
INTRODUCTION
Glial cells are the major central nervous system-resident cells, and represent critical effectors of central nervous system inflammation [1, 2]. Any disruption in the normal function of glial cells may have drastic consequences on brain function [3, 4]. Neuroinflammation is initiated upon recognition of the invading pathogen(s). Germ-line pathogen recognition receptors recognize pathogen-associated molecular patterns and initiate a host immune response against the invaders [5, 6].
Polyinosinic-polycytidylic acid (poly(I:C)) is a double-stranded RNA immune stimulant that is recognized by pathogen recognition receptors, and results in the production and release of several inflammatory cytokines [7]. In the central nervous system, glial cells can recognize poly(I:C), resulting in marked gliosis [8, 9]. Glial cells recognize poly(I:C) in two different ways [10]. In one way, poly(I:C) is internalized by endocytosis and recognized by toll-like receptor 3. In another way, poly(I:C) is recognized by melanoma differentiation-associated gene 5 (MDA-5) and retinoic acid inducible gene I [10]. Stimulation of toll-like receptor 3 by naked poly(I:C) results in nuclear factor-kappa B (NF-κB) activation, which in turn leads to production of inflammatory cytokines such as tumor necrosis factor (TNF)-α, chemokine (C-C motif) ligand 5 (CCL5), and interleukin (IL)-8 [11, 12]. On the other hand, expression of both retinoic acid inducible gene I and MDA-5 is increased in poly(I:C)-treated glial cells, but only MDA-5 is believed to play a central role in its recognition and the subsequent immune activation [13, 14]. Following recognition of poly(I:C) by MDA-5, MDA-5 associates with mitochondrial antiviral signaling protein (MAVS), and this interaction is tightly regulated. The MDA-5/MAVS complex then recruits both the IKKε/TBK1 and IKK α/β/γ complexes, which subsequently activate interferon regulatory factor 3 (IRF3) and NF-κB pathways [15–18].
MicroRNAs (miRNAs) are critical regulators of gene expression that utilize sequence complementarity to bind to and decrease the stability or translation efficiency of target mRNAs [19]. Recent studies have revealed that miRNAs participate in various biological processes such as organogenesis, cellular proliferation and differentiation, apoptosis, innate and adaptive immunity, inflammation, and tumorigenesis [20–25]. Accumulating evidence also suggests a decisive role for miRNAs in neuroinflammation. For instance, miR-155 targets suppressor of cytokine signaling 1 and modulates cytokine production in microglia [26], and it also negatively regulates blood-brain barrier function [27]. Recently, miR-200b and miR-210 were reported to reduce neuroinflammation [28, 29]. Critical roles for miR-181 and miR-146 in astrocyte-mediated inflammation have also been described [30, 31]. We have also previously reported a role for miR-206 in lipopolysaccharide-mediated inflammatory cytokine production in astrocytes [32]. Hence, several miRNAs mediate neuroinflammation in glial cells. However, the details of miRNA-mediated regulation of neuroinflammation remain unclear.
In the present study, we investigated that miR-22 is a negative regulator of type I interferon and inflammatory cytokine production in poly(I:C)-treated human glial cells. To the best of our knowledge, we demonstrated for the first time that the suppressive functions of miR-22 are achieved through targeting MAVS.
RESULTS
miR-22 level is upregulated in poly(I:C)-treated cells
Poly(I:C) induces increased expression of several miRNAs that control immune responses [33–35]. Of these, we identified miR-22 as a potentially neuroprotective miRNA based on its predicted regulation of targets implicated in immune responses. To delineate the kinetics of miR-22 in poly(I:C)-mediated immune responses, the expression of miR-22 was determined in poly(I:C)-treated U251 cells. The results revealed that miR-22 expression was significantly up-regulated in a time- (Figure 1A) and dose-dependent manner (Figure 1B). Similar to the results for mature miR-22, primary miR-22 transcripts (pri-miR-22) and miR-22 precursors (pre-miR-22) were also found to be up-regulated in poly(I:C)-stimulated U251 cells (Figure 1C and 1D). We also investigated the expression of miR-22 in poly(I:C)-treated SH-SY5Y cells (Supplementary Figure S1A and S1B), and the results were concordant with poly(I:C)-stimulated U251 cells. These findings suggest that miR-22 expression is up-regulated in poly(I:C)-treated cells.
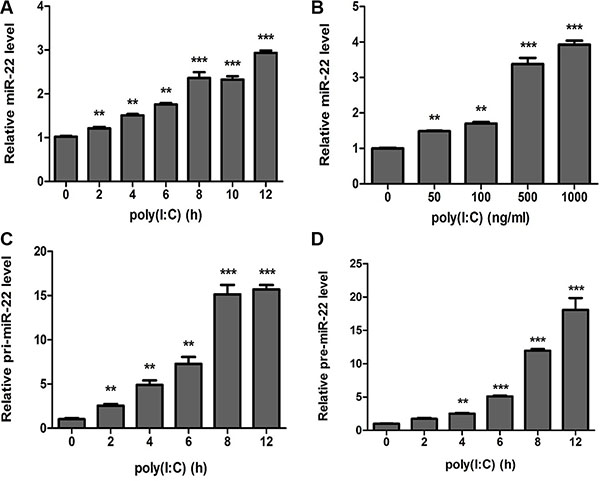
Figure 1: miR-22 is upregulated in poly(I:C)-treated glial cells. (A and B) Human U251 cells were transfected with 100 ng/ ml poly(I:C) for different periods (A) or with different concentrations for 6 h (B), and miR-22 level was determined with quantitative real-time PCR. (C and D) U251 cells were transfected with 100 ng/ml poly(I:C) for different periods, and the level of each of pri-miR-22 (C) and pre-miR-22 (D) was detected with quantitative real-time PCR. All data are representative of at least three independent experiments. (**p < 0.01; ***p < 0.001).
miR-22 reduces poly(I:C)-triggered type I interferon and inflammatory cytokine production
Stimulation of cells with poly(I:C) results in a type I interferon immune response along with the production of other inflammatory cytokines [3, 36]. To identify whether poly(I:C)-induced miR-22 mediates poly(I:C)-triggered immune responses in U251 cells, we investigated the role of miR-22 in type I interferon production following stimulation of U251 cells with poly(I:C). Quantitative real-time PCR results revealed that overexpression of miR-22 significantly decreased poly(I:C)-triggered IFN-β production (Figure 2A), whereas inhibition of miR-22 expression increased poly(I:C)-triggered IFN-β production (Figure 2B). We next investigated the function of miR- 22 in IFN-β promoter activation. U251 cells were co- transfected with miR-22 mimics and the IFN-β luciferase reporter plasmids together with the internal control plasmid pRL-TK and then transfected with poly(I:C). Consistent with the effect of miR-22 on IFN-β production, miR-22 significantly suppressed IFN-β promoter activity in response to stimulation with poly(I:C) (Figure 2C). In contrast, inhibition of endogenous miR-22 expression enhanced IFN-β promoter activity (Figure 2D).
