical (squamous cell carcinoma) |
2q, 3p, 3q, 4p, 4q, 6p, 6q, 8p, 10p, 10q, 11p, 11q, 13q, 17p,19p |
||
Cervical (adenocarcinoma) |
4p, 4q, 11q, 11p, 16q, 16p, 18q, 19p |
[71] |
|
HNSCC |
2q37.3, 3p13, 3p14.1, 3p24.1, 6p25.3, 6p21.33, 7q36.1, 7p22.3, 11q23.3, 11q14.2, 1314.2, 16q13, 17q25.3, 19p13.3, |
1p36.13, 1p13.2, 2q21.2, 2q36.2, 2q22.1, 3p25.3, 3p14.2, 3p12.2, 4q22.1, 4q35.2, 5q11.2, 5q15, 5q35.3, 6p25.3, 7q36.1, 8p23.2, 9p24.1, 9p21.3, 9p13.1, 9q34.3, 10p11.21, 10q23.31, 11q14.1, 11q23.1, 13q12.11, 14q11.2, 14q32.32, 16q12.1, 16q23.3, 17q25.3, 18q21.2, 18q23, 19p13.3, 19q13.43, Xp21.3, Xp11.3, Xq21.33, |
|
*Amplifications correlate with integration sites of HPV, bold indicates regions that are statistically significant.
Genomic, epigenomic and transcriptomic landscape of HPV-driven cancers
As mentioned above, HPV integration into the host genome has been associated with deregulated gene expression, with or without copy number alterations, in cervical cancers and HNSCCs [13, 60]. HPV integration has also been found to increase the expression of E6/E7 and to induce the development of numerical and structural chromosomal alterations [78], which donate a selective growth advantage to the cells affected [55].
DNA copy number alterations and mutations
Focal amplifications of the 2q11.2, 2q31.2 (NFE2L2), 7p11.2 (EGFR), 7q22.1, 8q24.21 (c-MYC), 11q13.3, 14q11.2 and 15q13.3 (FADD) regions are significantly higher in HPV-negative than HPV-positive HNSCCs, as well as focal deletions of the 4q35.2 (FAT1), 9p24.1 (PTPRD), 9p21.3 (CDKN2A), 9q34.3 (NOTCH1), 10q23.32 (PTEN), 18q21.2 (SMAD4) and Xp11.3 (KDM6A) regions [34]. In fact, both common and unique genomic alterations have been encountered in HPV-positive and negative HNSCCs. For example, focal amplifications of the 3q28 (PIK3CA and TP63) and 20q11.22 (E2F1) regions predominantly occur in HPV-positive HNSCCs (27.8 %), but do also occur in HPV-negative tumors (20.2%). Similarly, mutations and/or amplifications of PIK3CA (56%), FGFR2 (4%) and FGFR3 (11-14%), and deletions or truncating mutations in TRAF3 have been observed mostly in HPV-positive HNSCCs [34, 79], whereas mutations in PIK3CA have also been found in HPV-negative (18.5%) cases. In addition, aberrations in the DNA repair genes BRCA1, BRCA2, ATM, FANCG, FANCA, FANCD2 and RAD51B have been found to occur mostly in HPV-positive HNSCCs [34]. It has also been found that some of genes exhibit mutations only in HPV-negative HNSCCs, such as the CDKN2A (25.9%), FAT1 (26.3) and AJUBA (7.0%) genes [34]. It is intriguing that there exists an inverse correlation between HPV status and inactivating mutations in the CDKN2A and TP53 genes in HNSCCs [29, 80], i.e., mutations in TP53 (84-87%) are more frequent in HPV-negative HNSCs than in HPV-positive (3-16%) cases [33, 34]. Of note, some tumor-suppressor genes such as CDKN2A have been found to be inactivated by multiple mechanisms, i.e., mutation (25.9%), deletion (homo- or heterozygous; 43.6%), abnormal splicing (2.1%) and hypermethylation (18.5%) in HPV-negative HNSCCs [34].
