
INTRODUCTION
Precision oncology entails therapeutic decision-making on the basis of an individual patient’s molecular tumor profile. To that end, it is imperative to develop strategies to rapidly identify clinically relevant patient subgroups. While next-generation sequencing technologies have greatly advanced molecular classification, they are not routinely used for prostate cancer and may be costly. Because histological variants can correlate with genomic alterations in other malignancies (e.g. colorectal carcinoma, acute myelogenous leukemia), we hypothesized that distinct prostate cancer histologies may also associate with underlying molecular aberrations – allowing for the rapid identification of patients for further screening [1–5]. In this study, we sought to determine if ductal prostate cancer (dPC) was associated with clinically actionable molecular features.
Ductal prostatic adenocarcinomas (dPC) are an aggressive histopathologic variant of prostate cancer, characterized by large glands lined by tall, pseudostratified, columnar neoplastic epithelial cells [6]. Approximately 3% of all prostate cancers have at least a component of ductal histology, with only 0.2% having pure ductal histology [7]. Clinically, dPCs tend to have a more aggressive course – behaving similarly to Gleason 4 + 4 = 8 carcinomas [8]. Tumors with >10% ductal component are associated with a higher stage, are more likely to present with metastatic disease, and may be less responsive to androgen deprivation [7].
While the more aggressive clinical course associated with dPC has been well documented, little is known about the molecular features underlying this histologic subtype. Studies using fluorescence in situ hybridization have reported the prevalence of TMPRSS2:ERG fusions in ductal cases to range from approximately 10–50%, which is not substantially different than typical acinar carcinomas [9, 10]. Otherwise, gene expression profiling studies reveal extensive similarities between ductal and acinar adenocarcinomas. In one study comparing the transcriptional profile of eight ductal tumors to 11 acinar adenocarcinomas, differences in gene expression profiles encompassed only 25 genes [11].
Given that little is known regarding the underlying genomic abnormalities associated with the ductal histologic phenotype, we sequenced consecutive cases of dPC using the UW-OncoPlex platform – a targeted next-generation sequencing panel that includes genes with actionable or potentially actionable mutations [12].
RESULTS
Patient characteristics
From January 2015 to April 2016, ten consecutive patients with dPC were identified and their tumors were sequenced (Figure 1). The median age at diagnosis was 59 years (range, 40 to 73). Four (40%) patients had metastatic disease at the time of presentation. Additional details regarding the patients included in this study and their tumor samples are provided in Table 1.
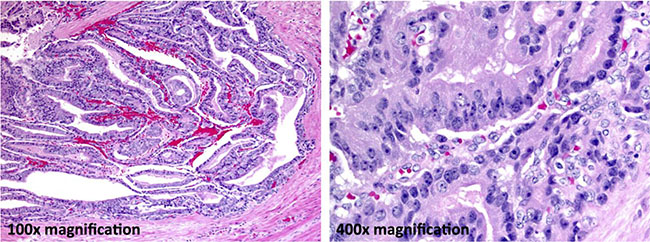
Figure 1: Ductal adenocarcinoma component. In this case, approximately 65% of the carcinoma is ductal. Large tumor cell aggregates have a tubulopapillary architecture (100× final magnification). Forming a pseudostratified columnar epithelium the tumor cells have markedly atypical nuclei with clumped chromatin and prominent nucleoli (400× final magnification).
Table 1: Demographics
Subject number |
Age at diagnosis |
Gleason |
Disease state at presentation |
Disease state at Time of Tissue Acquisition |
Source of tissue for UW-OncoPlex |
Clinical state at last follow up |
Time from diagnosis to last follow up (months) |
|---|---|---|---|---|---|---|---|
1 |
72 |
9 |
Localized |
Localized |
Prostatectomy |
NED |
34.8 |
2 |
69 |
9 |
Metastatic |
mHSPC |
Needle Biopsy |
mHSPC |
8.2 |
3 |
52 |
8 |
Localized |
Localized |
Prostatectomy |
NED |
16.6 |
4 |
66 |
9 |
Localized |
Localized |
Prostatectomy |
Death |
10.3 |
5 |
73 |
7 |
Localized |
Localized |
Prostatectomy |
Biochemical recurrence |
28.1 |
6 |
51 |
8 |
Localized |
Localized |
Prostatectomy |
NED |
28.7 |
7 |
40 |
9 |
Metastatic |
mCRPC |
Prostatectomy |
mHSPC |
1.0 |
8 |
61 |
9 |
Metastatic |
mHSPC |
Needle Biopsy |
mHSPC |
29.6 |
9 |
58 |
9 |
Localized |
mHSPC |
Needle Biopsy |
NED |
15.3 |
10 |
54 |
7 |
Metastatic |
Localized |
Soft Tissue Met |
mCRPC |
16.5 |
mHSPC, metastatic hormone-sensitive prostate cancer; NED, no evidence of disease; mCRPC, metastatic castration-resistant prostate cancer.