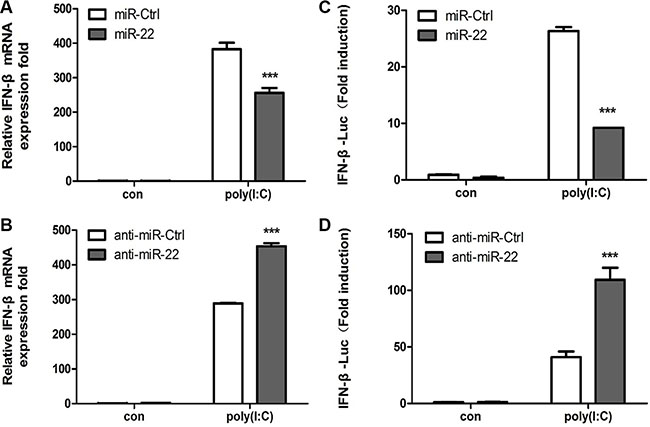
Figure 2: miR-22 negatively regulates poly(I:C)-triggered type I interferon production in human U251 cells. (A and B) U251 cells were transfected with miR-22 mimics (A), miR-22 inhibitors (B), or the corresponding control oligo (final concentration, 50 nM) for 24 h and then transfected with 100 ng/ml poly(I:C) for 8 h. IFN-β mRNA levels were determined with quantitative real-time PCR and normalized to the expression of β-actin in each sample. (C and D) miR-22 mimics (C), miR-22 inhibitors (D), or the corresponding control oligo (final concentration, 50 nM) were co-transfected with IFN-β-Luc as well as pRL-TK (internal control). After 24 h, the cells were transfected with 100 ng/ml poly(I:C). Luciferase activity was measured 8 h later, and Renilla reniformis luciferase activity was normalized to the firefly luciferase activity. (***p < 0.001). All data are representative of at least three independent experiments.
Next, the effects of miR-22 on related inflammatory cytokines were examined. In agreement with previous results, miR-22 overexpression reduced poly(I:C)-triggered production of TNF-α, IL-1β, IL6, and CCL5 (Figure 3A– 3D), whereas inhibition of miR-22 expression resulted in increased production of inflammatory cytokines (Figure 4A–4D). Taken together, these data strongly demonstrate that miR-22 reduces the production of type I interferon and inflammatory cytokines in poly(I:C)-treated cells.
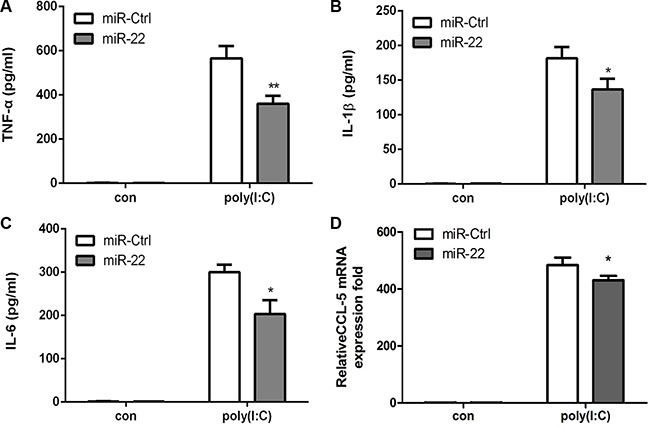
Figure 3: miR-22 suppresses poly(I:C)-triggered production of inflammatory cytokines. (A–D) U251 cells were transfected with miR-22 mimics or control miRNA (final concentration, 50 nM) for 24 h and then transfected with 100ng/ml poly(I:C) for 8 h. The protein levels of TNF-α, IL-1β, and IL6 were analyzed by ELISA. Data represent means ± SD from three independent experiments performed in duplicate. (*p < 0.05; **p < 0.01). CCL5 mRNA levels were determined with quantitative real-time PCR and normalized to the expression of β-actin in each sample. All data are representative of at least three independent experiments. (*p < 0.05).
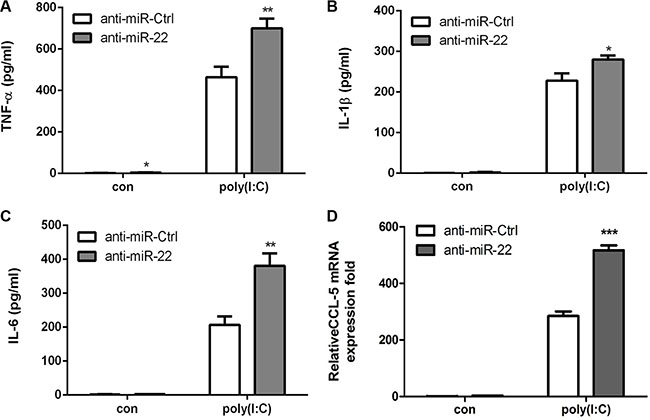
Figure 4: Inhibition of miR-22 increases poly(I:C)-triggered production of inflammatory cytokines. (A–D) U251 cells were transfected with miR-22 inhibitors or control oligos (final concentration, 50 nM) for 24 h and then transfected with 100 ng/ml poly(I:C) for 8 h. The protein levels of TNF-α, IL-1β, and IL6 were analyzed by ELISA. Data represent means ± SD from three independent experiments performed in duplicate. (*p < 0.05; **p < 0.01). CCL5 mRNA levels were determined with quantitative real-time PCR and Zormalized to the expression of β-actin in each sample. All data are representative of at least three independent experiments. (***p < 0.001).
miR-22 targets human MAVS
We used the publically available miRNA target-prediction algorithms TargetScan, Pictar, PITA, miRBase, and RNAhybrid to identify miR-22 targets with potential relevance to control of type I interferon and inflammatory cytokine production. Among them, we selected MAVS as a potential target for miR-22. The crucial role of MAVS in the host innate immune response is well established [15, 16]. Both retinoic acid–inducible gene I and MDA- 5 recognize viral RNA and signal through a common adaptor molecule, i.e., MAVS, to activate the downstream IRF3 and NF-κB signaling pathways [36]. Therefore, we hypothesized that miR-22-mediated reduction of type I interferon and inflammatory cytokines may achieve through targeting MAVS. Our preliminary results using TargetScan, Pictar, PITA, miRBase, and RNAhybrid showed that MAVS had potential seed matches for miR-22 and thus, was further investigated. The predicted miR-22 target sequences in the 3′-UTR of MAVS are shown in Figure 5A. The MAVS 3′-UTR containing a potential seed match region was cloned into the dual-luciferase reporter plasmid (Figure 5A), and 293T cells were co-transfected with this reporter plasmid and miR-22 mimics or inhibitors. The luciferase signal decreased significantly following transfection of miR- 22 mimics. By contrast, luciferase activity increased following treatment with miR-22 inhibitors (Figure 5B). To confirm that this reduction in luciferase activity was indeed due to interaction of miR-22 with the 3′-UTR of MAVS, a mutant dual luciferase reporter containing four base pair mutations in the seed region was also co- transfected into 293T cells together with miR-22 mimics or inhibitors. As expected, no significant effect of either miR-22 mimics or inhibitors was observed (Figure 5B). To further probe the impact of the interaction between miR-22 and the MAVS 3′-UTR, expression of endogenous MAVS was measured in U251 cells treated with miR-22 mimics or inhibitors. As predicted, ectopic expression of miR-22 resulted in marked reduction in MAVS both at the transcriptional and post-transcriptional levels, whereas miR-22 inhibitors increased the expression of MAVS (Figure 5C and 5D). Thus, these results suggest that MAVS is a direct target of miR-22, and its expression is repressed by miR-22.