The mutation rate of HPV-negative tumors (4.83 mutations/megabase) has been reported to be almost twice as high as that of HPV-positive cases (2.28 mutations/megabase) [81]. Additionally, it has been found that transversions at CpG sites are more frequent in HPV-negative cases, whereas TpC mutations appear to be predominant in HPV-positive cases [34]. In fact, tobacco use is the main cause of HPV-negative HNSCCs and smoking HNSCC patients exhibit a 3.2 fold higher mutation rate than nonsmokers [81]. Consistent with this notion, genomic instability, including deletions and amplifications, are significantly higher in HPV-negative tumors than in HPV-positive cases (Table 2) [34]. Oropharyngeal squamous cell carcinomas (OSCC) are prevalent among HPV-positive HNSCCs, and are almost exclusively associated with HPV16. Similarly, CCND1 amplifications (55%) and CDKN2A/B deletions (40%), and TP53 mutations (100%) have been found to be specifically associated with HPV-negative OSCCs, similar to HNSCCs [34, 82].
Mutations in PIK3CA (14%), PTEN (6%), STK11 (4%), EP300 (16%), FBXW7 (15%), HLA-B (9%), MAPK1 (8%), NFE2L2 (4%), TP53 (9%), and ERBB2 (5%), are common in cervical squamous cell carcinomas, while mutations in ELF3 (13%) and CBFB (8%) are frequent in adenocarcinomas of the cervix. Interestingly, mutations in PIK3CA, EP300 and FBXW7 have also been reported in HPV-positive HNSCC [83]. The rate of somatic mutation is greater in squamous cell carcinomas than in adenocarcinomas within the Tp*C dinucleotide context [71]. Interestingly, it has been noted that DNA copy number profiles are different among HPV types in cervical cancers, i.e., DNA copy number changes are more frequent in non-HPV16 (HPV18-, HPV31-, HPV45- and HPV52-positive) than in HPV16-positive cervical cancers. In particular, DNA copy number losses are more common in non-HPV16 cases than in HPV16-positive cases. In addition, DNA copy number losses in the chromosomal regions 2q, 4p, 4q,6p, 6q, 8q and 17p (TP53 region), and copy number gains in chromosome 1 are more common in HPV31-positive than in HPV16-positive cervical cancers, whereas DNA copy number gains in chromosome 3q (PIK3CA region) are more common in HPV16-positive than HPV31-positive cervical cancers [84]. These data indicate that each HPV type may induce a distinct genomic profile in cervical cancers, and thus need to be evaluated separately rather than collectively. On the other hand, it has been found that both squamous cell carcinomas and adenocarcinomas of the cervix share a copy number gain at 20q [60]. DNA copy number gains in chromosome 3q are more frequent in squamous cell carcinomas than in adenocarcinomas of cervix, whereas copy number losses in chromosome 13q are more frequent in squamous cell carcinomas of the cervix and in HNSCC [60]. This raises the question whether different HPV types induce the same or different chromosomal aberrations and/or even distinct disease subtypes. The most significant copy number alterations and mutations in cervical cancers and HNSCCs are listed in Table 2.
Deregulated pathways
HPV-driven cancers (HNSCCs and cervical cancers) show shared genomic alterations and shared deregulated cellular pathways, with different frequencies [13, 85]. For example, the Notch pathway has been found to be deregulated in both HNSCCs and cervical cancers, with NOTCH1 mutations predominant in HPV-negative HNSCCs (20%) and infrequent in HPV-positive cases (8%). The same pathway has been found to be deregulated by mutations in other genes, such as FBXW7, in cervical cancers [86, 87]. In fact, even changes including mutations, deletions and amplification in the RTK-RAS-PI3K, NOTCH and NFKB cell cycle pathways, as well as other pathways, have been reported to occur in both HPV-positive and HPV-negative cases, but their rates and the genes involved in these pathways, including their activation and inactivation mechanisms, differ between the HPV-negative and HPV-positive groups. For instance, inactivation of the wild type p53 and RB1 proteins by the HPV proteins E6 and E7 that control cell cycle progression has been found to be common in HPV-positive cases, while inactivation of TP53 by mutations is common in HPV-negative cases. Disruption of the PIK3CA, FGFR3, TRAF3 and E2F1 genes is predominant in HPV-positive tumors, while aberrations of the EGFR, PTEN, FGFR1, CCND1, FADD, CASP8, FAT1, NOTCH1 and NFE2L2 genes are more common in HPV-negative cases [34]. Consistent with this notion, it has been found that mutations and/or copy number alterations occur frequently within the PI3K signaling pathway in HPV-positive HNSCCs [PIK3CA (30-56%), PTEN (6%) and RICTOR (4-6%)] as well as in HPV-negative cases [PIK3CA (16-34%), PTEN (12%), AKT (5%), RICTOR (4-9%) and mTOR (2%)] [33, 34]. Even though the RTK-RAS-PI3K (62% in HPV-negative versus 61% in HPV-positive), cell death (44% in HPV-negative versus 31% in HPV-positive), immunity (7% in HPV-negative versus11% in HPV-positive), differentiation (64% in HPV-negative versus 44% in HPV-positive) and oxidative stress (22% in HPV-negative versus 3% in HPV-positive) pathways have been found to be deregulated in both groups with different frequencies (Figure 2), and, the prognosis has been found to be better in HPV-positive HNSCC cases [34]. Similar to HNSCCs, it has been found that PI3K-AKT pathway genes are activated by mutations in 31% of the cervical squamous carcinomas and 24% of the adeno- and adeno-squamous cases. Interestingly, mutations in the most frequently activated gene, PIK3CA, are more common in HPV16-positive than in HPV18- and HPV45-positive cervical cancers [88].