Sequencing results
To characterize the molecular features of dPC, we sequenced 10 prostate cancers with prominent dPC components: nine samples from FFPE archival tissue (radical prostatectomy or prostate needle biopsy specimens), and one frozen tissue biopsy from a metastasis. Nine of 10 samples had sufficient material for UW-OncoPlex testing. The tumors from four (40%) patients had an alteration predicted to be pathogenic in one of the mismatch repair (MMR) genes (2 in MSH2, 1 in MSH6 and 1 in MLH1), of which 3 (75%) had evidence of hypermutation associated with microsatellite instability (MSI). The 3 patients with hypermutated tumors had evidence of bi-allelic MMR mutation. Other genomic alterations common to prostate cancer were also detected, including alterations in genes involved in homologous recombination repair (i.e. BRCA2, CHEK2) (N = 2), androgen receptor (AR) (N = 1), TMPRSS2:ERG rearrangements (N = 3) and alteration in the PI3K/Akt/mTOR signaling pathway (N = 5) (Table 2).
Table 2: Summary of somatic alterations identified in ductal prostate cancer cases
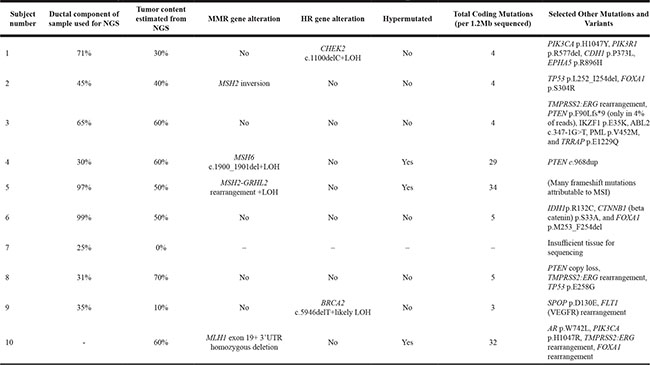
All mismatch repair (MMR) gene and homologous recombination (HR) gene alterations were known or predicted to be pathogenic. Note: metastatic tissue from subject 10 was sequenced. LOH, loss of heterozygosity; NGS, next generation sequencing.
Histopathology of hypermutated prostate cancer
To determine the histopathologic features of hypermutated prostate cancer, we reviewed the pathology of known hypermutated cases from the University of Washington rapid autopsy program. We previously reported 5 prostate cancer patients who participated in this program and were found to have hypermutated tumors with complex MMR gene alterations [13]. Since that publication, we have identified 3 additional hypermutated prostate cancer cases using similar methods. Of the now 8 hypermutated prostate cancer cases in the autopsy series, 2 had untreated primary prostate cancer tissue available for pathology review. Both of these cases had a ductal adenocarcinoma component. The first subject (Autopsy Patient: 05-165) was previously reported to have an MSH2-C2orf61 343 kb inversion, MSH2-KCNK12 74 kb inversion, and MSH2-KCNK12 40 kb inversion [13]. The second subject (Autopsy Patient: 01-002), who was not included in our previous publication, had a germline MSH2 exon 1–8 deletion with loss of heterozygosity in tumor tissue.