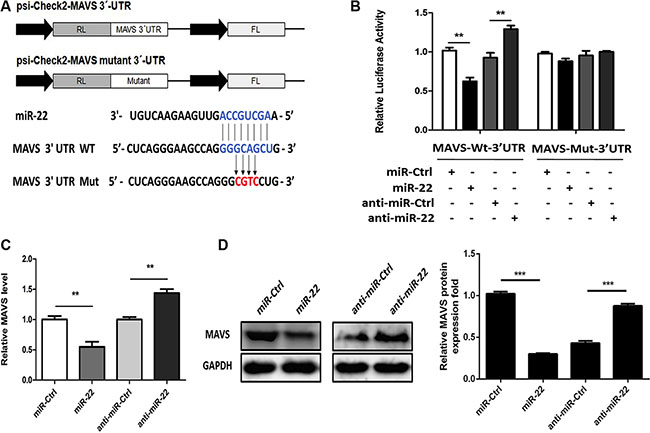
Figure 5: miR-22 targets human MAVS. (A) The predicted miR-22 target sequence in the 3′-UTR of MAVS was downloaded from TargetScan. The mutant construct that contains four base pair mutations is shown below. The upper panel is the schematic diagram showing dual-luciferase reporter constructs harboring 3′-UTR of MAVS. (B) 293T cells were co-transfected with miR-22 mimics, miR-22 inhibitors, or the corresponding control oligo (final concentration, 50 nM) together with a wild-type (Wt) or mutated (Mut) MAVS 3′- UTR dual-luciferase reporter plasmid, and Renilla luciferase activity was measured and normalized to firefly luciferase activity after 24 h. (C and D) U251 cells were transfected with miR-22 mimics, miR-22 inhibitors, or the corresponding control oligo (final concentration, 50 nM), and then MAVS mRNA (C) and protein levels (D) were determined after 48 h with quantitative real-time PCR and immunoblotting, respectively. Protein levels were quantified with immunoblot scanning and normalized to the amount of GAPDH expression. All data are representative of at least three independent experiments. (**p < 0.01; ***p < 0.001).
MAVS expression is downregulated in poly(I:C)-treated cells
To study the effect of poly(I:C) on MAVS, time-dependent and dose-dependent expression pattern of MAVS mRNA (Figure 6A and 6C) and protein (Figure 6B and 6D) in U251 cells following poly(I:C) treatment was studied. Significant downregulation of MAVS mRNA and protein levels were observed in poly(I:C)-treated cells. Furthermore, MAVS mRNA and protein expression levels were also determined in poly(I:C)-stimulated SH-SY5Y cells (Supplementary Figure S1C and S1D). The results were similar to those as were observed in U251 cells. Thus, these data demonstrate that MAVS expression is downregulated upon poly(I:C) treatment.
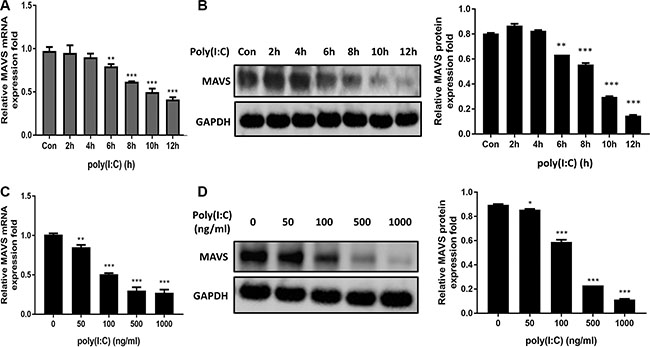
Figure 6: MAVS expression is downregulated in poly(I:C)-treated glial cells. (A and B) Human U251 cells were transfected with 100ng/ml poly(I:C) for different periods, and MAVS mRNA (A) and protein (B) level was determined with quantitative real-time PCR and immunoblotting, respectively. (C and D) U251 cells were transfected with poly(I:C) with different concentrations for 6h, and and MAVS mRNA (C) and protein (D) level was determined with quantitative real-time PCR and immunoblotting, respectively. Data represent means ± SD from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. Protein levels were quantified with immunoblot scanning and normalized to the amount of GAPDH expression.
miR-22 reduces poly(I:C)-triggered IFN-β and inflammatory cytokine production via targeting MAVS
Overexpression of MAVS is sufficient to activate the IRF3/7 and NF-κB pathways to induce type I interferon and inflammatory cytokines production, respectively [15]. We therefore determined whether the MAVS expression plasmid could rescue the inhibition of type I interferon and inflammatory cytokine production mediated by miR- 22. U251 cells were co-transfected with miR-22 mimics and the MAVS expression plasmid, and then transfected with poly(I:C). We found that overexpression of MAVS fully rescued the suppression of type I interferon (Figure 7A) and inflammatory cytokines (Figure 7B–7E) observed upon miR-22 overexpression.
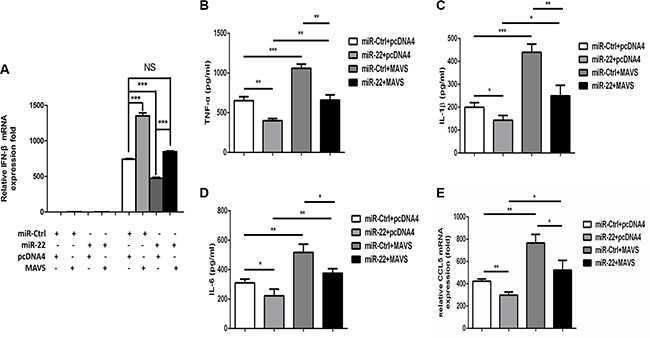
Figure 7: Overexpression of MAVS enhances the poly(I:C)-triggered production of IFN-β and inflammatory cytokines. (A) U251 cells were co-transfected with miR-22 mimics or control mimics and pCDNA4 (200 ng) or pCDNA4-MAVS (200 ng) for 24 h and either left untransfected or transfected with 100 ng/ml poly(I:C). After 8 h, total RNA was extracted, and IFN-β level was determined with quantitative real-time PCR. (B–E) U251 cells were co-transfected with miR-22 mimics or control mimics and pCDNA4 (200 ng) or pCDNA4-MAVS (200ng) for 24 h and either left untransfected or transfected with 100ng/ml poly(I:C) for 8 h. The protein levels of TNF-α, IL-1β, and IL6 were analyzed by ELISA. Data represent means ± SD from three independent experiments performed in duplicate. (*p < 0.05; **p < 0.01). CCL5 mRNA levels were determined with quantitative real-time PCR and normalized to the expression of β-actin in each sample. All data are representative of at least three independent experiments. (*p < 0.05; **p < 0.01).