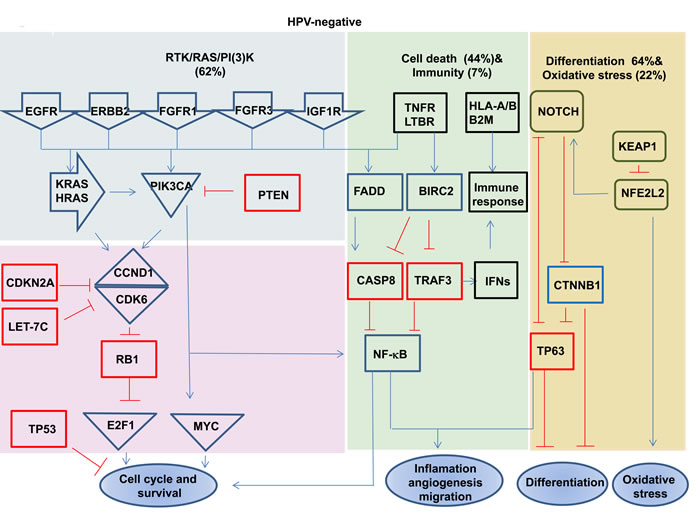
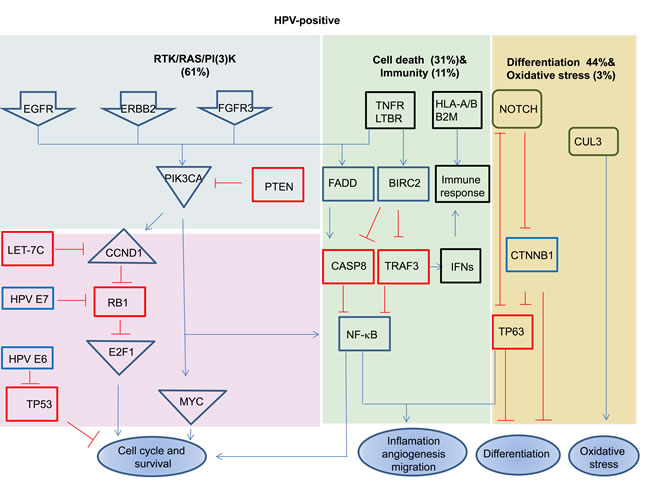
Figure 2: Signaling pathways deregulated in HPV-positive and negative HNSCC. Red boxes highlight the tumor suppressor genes, and blue boxes highlight the oncogenes. Deletion, amplification, and somatic mutations are included in pathway alterations. Data are based on results from TCGA [34]. The present of tumors showing the indicated alterations.
Epigenetic alterations
In contrast to the genomic changes mentioned above, HPV-positive HNSCCs appear to exhibit distinct epigenetic profiles compared to HPV-negative cases [34, 58, 82, 89]. For example, the tumor-suppressor genes BARX2 and IRX4 have been found to be hyper-methylated and, consequently, expressed at lower levels in HPV integration-positive HNSCCs compared to integration-negative cases, whereas SIM2 and CTSE genes have been found to be hypo-methylated and, consequently, expressed at higher levels in HPV integration-negative HNSCCs compared to integration-positive cases [58]. Similarly, IRX4 has been found to be hyper-methylated and expressed at a lower level in HPV integration-positive oropharyngeal tumors compared to non-integrated tumors [90].