The tumor of a third patient with known hypermutated prostate cancer (determined through previously described methods) being followed in our clinic was histologically reviewed [14]. There was no ductal adenocarcinoma component in his tumor. It is notable, however, that this patient had a PSA decline following treatment with the immune checkpoint inhibitor pembrolizumab (i.e. anti-PD1) despite previously progressing on abiraterone, enzalutamide, docetaxel, carboplatin and cabazitaxel (Figure 2).
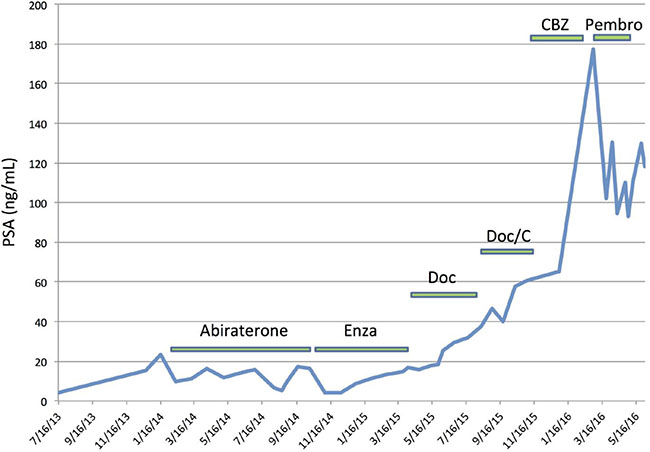
Figure 2: PSA response to checkpoint blockade immunotherapy in a patient with hypermutated prostate cancer. Prior to initiating pembrolizumab, this patient had bone, adrenal and lymph node metastases, and a baseline PSA of 177.35 ng/mL. A total of 3 cycles of pembrolizumab were administered before stopping due to an immune related adverse event (anasarca) requiring corticosteroids. He expired in June 2016. Note: this patient did not have ductal histopathologic features. Enza, enzalutamide; Doc, docetaxel; C, carboplatin; CBZ, cabazitaxel; Pembro, pembrolizumab.
DISCUSSION
This series of consecutive patients with dPC represents the largest next-generation sequencing study focused on this rare prostate cancer subset to date. Consistent with other published reports, patients in our series had aggressive clinical features, including young age at diagnosis and a high proportion of metastatic disease at presentation [6–8, 15]. Surprisingly, we found that alterations in MMR genes and associated hypermutation were far more prevalent in dPC compared to prostate cancers not selected by histologic subtype [13, 14]. Providing further support for an association between ductal histology and MMR deficiency, we found that two of three patients with MMR-deficient hypermutated metastatic prostate cancer whose primary tumors were available for review had dPC.
Hypermutated prostate cancers have only recently been described, with initial reported incidence ranging from approximately 3% to 12% in men with metastatic castration-resistant prostate cancer (mCRPC) [13, 14]. Although further validation to establish prevalence through larger systematic studies is needed, our findings are intriguing because they suggest a potential histologic association between the hypermutated genotype and a ductal histopathologic phenotype. More broadly, this finding supports an argument for sequencing rare histologic subtypes, as histology may provide insights into a tumor’s underlying molecular features. Indeed, it is notable that a similar genotype-phenotype correlation in hypermutated MSI colorectal cancer has also been described – lending credence to the possibility that hypermutated cancers may have distinct histology compared to matched microsatellite stable cases [1, 3].
Determining which patients have hypermutated prostate tumors may have important implications for future precision oncology trials, as mutational burden has been shown to correlate with response to immune checkpoint blockade in several tumor types (e.g. anti-CTLA4, anti-PD1, anti-PDL1) [16–18]. Although objective responses to immune checkpoint inhibition have initially been generally disappointing in patients with prostate cancer, most have a relatively low mutational load [19, 20]. A recent Phase II study testing pembrolizumab (anti-PD1 therapy) in patients with metastatic colorectal carcinoma with and without MMR deficiency reported that 40% of hypermutated colorectal cancer patients had an immune-related objective response (irOR) compared to 0% of patients without MSI-high tumors. Moreover, a 50% response rate to pembrolizumab in hypermutated non-colorectal gastrointestinal malignancies has been observed – supporting the hypothesis that mutational load may be a predictive biomarker for response to immune checkpoint blockade in prostate cancer [17]. The observation that one of the hypermutated patients followed in our clinic had a dramatic response to anti-PD1 therapy in spite of being heavily pretreated further bolsters the hypothesis that hypermutation may be predictive of response to PD1/PDL1 pathway inhibition.