To further examine whether the observed effects of miR-22 on type I interferon and inflammatory cytokines in response to poly(I:C) were, at least partially, mediated through MAVS, we analyzed the effects of silencing of MAVS expression by siRNA in U251 cells. The cells were co-transfected with miR-22 inhibitors and MAVS-specific siRNA, and then transfected with poly(I:C). First, we confirmed that the siRNA significantly inhibited MAVS in U251 cells at both the mRNA and protein levels (Figure 8A and 8B). Knockdown of MAVS significantly decreased the expression level of IFN-β and inflammatory cytokines, which means that MAVS silencing produces effects similar to those of miR-22 overexpression (Figure 8C and 8D–H). Thus, these data demonstrate that reduction of poly(I:C)-triggered IFN-β and inflammatory cytokines by miR-22 was achieved through MAVS.
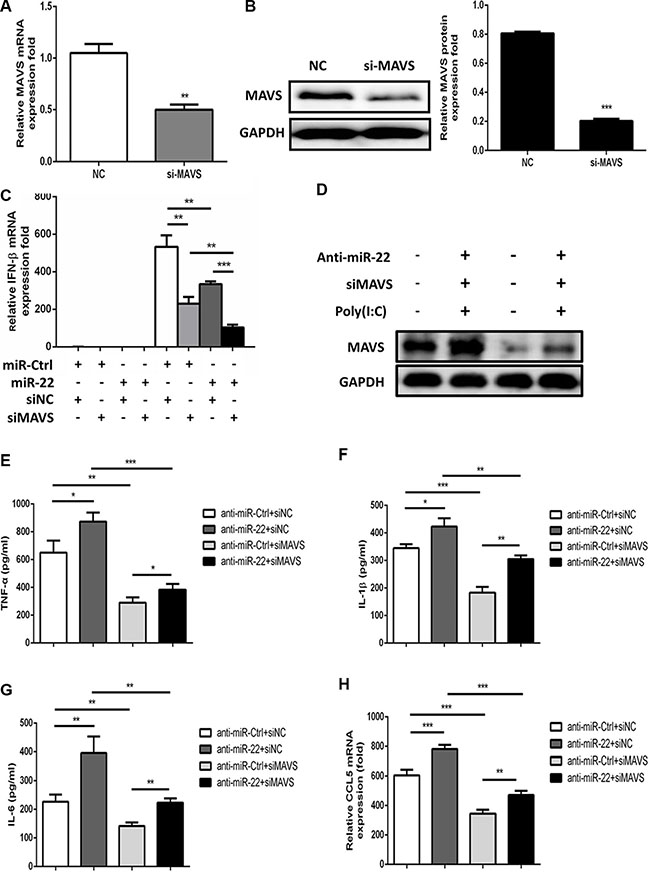
Figure 8: Silencing of MAVS reduces the poly(I:C)-triggered production of IFN-β and inflammatory cytokines. (A and B) U251 cells were transfected with siMAVS or nonspecific control siRNA (final concentration, 50 nM) for 24 h, and then MAVS mRNA (A) and protein (B) levels were measured with quantitative real-time PCR and immunoblotting, respectively. Protein levels were quantified with immunoblot scanning and normalized to the amount of GAPDH expression. (C) U251 cells were co-transfected with miR-22 inhibitors or control oligos and siMAVS or non-specific control siRNA (final concentration, 50 nM) for 24 h, and then either left untransfected or transfected with 100 ng/ml poly(I:C). After 8 h, total RNA was extracted, and IFN-β level was determined with quantitative real-time PCR. (D–H) U251 cells were co-transfected with miR-22 inhibitors or control oligos and siMAVS or non-specific control siRNA (final concentration, 50 nM) for 24 h, and then either left untransfected or transfected with 100 ng/ml poly(I:C) for 8 h. (D) MAVS protein levels were determined by immunoblotting. Protein levels were normalized to the amount of GAPDH expression. (E–H) The protein levels of TNF-α, IL-1β, and IL6 were analyzed by ELISA. Data represent means ± SD from three independent experiments performed in duplicate. (*p < 0.05; **p < 0.01; ***p < 0.001). CCL5 mRNA levels were determined with quantitative real-time PCR and normalized to the expression of β-actin in each sample. All data are representative of at least three independent experiments. (**p < 0.01; ***p < 0.001).
miR-22 suppresses the interferon pathway downstream of MAVS
Activated MAVS can recruit TNF receptor–associated factor family proteins, which leads to production of type I interferon and inflammatory cytokines through activation of the IRF3 and NF-κB [16, 17]. To further define the mechanisms by which miR-22 regulates the production of type I interferon and inflammatory cytokines, the impact of miR-22 on activation of IRF3 and NF-κB was determined in U251 cells treated with poly(I:C). First, we investigated the effect of miR-22 on the activity of IRF3 and NF-κB. U251 cells were co-transfected with miR-22 mimics and the luciferase reporter plasmids harboring the IRF3 or NF-κB binding sites together with the internal control plasmid pRL- TK, and then transfected with poly(I:C). Consistent with the effect of miR-22 on IFN-β promoter activity, miR-22 significantly suppressed the activity of IRF3 and NF-κB upon stimulation with poly(I:C) (Figure 9A and 9B). In contrast, inhibition of endogenous miR-22 expression enhanced the activities of both transcriptional factors (Figure 9C and 9D). It has been well established that translocation of NF-κB from the cytoplasm to the nucleus is a key determinant of NF-κB activation [37, 38]. Therefore, it was of interest to evaluate the effect of miR- 22 on NF-κB and IRF3 activation in poly(I:C)-treated cells. Nuclear translocation of NF-κB (p65) and IRF3 was detected with immunoblotting. Transfection of miR- 22 mimics inhibited the translocation of IRF3 and NF-κB from the cytoplasm to the nucleus (Figure 10A and Figure 11A). In contrast, treatment of cells with miR-22 inhibitors significantly increased the nuclear translocation of IRF3 and NF-κB in poly(I:C)-stimulated U251 cells (Figure 10B and Figure 11B). Taken together, these findings demonstrate that miR-22 appears to regulate the signaling pathway downstream of MAVS.