Trancriptomic alterations
In concordance with the epigenetic profiles described above, gene expression array studies have revealed similarities and differences between HPV-positive and HPV-negative cancers, as well as similarities between HPV-positive HNSCCs and cervical cancers [34, 71, 85, 91]. Different genes acting in the cell cycle pathway have been found to be activated in HPV-negative HNSCCs compared to HPV-positive cases. For example, CCND1, CCND2 and CCNA1 were found to be up-regulated in HPV-negative HNSCC, while CDKN2A, CCNE2, CCNB1 and MCMs were found to be over-expressed in HPV-positive cases. In addition, multiple genes that regulate DNA replication and cell proliferation, including PCNA, E2Fs, CDC2, and CDC7, were found to be significantly up-regulated in HPV-positive HNSCCs and cervical cancers compared to HPV-negative HNSCCs. Interestingly, three testis-specific genes SYCP2, TCAM1 and STAG3, were found to be over-expressed in HPV-positive HNSCCs and cervical cancers. Normally, these genes are not expressed in somatic cells [85].
Deregulated miRNAs
Based on microRNA (miRNA) profiling, HPV-positive and -negative HNSCCs can be clustered into two distinct subgroups, whereas significant similarities between HPV-positive HNSCC and cervical cancers have reported [34, 71, 91]. The expression of miR15a (4.19 fold) and miR-20b (3.42 fold) has been found to be higher in HPV-positive HNSCCs than in HPV-negative cases, while the expression of miR-139-3p (2.26 fold) and miR-145 (2.19 fold), among others, has been found to be lower in HPV-positive than in HPV-negative HNSCCs. Also, the expression of miR-145 has been found to be lower in HPV-positive cervical cancers [92] and this miRNA has been reported to suppress the growth of cervical cancer cells in vitro [93]. Notably, five significantly deregulated miRNAs, i.e., miR-9 (1q23.2), miR-15b (3q25.32), miR-28-5p (3q27.3), miR-100 and miR-125b (11q24.1) have been found to be associated with recurrent chromosomal alterations [92]. MiR-9 is a potential oncogene, and its over-expression has been associated with increased copy numbers of chromosome 1q and increases cell viability, anchorage-independent growth and migration in vitro [94]. In concordance, the expression of miR-34s was found to be deregulated after HPV infection by the viral oncoprotein E6 [91]. It has also been found that the expression of miR-20b, miR-9, miR-9*, miR-492, miR-545, miR-591, miR-422a, miR-142-3p, miR-383,miR-520g, miR-101, miR-381, miR146a, miR34a, miR412, miR-155, miR-554, miR380-5p, miR-16, miR-219, miR-29c, let-7g, miR-30e-5p and miR-139 is significantly higher in HPV-positive OSCCs than in HPV-negative cases, whereas the expression of miR-193b, miR-107, miR-23a, miR-365, miR-452, miR-198, miR-196a, miR-181d, miR-324-5p, miR-550, miR-487b, miR-599, miR-382, miR-221 and miR-22 is lower in HPV-positive versus HPV-negative OSCC cases [95]. Interestingly, the expression of miR-34 has been found to be down-regulated in cervical cancers [91], while it is has been found to be over-expressed in HPV-positive oropharyngeal carcinomas [95], suggesting that expression of miR-34 may be tissue dependent, and may not be related to HPV status.