Consistent with our prior observations, we found that somatic loss-of-function mutations in MSH2 and MSH6 were the primary cause of microsatellite instability in patients with prostate cancer [13].This is in contrast to colorectal cancer where hypermutation has been found to be associated with epigenetic silencing of MLH1, which occurs in nearly 2/3 of the cases [21]. Interestingly, the tumor of Subject #2 showed evidence of MSH2 inversion without clear evidence of hypermutation or MSI. Whether the MSH2 loss-of-function alteration represents an early event and hypermutation is a later consequence in the disease course or follows selective pressures of treatment will need to be further examined. Given that the mechanisms underlying prostate cancer hypermutation appear distinct from colorectal cancer, patterns of MSI may also be divergent and a tailored approach to MSI testing of prostate cancer may be needed. However, our findings suggest that ductal histology may be a cue to investigate further for evidence of MMR deficiency and hypermutation.
The finding that prostate cancers with ductal histologic features may be enriched for somatic hypermutation is intriguing; however, our small sample size limits our ability to draw definitive conclusions regarding this genotype-histologic phenotype relationship. If this finding is confirmed, however, the presence of ductal adenocarcinoma histology could be a means to prioritize patients for additional studies to assess mutational burden, which may have clinical implications, as hypermutation appears to predict for response to immune checkpoint blockade in several cancer types, including early signals in prostate cancer [22]. Future efforts to define the landscape of genomic alterations in patients with this prostate cancer variant will likely require multi-institutional studies. Such studies may facilitate the promise and rapid completion of precision oncology approaches for targeting this molecular subset of prostate cancer.
MATERIALS AND METHODS
Patients
All patients carried a diagnosis of prostate cancer and were followed by a medical oncologist at the University of Washington Medical Center or Seattle Cancer Care Alliance (both in Seattle, Washington). Consecutive patients with a component of ductal adenocarcinoma were identified by the treating medical oncologist and offered tumor sequencing. After obtaining written informed consent, tumor samples were tested on the UW-OncoPlex platform [12].The original diagnoses of dPC, made by genitourinary (GU) pathologists (M.S.T., F.V.L.), were independently verified by a third GU pathologist (L.T.).
Ethics statement
This study was performed in accordance with the declaration of Helsinki guidelines and with ethics approval from the Institutional Review Board at the Fred Hutchinson Cancer Research Center/University of Washington Comprehensive Cancer Consortium.
Macrodissection of tumor tissue
Hematoxylin and eosin stained sections of the tumors were reviewed by an anatomic and molecular pathologist. Ten-micron unstained recut sections were cut from the FFPE block, which were determined to contain the maximum amount of ductal adenocarcinoma. The dPC component, which ranged from 20% to 99% of the cells by visual estimate of each tumor, was macrodissected prior to deparaffinization and DNA extraction.
Next-generation sequencing (NGS) testing
DNA was extracted from FFPE samples as previously described [12]. Fresh tumor samples were snap frozen and unselected tissue was submitted for DNA extraction. UW-OncoPlex was performed according to previously published methods [12]. Microsatellite instability (MSI) testing was performed directly on NGS data using the mSINGS method [23]. Total mutation burden was estimated from targeted NGS data as previously described, with hypermutation defined as > 12 mutations/megabase [24].
ACKNOWLEDGMENTS
We thank all the men who generously participated in this research. We are grateful to Agnes Gawne, Lori Kollath and Hiep Nguyen for their assistance in this work. We acknowledge funding support from PNW Prostate SPORE CA097186; DOD awards PC131820, W81XWH-15-1-0562 and W81XWH-15-1-0430; NCI Cancer Center Support Grant 5P30 CA015704-40; the Prostate Cancer Foundation (including Young Investigator Awards to MTS, HHC, and CCP); and an award from the FHCRC Solid Tumor Translational Research Program.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Bessa X, Alenda C, Paya A, Alvarez C, Iglesias M, Seoane A, Dedeu JM, Abuli A, Ilzarbe L, Navarro G, Pellise M, Balaguer F, Castellvi-Bel S, et al. Validation microsatellite path score in a population-based cohort of patients with colorectal cancer. J Clin Oncol. 2011; 29:3374–80. doi: 10.1200/jco.2010.34.3947.
2. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008; 111:2505–15. doi: 10.1182/blood-2007-07-102798.
3. Shia J, Ellis NA, Paty PB, Nash GM, Qin J, Offit K, Zhang XM, Markowitz AJ, Nafa K, Guillem JG, Wong WD, Gerald WL, Klimstra DS. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am J Surg Pathol. 2003; 27:1407–17.
4. Fenton PA, Clarke SE, Owen W, Hibbert J, Hodgson SV. Cribriform variant papillary thyroid cancer: a characteristic of familial adenomatous polyposis. Thyroid. 2001; 11:193–7. doi: 10.1089/105072501300042965.
5. Sainty D, Liso V, Cantu-Rajnoldi A, Head D, Mozziconacci MJ, Arnoulet C, Benattar L, Fenu S, Mancini M, Duchayne E, Mahon FX, Gutierrez N, Birg F, et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements. Blood. 2000; 96:1287–96.
6. (2016). WHO Classification of Tumoours of the Urinary System and Male Genital Organs: World Health Organization.
7. Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012; 60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x.
8. Brinker DA, Potter SR, Epstein JI. Ductal adenocarcinoma of the prostate diagnosed on needle biopsy: correlation with clinical and radical prostatectomy findings and progression. Am J Surg Pathol. 1999; 23:1471–9.
9. Han B, Mehra R, Suleman K, Tomlins SA, Wang L, Singhal N, Linetzky KA, Palanisamy N, Zhou M, Chinnaiyan AM, Shah RB. Characterization of ETS gene aberrations in select histologic variants of prostate carcinoma. Mod Pathol. 2009; 22:1176–85. doi: 10.1038/modpathol.2009.79.
10. Morais CL, Herawi M, Toubaji A, Albadine R, Hicks J, Netto GJ, De Marzo AM, Epstein JI, Lotan TL. PTEN loss and ERG protein expression are infrequent in prostatic ductal adenocarcinomas and concurrent acinar carcinomas. Prostate. 2015; 75:1610–9. doi: 10.1002/pros.23042.
11. Sanati S, Watson MA, Salavaggione AL, Humphrey PA. Gene expression profiles of ductal versus acinar adenocarcinoma of the prostate. Mod Pathol. 2009; 22:1273–9. doi: 10.1038/modpathol.2009.103.
12. Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, Wu D, Lee MK, Dintzis S, Adey A, Liu Y, Eaton KD, Martins R, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014; 16:56–67. doi: 10.1016/j.jmoldx.2013.08.004.
13. Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, Salipante SJ, Milbank J, Yu M, Grady WM, Tait JF, Corey E, Vessella RL, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014; 5:4988. doi: 10.1038/ncomms5988.
14. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–28. doi: 10.1016/j.cell.2015.05.001.
15. Morgan TM, Welty CJ, Vakar-Lopez F, Lin DW, Wright JL. Ductal adenocarcinoma of the prostate: increased mortality risk and decreased serum prostate specific antigen. J Urol. 2010; 184:2303–7. doi: 10.1016/j.juro.2010.08.017.
16. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189–99. doi: 10.1056/NEJMoa1406498.
17. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20. doi: 10.1056/NEJMoa1500596.
18. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016; 387:1909–20. doi: 10.1016/s0140-6736(16)00561-4.
19. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54. doi: 10.1056/NEJMoa1200690.
20. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–58. doi: 10.1126/science.1235122.
21. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7. doi: 10.1038/nature11252.
22. Graff JN, Alumkal JJ, Drake CG, Thomas GV, Redmond WL, Farhad M, Cetnar JP, Ey FS, Bergan RC, Slottke R, Beer TM. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget. 2016; 7:52810–17. doi: 10.18632/oncotarget.10547.
23. Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014; 60:1192–9. doi: 10.1373/clinchem.2014.223677.
24. Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, Pritchard CC. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014; 147:1308–16.e1. doi: 10.1053/j.gastro.2014.08.041.