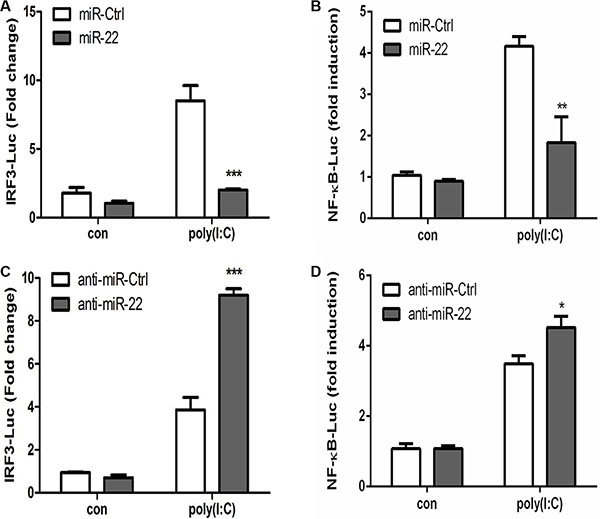
Figure 9: miR-22 negatively regulates the activity of transcriptional factors IRF3 and NF-κB. (A and B) miR-22 mimics or the corresponding control oligo (final concentration, 50 nM) were co-transfected with IRF3-Luc or NF-κB-Luc, as well as pRL-TK (internal control). After 24 h, the cells were transfected with 100 ng/ml poly(I:C). Luciferase activity was measured 8 h later, and the Renilla reniformis luciferase activity was normalized to firefly luciferase activity. (C and D) miR-22 inhibitors or the corresponding control oligo (final concentration, 50 nM) were co-transfected with IRF3-Luc or NF-κB-Luc, as well as pRL-TK (internal control). After 24 h, the cells were transfected with 100 ng/ml poly(I:C). Luciferase activity was measured 8 h later, and the Renilla reniformis luciferase activity was normalized to firefly luciferase activity. *p < 0.05; **p < 0.01; ***p < 0.001.
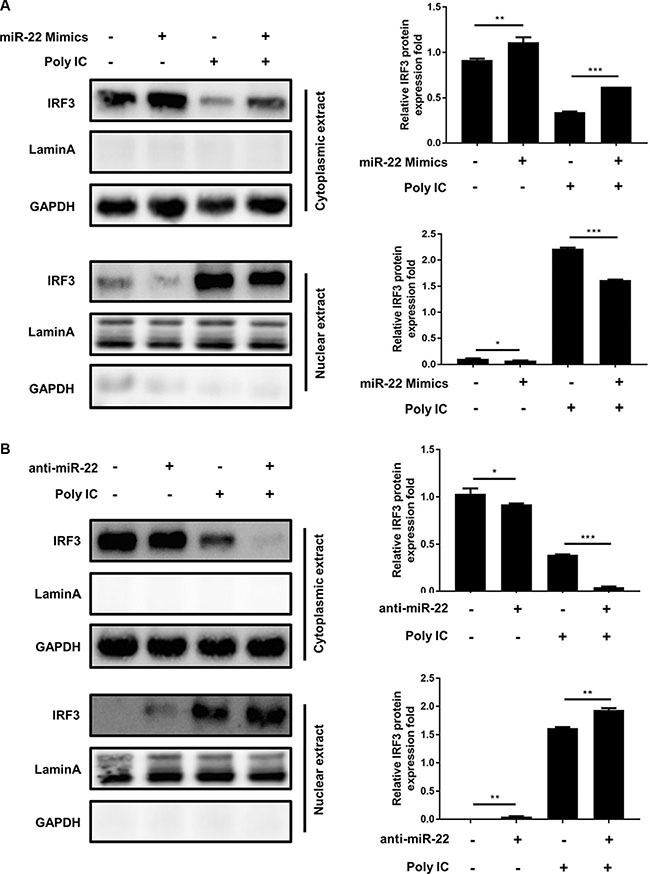
Figure 10: miR-22 suppresses the nuclear translocation of IRF3. (A and B) U251 cells were transfected with miR-22 mimics (A), miR-22 inhibitors (B), or the corresponding control oligo (final concentration, 50 nM) for 24 h and then transfected with poly(I:C) for another 8 h. The cytosolic extracts (upper panel) and nuclear extracts (lower panel) were isolated and subjected to immunoblotting with antibodies against IRF3, LaminA, and GAPDH. LaminA was used as a marker for nuclei. GAPDH and LaminA were used as the loading control. Protein levels were quantified with immunoblot scanning and normalized to the amount of GAPDH or LaminA expressions. *p < 0.05; **p < 0.01; ***p < 0.001.
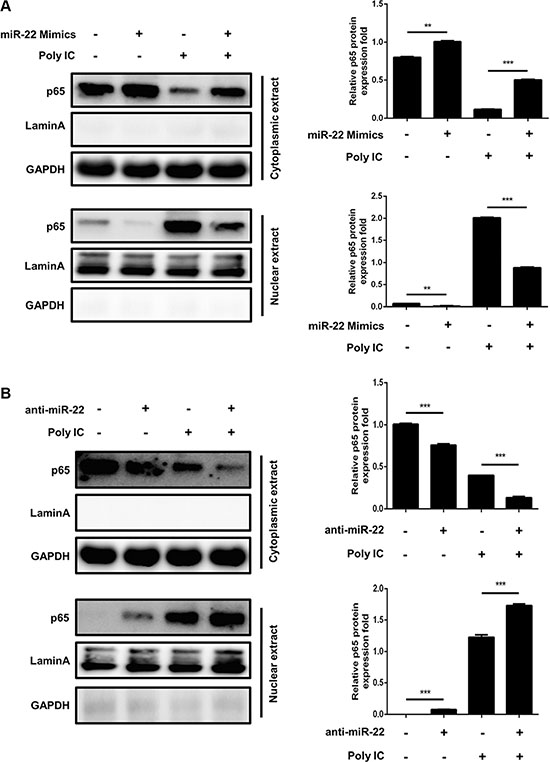
Figure 11: miR-22 inhibits the translocation of NF-κB from the cytoplasm to the nucleus. (A and B) U251 cells were transfected with miR-22 mimics (A), miR-22 inhibitors (B), or the corresponding control oligo (final concentration, 50 nM) for 24 h and then transfected with poly(I:C) for another 8 h. The cytosolic extracts (upper panel) and nuclear extracts (lower panel) were isolated and subjected to immunoblotting with antibodies against NF-κB (p65), LaminA, and GAPDH. LaminA was used as a marker for nuclei. GAPDH and LaminA were used as the loading control. Protein levels were quantified with immunoblot scanning and normalized to the amount of GAPDH or LaminA expressions. **p < 0.01; ***p < 0.001.