Conclusion and future direction
Recent efforts of The Cancer Genome Atlas (TCGA) network together with other NGS and ‘omics’ studies in virus-associated tumors have provided detailed information on the etiology of HPV-driven cancers. It is important to stress that integration of NGS data with other ‘omics’ data is crucial to better understand the pathophysiology of each individual disease and, based on this, to select targets and effective personalized treatment options. HPV-positive and -negative cases may exhibit both similar and different genomic alterations, whereas the epigenomic and transcriptomic profiles may be distinct between these groups. For example, the expression of cell cycle regulatory and testis-specific genes distinguishes HPV-positive from HPV-negative HNSCCs. Similarly, over-expression of CCND1 is frequently observed in HPV-negative cases, whereas up-regulation of E2F1 is common in HPV-positive cases. In contrast, mutation and/or amplification of PIK3CA are a common feature of both HPV-positive and -negative HNSCCs and cervical cancers [34]. These data indicate that for the design of combination therapies both shared alterations (in genes or pathways) between groups, and alterations that are specific to each group may be important to bear in mind. It is also clear that for selecting targets and therapeutic agents, and thus for designing more effective therapies, further interrogations are required. To this end, it is considered critical to integrate NGS and ‘omics’ data with clinical data in order to evaluate its functional consequences. In addition, both proteomics and epigenemics studies in HPV-associated cancers have so far been limited. In particular ‘omics’ studies such as glycomics and metabolomics require further interrogation. Also, the (anomalous) localization of aberrant proteins in cancer cells and its functional consequences requires further clarifications. Finally, multicenter studies may be required to increase sample size, especially for HPV-positive HNSCCs, to uncover novel biomarkers, therapeutic targets and, ultimately, potentially effective treatment options.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
1. zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002; 2: 342-350.
2. Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006; 118: 3030-3044.
3. zur Hausen H. Oncogenic DNA viruses. Oncogene. 2001; 20:7820-7823.
4. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008; 319: 1096-1100.
5. Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A. 1982; 79: 2031-2035.
6. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994; 266: 1865-1869.
7. Epstein MA, Achong BG, Barr YM: Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet. 1964; 1: 702-703.
8. Salyakina D, Tsinoremas NF. Viral expression associated with gastrointestinal adenocarcinomas in TCGA high-throughput sequencing data. Hum Genomics. 2013; 7: 23.
9. Wang WY, Lin TY, Twu CW, Tsou HH, Lin PJ, Liu YC, Huang JW, Hsieh HY, Lin JC. Long-term clinical outcome in nasopharyngeal carcinoma patients with post-radiation persistently detectable plasma EBV DNA. Oncotarget. 2016; 7:42608-42616. doi: 10.18632/oncotarget.9323.
10. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981; 2: 1129-1133.
11. Hobbs JA, Adamson-Small LA. Parvovirus and Thyroid Cancer. Semin Oncol. 2015; 42: 304-308.
12. Wentzensen N, Ridder R, Klaes R, Vinokurova S, Schaefer U, Doeberitz M. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene. 2002; 21: 419-426.
13. Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013; 4: 2513.
14. White FA, 3rd, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992; 66: 5726-5734.
15. Fanci R, De Santis R, Zakrzewska K, Paci C, Azzi A. Presence of TT virus DNA in bone marrow cells from hematologic patients. The new microbiologica. 2004; 27: 113-117.
16. Bando M, Takahashi M, Ohno S, Hosono T, Hironaka M, Okamoto H, Sugiyama Y. Torque teno virus DNA titre elevated in idiopathic pulmonary fibrosis with primary lung cancer. Respirology. 2008; 13: 263-269.
17. Zhong S, Yeo W, Tang M, Liu C, Lin XR, Ho WM, Hui P, Johnson PJ. Frequent detection of the replicative form of TT virus DNA in peripheral blood mononuclear cells and bone marrow cells in cancer patients. Journal of medical virology. 2002; 66: 428-434.
18. Singh S, Kaye S, Francis N, Peston D, Gore M, McClure M, Bunker C. Human endogenous retrovirus K (HERV-K) rec mRNA is expressed in primary melanoma but not in benign naevi or normal skin. Pigment cell & melanoma research. 2013; 26: 426-428.
19. Goering W, Schmitt K, Dostert M, Schaal H, Deenen R, Mayer J, Schulz WA. Human endogenous retrovirus HERV-K(HML-2) activity in prostate cancer is dominated by a few loci. The Prostate. 2015; 75: 1958-1971.
20. Kraus B, Monk B, Sliva K, Schnierle BS. Expression of human endogenous retrovirus-K coincides with that of micro-RNA-663 and -638 in germ-cell tumor cells. Anticancer Res. 2012; 32: 4797-4804.
21. Serafino A, Balestrieri E, Pierimarchi P, Matteucci C, Moroni G, Oricchio E, Rasi G, Mastino A, Spadafora C, Garaci E, Vallebona PS. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Experimental cell research. 2009; 315: 849-862.