Japanese encephalitis virus (JEV) infection upregulates miR-22 expression and downregulates MAVS expression
Because poly(I:C) is used as analog of viral double-stranded RNA, we hypothesized that upregulation of miR-22 expression in glial cells would facilitate the replication of neurotropic viruses such as JEV which can replicate effectively in glial cells. To study the effect of JEV infection on miR-22 and MAVS, expression pattern of miR-22 and MAVS in U251 cells following JEV infection was studied. The results revealed that miR- 22 was significantly up-regulated in a time-dependent (Figure 12A) and dose-dependent manner (Figure 12B). Significant down-regulation of MAVS mRNA and protein levels at 12, 24, and 36 h post- infection was also observed (Figure 12C and 12D). Furthermore, we found that treatment of cells with miR-22-specific mimics enhanced the JEV replication, whereas miR-22-specific inhibitors exhibited an antiviral activity (Figure 12E). These findings suggest that JEV infection upregulates miR-22 expression and downregulates MAVS expression in glial cells.
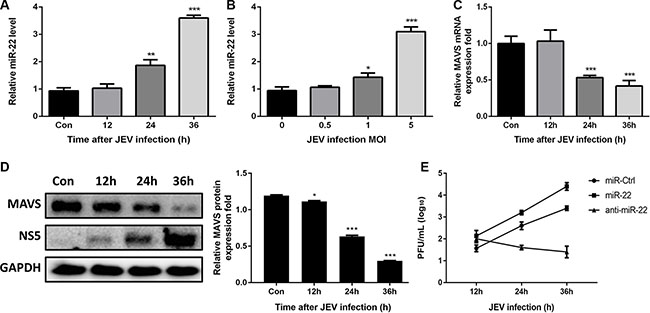
Figure 12: JEV infection upregulates miR-22 expression and downregulates MAVS expression. (A and B) U251 cells were infected with JEV at MOI of 5 for the indicated times (A), or at indicated MOIs for 36 h (B). The levels of miR-22 were detected by quantitative real-time-PCR. (C and D) U251 cells were infected with JEV at MOI of 5 for the indicated times, and then MAVS mRNA (C) and protein (D) expression levels were determined by quantitative real-time PCR and immunoblotting, respectively. Data represent means ± SD from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. Protein levels were quantified with immunoblot scanning and normalized to the amount of GAPDH expression. (E) The transfected U251 cells were infected with JEV at MOI of 5. Cells were collected at the indicated time points, and titers of infectious virus in the culture supernatants were determined by plaque assay. The data represent three independent experiments with identical results.
DISCUSSION
Innate immunity is a vital component of central nervous system, and entrusts mainly on resident glial cells [39]. Glial cells exhibits an array of receptors associated with innate immunity such as Toll like receptors, double-stranded RNA-dependent protein kinase, nucleotide-binding oligomerization domains, mannose receptors, scavenger receptors, and components of the complement system [40–44]. Inflammation in the central nervous system is characterized by marked activation of these glial cells [45]. Upon activation, glial cells are endowed with the ability to produce type I interferons, inflammatory cytokines, and chemokines [39, 46]. Multiple miRNAs can regulate human innate immune signaling pathways and mediate inflammatory responses. Recently, numerous miRNAs have been reported to play pivotal roles in neuroinflammation. For example, miR-155 modulates JEV–induced neuroinflammation by targeting Src homology 2–containing inositol phosphatase 1 [47]. Another miRNA, miR-29b, regulates JEV–induced microgliosis by targeting TNF-α–induced protein 3 [48]. Recently, we also reported the roles of miR-15b and miR-19b-3p in JEV-mediated neuroinflammation in glial cells by targeting ring finger protein 125 and 11, respectively [38, 49]. However, the role of miRNAs in reducing neuroinflammation is largely unknown. Here, we demonstrated that miR-22 is a negative regulator of poly(I:C)-induced neuroinflammation in human glial cells. Briefly, miR-22 inhibited MAVS expression and negatively regulated poly(I:C)-triggered production of type I interferon and inflammatory cytokines in U251 cells.
miR-22 has been found to be ubiquitously expressed in various tissues [50, 51]. Evolutionary clustering suggests that miR-22 is highly conserved in vertebrate evolution, indicating its functional importance in vertebrate species. It had been deduced from the statistical analysis of 3′-UTR in transcriptome that miR-22 is involved in the regulation of many target genes [52]. Previous studies have suggested that miR-22 functioned in multiple cellular processes such as proliferation, differentiation, apoptosis, senescence, and its deregulation is a hallmark of cancer [53–55]. However, the neuroprotective activity of miR-22 remains poorly understood. In the present study, we demonstrated that miR-22 was upregulated in U251 cells treated with poly(I:C) and repressed the production of type I interferon and inflammatory cytokines by targeting MAVS. Poly(I:C) activates MDA5 and its downstream mediator MAVS, which subsequently results in activation of IRF3 and NF-κB signaling pathways [10]. Interestingly, overexpression of miR-22 markedly inhibited the expression of MAVS in U251 cells. Also, our findings demonstrated that miR-22 inhibited the translocation of IRF3 and NF-κB from the cytoplasm to the nucleus, which in turn led to reduced activation of IFN-β and inflammatory cytokine genes. Knockdown of MAVS with siRNA induced the same effects on IFN-β and inflammatory cytokines levels as was observed following miR-22 overexpression, whereas overexpression of MAVS abrogated the miR-22–mediated inhibitory effects on IFN-β and inflammatory cytokine production in poly(I:C)-treated U251 cells. Taken together, these results indicate that poly(I:C)-induced miR-22 acts as a potent anti-neuroinflammatory miRNA in human glial cells.
Previously, it has been reported that MAVS expression could be downregulated by virus infection via ubiquitin-proteasome-dependent pathways and mRNA instability [56, 57]. However, in this study, we proposed that MAVS expression could be modulated via miR- 22, suggesting a new mechanism of miRNA-mediated downregulation of MAVS.
Many viruses can take advantage of host-encoded miRNAs to modulate the host innate immune response, resulting in increased viral replication. miR-146a negatively regulates vesicular stomatitis virus–triggered production of type I interferon by targeting TNF receptor–associated factor 6 and the kinases IRAK1 and IRAK2 in macrophages, thus promoting vesicular stomatitis virus replication [58]. miR-146a also promotes replication of other important viruses such as Hendra virus, dengue virus, and enterovirus 71 [59–61]. miR- 21 suppresses hepatitis C virus–triggered production of type I interferon by targeting MyD88 and IRAK1 in hepatocytes, which results in increased hepatitis C virus replication [62]. Because poly(I:C) is used as analog of viral double-stranded RNA and miR-22 represses poly(I:C)-triggered production of type I interferon and inflammatory cytokines, we predict that upregulation of miR-22 expression in glial cells would facilitate the replication of neurotropic viruses which can replicate effectively in glial cells. To confirm this prediction, we determined the expression pattern of miR-22 and MAVS in JEV-infected glial cells, and the results showed that JEV infection upregulated the expression pattern of miR-22 and downregulated the levels of MAVS. We also observed that treatment of cells with miR-22 mimics resulted in enhanced JEV replication, whereas inhibitors had the opposite results. From these findings, we propose that other neurotropic viruses may utilize the similar strategy to invade host immune response. To validate this prediction, further studies are needed.