22. Wentzensen N, Coy JF, Knaebel HP, Linnebacher M, Wilz B, Gebert J, von Knebel Doeberitz M. Expression of an endogenous retroviral sequence from the HERV-H group in gastrointestinal cancers. Int J Cancer. 2007; 121: 1417-1423.
23. Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR, Strong TV. Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin Cancer Res. 2001,; 7: 1553-1560.
24. Wang-Johanning F, Liu J, Rycaj K, Huang M, Tsai K, Rosen DG, Chen DT, Lu DW, Barnhart KF, Johanning GL. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int J Cancer. 2007; 120: 81-90.
25. Deligdisch L, Marin T, Lee AT, Etkind P, Holland JF, Melana S, Pogo BG. Human mammary tumor virus (HMTV) in endometrial carcinoma. International journal of gynecological cancer. 2013; 23: 1423-1428.
26. Pogo BG, Holland JF, Levine PH. Human mammary tumor virus in inflammatory breast cancer. Cancer. 2010; 116: 2741-2744.
27. Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S. Global burden of human papillomavirus and related diseases. Vaccine. 2012; 30 Suppl: F12-23.
28. Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999; 189: 12-19.
29. Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000; 92: 709-720.
30. Klussmann JP, Weissenborn SJ, Wieland U, Dries V, Kolligs J, Jungehuelsing M, Eckel HE, Dienes HP, Pfister HJ, Fuchs PG. Prevalence, distribution, and viral load of human papillomavirus 16 DNA in tonsillar carcinomas. Cancer. 2001; 92: 2875-2884.
31. Mork J, Lie AK, Glattre E, Hallmans G, Jellum E, Koskela P, Moller B, Pukkala E, Schiller JT, Youngman L, Lehtinen M, Dillner J. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med. 2001; 344: 1125-1131.
32. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Axelrod R, Silverman CC, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010; 363: 24-35.
33. Chung CH, Guthrie VB, Masica DL, Tokheim C, Kang H, Richmon J, Agrawal N, Fakhry C, Quon H, Subramaniam RM, Zuo Z, Seiwert T, Chalmers ZR, et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann Oncol. 2015; 26: 1216-1223.
34. The Cancer Genome Atlas Network: Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517: 576-582.
35. Smith JS, Backes DM, Hoots BE, Kurman RJ, Pimenta JM. Human papillomavirus type-distribution in vulvar and vaginal cancers and their associated precursors. Obstet Gynecol. 2009; 113: 917-924.
36. Serrano B, de Sanjose S, Tous S, Quiros B, Munoz N, Bosch X, Alemany L: Human papillomavirus genotype attribution for HPVs 6, 11, 16, 18, 31, 33, 45, 52 and 58 in female anogenital lesions. Eur J Cancer. 2015; 51: 1732-1741.
37. Hoots BE, Palefsky JM, Pimenta JM, Smith JS. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int J Cancer. 2009; 124: 2375-2383.
38. de Sousa ID, Vidal FC, Branco Vidal JP, de Mello GC, do Desterro Soares Brandao Nascimento M, Brito LM. Prevalence of human papillomavirus in penile malignant tumors: viral genotyping and clinical aspects. BMC Urol. 2015; 15: 13.
39. Lohavanichbutr P, Houck J, Fan W, Yueh B, Mendez E, Futran N, Doody DR, Upton MP, Farwell DG, Schwartz SM, Zhao LP, Chen C. Genomewide gene expression profiles of HPV-positive and HPV-negative oropharyngeal cancer: potential implications for treatment choices. Arch Otolaryngol Head Neck Surg. 2009; 135: 180-188.
40. Galloway DA, Laimins LA. Human papillomaviruses: shared and distinct pathways for pathogenesis. Current opinion in virology. 2015; 14: 87-92.
41. Kocjan BJ, Sterbenc A, Hosnjak L, Chouhy D, Bolatti E, Giri AA, Poljak M. Genome announcement: complete genome sequence of a novel Mupapillomavirus, HPV204. Acta dermatovenerologica Alpina, Pannonica, et Adriatica. 2015; 24: 21-23.
42. Arbyn M, Tommasino M, Depuydt C, Dillner J. Are 20 human papillomavirus types causing cervical cancer? J Pathol. 2014; 234: 431-435.
43. Giuliani L, Ronci C, Bonifacio D, Di Bonito L, Favalli C, Perno CF, Syrjanen K, Ciotti M. Detection of oncogenic DNA viruses in colorectal cancer. Anticancer Res. 2008; 28: 1405-1410.
44. Chen TH, Huang CC, Yeh KT, Chang SH, Chang SW, Sung WW, Cheng YW, Lee H. Human papilloma virus 16 E6 oncoprotein associated with p53 inactivation in colorectal cancer. World J Gastroenterol. 2012; 18: 4051-4058.
45. de Villiers EM, Gissmann L, zur Hausen H. Molecular cloning of viral DNA from human genital warts. J Virol. 1981; 40: 932-935.
46. Boshart M, Gissmann L, Ikenberg H, Kleinheinz A, Scheurlen W, zur Hausen H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. Embo J. 1984; 3: 1151-1157.
47. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011; 11: 9-22.
48. Ganguly N, Parihar SP. Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosci. 2009; 34: 113-123.
49. Zheng ZM, Baker CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci. 2006; 11: 2286-2302.
50. DiMaio D, Mattoon D. Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene. 2001; 20: 7866-7873.
51. zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000; 92: 690-698.
52. Cullen AP, Reid R, Campion M, Lorincz AT. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J Virol. 1991; 65: 606-612.
53. Pirami L, Giache V, Becciolini A. Analysis of HPV16, 18, 31, and 35 DNA in pre-invasive and invasive lesions of the uterine cervix. J Clin Pathol. 1997; 50: 600-604.
54. Romanczuk H, Howley PM. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci U S A. 1992; 89: 3159-3163.
55. Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995; 92: 1654-1658.
56. Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, Liu H, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015; 47: 158-163.
57. Begum S, Cao D, Gillison M, Zahurak M, Westra WH. Tissue distribution of human papillomavirus 16 DNA integration in patients with tonsillar carcinoma. Clin Cancer Res. 2005; 11: 5694-5699.
58. Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova EV, Wilkerson MD, Protopopov A, Yang L, Seth S, et al: Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014; 111: 15544-15549.
59. Gradissimo Oliveira A, Delgado C, Verdasca N, Pista A. Prognostic value of human papillomavirus types 16 and 18 DNA physical status in cervical intraepithelial neoplasia. Clinical microbiology and infection. 2013; 19: E447-450.
60. Peter M, Stransky N, Couturier J, Hupe P, Barillot E, de Cremoux P, Cottu P, Radvanyi F, Sastre-Garau X. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J Pathol. 2010; 221: 320-330.
61. Wilson GA, Lechner M, Koferle A, Caren H, Butcher LM, Feber A, Fenton T, Jay A, Boshoff C, Beck S. Integrated virus-host methylome analysis in head and neck squamous cell carcinoma. Epigenetics. 2013; 8: 953-961.
62. Scheffner M, Romanczuk H, Munger K, Huibregtse JM, Mietz JA, Howley PM. Functions of human papillomavirus proteins. Curr Top Microbiol Immunol. 1994; 186: 83-99.
63. Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst. 2009; 101: 412-423.
64. Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. Embo J. 1989; 8: 3905-3910.
65. Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010; 10: 550-560.
66. Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Embo J. 1989; 8: 4099-4105.
67. Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol. 2013; 87: 6037-6043.
68. Cai X, Li G, Laimins LA, Cullen BR. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J Virol. 2006; 80: 10890-10893.
69. Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004; 64: 3878-3884.
70. Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, He D, Ried T, Symer DE, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014; 24: 185-199.
71. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevirio V, Vazquez-Santillan K, et al: Landscape of genomic alterations in cervical carcinomas. Nature. 2014; 506: 371-375.
72. Serber Z, Lai HC, Yang A, Ou HD, Sigal MS, Kelly AE, Darimont BD, Duijf PH, Van Bokhoven H, McKeon F, Dotsch V. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol. 2002; 22: 8601-8611.
73. Ferber MJ, Thorland EC, Brink AA, Rapp AK, Phillips LA, McGovern R, Gostout BS, Cheung TH, Chung TK, Fu WY, Smith DI. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene. 2003; 22: 7233-7242.