MATERIALS AND METHODS
Cell culture and treatment
U251 cells (human astrocytoma cell line), SH-SY5Y cells (human neuroblastoma cell line), and HEK 293T cells (human embryonic kidney epithelial cells) were cultured and maintained in Dulbecco’s Modified Eagle’s Medium that was supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin sulfate at 37°C in 5% CO2. U251 cells were plated in 12-well plates (1×105 cells/well) and grown to 80% confluency. Non-adherent cells were removed by washing with non-supplemented DMEM prior to further treatment. Cells were subsequently transfected with RNAs and/or plasmids using Lipofectamine 2000 (Invitrogen). After 24 h, cells were transfected with poly(I:C) for another 12 h. For JEV infection experiments, cells were infected with JEV at multiplicity of infection (MOI) of 5 for indicated times.
Reagents
Poly(I:C) was obtained from Sigma. Antibodies against IRF3 were obtained from Cell Signaling Technology. Antibodies against MAVS were purchased from Proteintech Technology. Antibodies against p65, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and LaminA were purchased from Abclonal Technology. Horseradish peroxidase–conjugated anti-mouse/rabbit secondary antibodies were from Boster. miR-22 mimics (double-stranded RNA oligonucleotides) and miR-22 inhibitors (single-stranded chemically modified oligonucleotides) from GenePharma were used for the overexpression and inhibition of miR-22 activity, respectively. Small interfering RNA (siRNA) oligonucleotide and control for MAVS were also purchased from GenePharma. Table 1 lists the sequences of the miR-22 mimics, miR-22 inhibitors, siMAVS oligonucleotides, and their controls.
Table 1: Sequences of RNAi oligonucleotides used in the present study
Name |
Sequence 5′→3′ |
|---|---|
si-MAVS |
UAGUUGAUCUCGCGGACGA-dTdT (sense) |
UCGUCCGCGAGAUCAACUA-dTdT (antisense) |
|
control mimic |
UUCUCCGAACGUGUCACGUTT (sense) |
ACGUGACACGUUCGGAGAATT (antisense) |
|
miR-22 mimic |
AAGCUGCCAGUUGAAGAACUGU (sense) |
AGUUCUUCAACUGGCAGCUUUU (antisense) |
|
control inhibitor |
CAGUACUUUUGUGUAGUACAA |
miR-22 inhibitor |
ACAGUUCUUCAACUGGCAGCUU |
microRNA target prediction
Putative microRNA target genes were identified using the microRNA databases (http://www.mirbase.org/) and target prediction tools PicTar (http://pictar.mdc-berlin.de/), TargetScan (http://www.targetscan.org/), PITA (https://omictools.com/pita-tool), and RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/). The potential targets of miR-22 predicted in these databases were filtered on the basis of conserved and poorly conserved sites for miR-22, aggregate PCT value, and potential relevance of predicted targets with inflammatory pathways. PCT value is the probability of conserved targeting for highly conserved miRNAs.
Constructs and plasmids
The psiCheck-2 dual-luciferase reporter vector (Promega) harboring the 3′-untranslated region (UTR) of MAVS was inserted into the XhoI and PmeI restriction sites at the 3′-end of Renilla, and was used to check the effect of miR-22 on Renilla luciferase activity. The 3′-UTR and the coding region of MAVS were amplified from U251 cell genomic DNA with specific primers and cloned into psiCheck-2 and pCDNA4, respectively (Life Technologies). The psiCheck-2 mutant MAVS 3′-UTR construct was generated by inducing point mutations with overlap-extension PCR method. Table 2 lists all primer sequences. All constructs were verified with sequencing.
Table 2: Sequences of the primers used in the present study
Primer |
Sequence 5′→3′ |
|---|---|
miR-22 stem-loop primer |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAGTT |
miR-22-F |
AAGCTGCCAGTTGAAGAACTGT |
Universal reverse primer |
GTGCAGGGTCCGAGGT |
pri-miR-22-F |
AGGAGTAGAAGGCTCAAACA |
pri-miR-22-R |
AGGAGGGTCAAGAAGGAA |
pre-miR-22-F |
GGCGAGCCGCAGAGC |
pre-miR-22-R |
GGCGGGGCCGTTCTT |
hCCL5-F |
CTGTCATCCTCATTGCTACTGC |
hCCL5-R |
ATGTACTCCCGAACCCATTTCT |
hIFN-β-F |
TTCCCAGGGCTTACACCG |
hIFN-β-R |
TAACCCAAGTTCCCGAGT |
hMAVS-3′-UTR-XhoI-F |
ACGCTCGAGAGCCCAGCCTGAGACCGT |
hMAVS-3′-UTR-PmeI-R |
CGGTTTAAACCTGCACCCTTGACCACTGTG |
Mutant-primer-2-R |
CTGACACGACCCCCTGGC |
Mutant-primer-3-F |
AGCCAGGGGGTCGTGTC |
hMAVS-KpnI-F |
CGAGGTACC ATGCCGTTTGCTGAAGACAAGACC |
hMAVS-XhoI-R |
ACGCTCGAGGTGCAGACGCCGCCGGT |
h: human.
Dual-luciferase reporter assays
For the MAVS 3′-UTR luciferase reporter assay, 239T cells were co-transfected with 200 ng psiCheck-2 dual-luciferase plasmid described above along with miR- 22 mimics, inhibitors, or controls (final concentration, 50 nM). After 36 h, luciferase activity was measured using the dual-luciferase reporter assay system (Promega). For the interferon (IFN)-β, NF-κB, and IRF3 luciferase reporter assays, U251 cells were co-transfected with 100 ng IFN-β, NF-κB or IRF3 luciferase reporter plasmids together with 10 ng of the internal control plasmids, miR-22 mimics, inhibitors, or controls (final concentration, 50 nM) for 24 h, and then transfected with poly(I:C). The data are expressed as relative Renilla luciferase activity normalized to the value of firefly luciferase, and are representative of three independent experiments.