74. Cheng L, Zhang J, Ahmad S, Rozier L, Yu H, Deng H, Mao Y. Aurora B regulates formin mDia3 in achieving metaphase chromosome alignment. Dev Cell. 2011; 20: 342-352.
75. Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012; 44: 765-769.
76. Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, Diao J, Yeung S, Jubb A. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2013; 22: 593-601.
77. Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, Ooi LL, Lee CG. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013; 34: 787-798.
78. Pett MR, Alazawi WO, Roberts I, Dowen S, Smith DI, Stanley MA, Coleman N. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res. 2004; 64: 1359-1368.
79. Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, Lawrence MS, Getz G, Bragelmann J. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015; 21: 632-641.
80. Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2008; 14: 366-369.
81. Sun W, Califano JA. Sequencing the head and neck cancer genome: implications for therapy. Annals of the New York Academy of Sciences. 2014; 1333: 33-42.
82. Lechner M, Frampton GM, Fenton T, Feber A, Palmer G, Jay A, Pillay N, Forster M, Cronin MT, Lipson D, Miller VA, Brennan TA, Henderson S. Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV- tumors. Genome Med. 2013; 5: 49.
83. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al: Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333: 1154-1157.
84. Bierkens M, Wilting SM, van Wieringen WN, van de Wiel MA, Ylstra B, Meijer CJ, Snijders PJ, Steenbergen RD. HPV type-related chromosomal profiles in high-grade cervical intraepithelial neoplasia. BMC Cancer. 2012; 12: 36.
85. Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007; 67: 4605-4619.
86. Schulein C, Eilers M, Popov N. PI3K-dependent phosphorylation of Fbw7 modulates substrate degradation and activity. FEBS Lett. 2011; 585: 2151-2157.
87. Siu KT, Rosner MR, Minella AC. An integrated view of cyclin E function and regulation. Cell Cycle. 2012; 11: 57-64.
88. Lou H, Villagran G, Boland JF, Im KM, Polo S, Zhou W, Odey U, Juarez-Torres E, Medina-Martinez I, Roman-Basaure E, Mitchell J, Roberson D, Sawitzke J, et al: Genome Analysis of Latin American Cervical Cancer: Frequent Activation of the PIK3CA Pathway. Clin Cancer Res. 2015; 21: 5360-70.
89. Lleras RA, Smith RV, Adrien LR, Schlecht NF, Burk RD, Harris TM, Childs G, Prystowsky MB, Belbin TJ. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin Cancer Res. 2013; 19: 5444-5455.
90. Kostareli E, Holzinger D, Bogatyrova O, Hielscher T, Wichmann G, Keck M, Lahrmann B, Grabe N, Flechtenmacher C, Schmidt CR, Seiwert T, Dyckhoff G, Dietz A, et al: HPV-related methylation signature predicts survival in oropharyngeal squamous cell carcinomas. J Clin Invest. 2013; 123: 2488-2501.
91. Wang X, Wang HK, McCoy JP, Banerjee NS, Rader JS, Broker TR, Meyers C, Chow LT, Zheng ZM. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. Rna. 2009; 15: 637-647.
92. Lajer CB, Garnaes E, Friis-Hansen L, Norrild B, Therkildsen MH, Glud M, Rossing M, Lajer H, Svane D, Skotte L, Specht L, Buchwald C, Nielsen FC. The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br J Cancer. 2012; 106: 1526-1534.
93. Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One. 2008; 3:e2557.
94. Wilting SM, Smeets SJ, Snijders PJ, van Wieringen WN, van de Wiel MA, Meijer GA, Ylstra B, Leemans CR, Meijer CJ, Brakenhoff RH, Braakhuis BJ, Steenbergen RD. Genomic profiling identifies common HPV-associated chromosomal alterations in squamous cell carcinomas of cervix and head and neck. BMC Med Genomics. 2009; 2: 32.
95. Hui AB, Lin A, Xu W, Waldron L, Perez-Ordonez B, Weinreb I, Shi W, Bruce J, Huang SH, O’Sullivan B, Waldron J, Gullane P, Irish JC. Potentially prognostic miRNAs in HPV-associated oropharyngeal carcinoma. Clin Cancer Res. 2013; 19: 2154-2162.