RNA extraction and quantitative real-time PCR
Total RNA in treated cells was extracted with TRIzol (Invitrogen), and 1 μg RNA was used to synthesize cDNA using a first-strand cDNA synthesis kit (TOYOBO). Quantitative real-time PCR was performed using a 7500 Real-time PCR System (Applied Biosystems) and SYBR Green PCR Master Mix (TOYOBO). Data were normalized to the level of β-actin expression in each sample. To quantify mature miRNA expression, a commercial Bulge-Loop miRNA quantitative reverse transcription (RT)-PCR detection method was used. Briefly, 1 μg total RNA was used as the template and reverse transcribed using a miR-22–specific RT primer. The resulting cDNA was used for quantitative real-time PCR with a universal reverse primer and a specific forward primer. Amplification was performed for 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 30 s. The relative expression of miRNAs was normalized to that of internal control U6 small nuclear RNA within each sample using the 2−ΔΔCt method. Expression was then standardized to the miRNA levels in mock or control miRNA–treated cells. Primers used are listed in Table 2.
Immunoblotting
Total cellular lysates were prepared using radioimmunoprecipitation assay buffer (Sigma) containing protease and phosphatase inhibitors (Roche). Radioimmunoprecipitation assay buffer contains 150 mM NaCl, 1.0% IGEPALTM CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris with pH 8.0. For preparation of total cell extracts, PBS-washed cells were treated with ice-cold radioimmunoprecipitation assay buffer for 20 min, and were subjected to vortex after every 5 min in an ice water bath. Later, the samples were centrifuged at 12,000 g for 20 min at 4°C. The cleared supernatant was collected and used as the whole cellular lysate. Cytosolic and nuclear extracts were prepared using NE-PEP Nuclear and Cytoplasmic Extraction Reagent (Thermo Scientific). Protein concentrations were measured using the BCA Protein Assay kit (Thermo Scientific). Samples were subjected to SDS-PAGE, and protein bands were transferred to a polyvinylidene fluoride membrane (Millipore) using a Mini Trans-Blot Cell (Bio-Rad). Blots were probed with relevant antibodies, and positive signals were detected using ECL reagents (Thermo Scientific).
Plaque assay
U251 cells were transfected with miR-22 mimics, inhibitors or their controls (final concentration, 50 nM) for 24 h, and subsequently infected with JEV at MOI of 5. At 12, 24, and 36 h post-infection, cell supernatants were harvested, serially diluted, and then used to inoculate monolayers of U251 cells. After removal of unbound JEV virus particles, U251 cells were further incubated for 3 to 5 days, and plaques identified. The visible plaques were counted and viral titers calculated. All data are expressed as the mean of triplicate samples.
ELISA
The culture supernatants were collected from the treated cells at the indicated time points and stored at –80°C. The protein levels of TNF-α, IL6, and IL-1β in cell cultures were determined by ELISA kits (eBioscience) following the manufacturer’s instructions.
Statistical analysis
All experiments were carried out at least three times with similar results. Analyses were conducted using Prism 5 (GraphPad Software, San Diego, CA). Results are expressed as the mean ± SD. Data were compared with two-way analysis of variance with subsequent t tests using the Bonferroni post-test for multiple comparisons, or with the Student’s t test. For all tests, p < 0.05 was considered significant.
CONFLICTS OF INTEREST
All authors report no potential conflicts of interest.
GRANT SUPPORT
This work was supported by the National Key Research and Development Program of China (2016YFD05004007), the National Natural Sciences Foundation of China (No.31172325, 31472221), Special Fund for Agro-scientific Research in the Public Interest (No.201203082), and Fundamental Research Funds for the Central University (No. 2013PY051, 2662016Q003).
REFERENCES
1. Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005; 6:626–640.
2. Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014; 15:43–53.
3. Aschner M. Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology. 1998; 19:269–281.
4. Choi SS, Lee HJ, Lim I, Satoh J, Kim SU. Human astrocytes: secretome profiles of cytokines and chemokines. PloS one. 2014; 9:e92325.
5. Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010; 58:253–263.
6. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006; 124:783–801.
7. Fortier ME, Kent S, Ashdown H, Poole S, Boksa P, Luheshi GN. The viral mimic, polyinosinic:polycytidylic acid, induces fever in rats via an interleukin-1-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2004; 287:R759–766.
8. Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005; 49:360–374.
9. Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J Immunol (Baltimore, Md : 1950). 2006; 176:3804–3812.
10. De Miranda J, Yaddanapudi K, Hornig M, Lipkin WI. Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J. 2009; 23:1064–1071.
11. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001; 413:732–738.
12. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (New York, NY). 2003; 301:640–643.
13. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006; 441:101–105.
14. Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008; 20:17–22.
15. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005; 6:981–988.
16. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB, IRF 3. Cell. 2005; 122:669–682.
17. Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006; 25:3257–3263.
18. Tang ED, Wang CY. TRAF5 is a downstream target of MAVS in antiviral innate immune signaling. PloS one. 2010; 5:e9172.
19. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297.
20. Gao F, Sun M, Gong Y, Wang H, Wang Y, Hou H. MicroRNA-195a-3p inhibits angiogenesis by targeting Mmp2 in murine mesenchymal stem cells. Mol Rep Devlop. 2016; doi: 10.1002/mrd.22638.
21. Shenoy A, Blelloch RH. Regulation of microRNA function in somatic stem cell proliferation and differentiation. Nat Rev Mol Cell Biol. 2014; 15:565–576.
22. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, Hua M, Li N, Yao H, Cao X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011; 12:861–869.
23. Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic acids Res. 2005; 33:1290–1297.
24. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009; 139:693–706.
25. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008; 40:43–50.
26. Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012; 135:73–88.
27. Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, Kay O, de Vries HE, Hirst MC, Sharrack B, Baker D, Male DK, Michael GJ, et al. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J. 2014; 28:2551–2565.
28. Jadhav SP, Kamath SP, Choolani M, Lu J, Dheen ST. microRNA-200b modulates microglia-mediated neuroinflammation via the cJun/MAPK pathway. J Neurochem. 2014; 130:388–401.
29. Qi J, Qiao Y, Wang P, Li S, Zhao W, Gao C. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. 2012; 586:1201–1207.
30. Hutchison ER, Kawamoto EM, Taub DD, Lal A, Abdelmohsen K, Zhang Y, Wood WH, 3rd, Lehrmann E, Camandola S, Becker KG, Gorospe M, Mattson MP. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013; 61:1018–1028.
31. Iyer A, Zurolo E, Prabowo A, Fluiter K, Spliet WG, van Rijen PC, Gorter JA, Aronica E. MicroRNA-146a: a key regulator of astrocyte-mediated inflammatory response. PloS one. 2012; 7:e44789.
32. Duan X, Zohaib A, Li Y, Zhu B, Ye J, Wan S, Xu Q, Song Y, Chen H, Cao S. miR-206 modulates lipopolysaccharide-mediated inflammatory cytokine production in human astrocytes. Cell Signal. 2015; 27:61–68.
33. Liu X, Zhan Z, Xu L, Ma F, Li D, Guo Z, Li N, Cao X. MicroRNA-148/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKIIalpha. J Immunol (Baltimore, Md: 1950). 2010; 185:7244–7251.
34. Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, Cao X, Wang Q. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1beta production in macrophages by targeting STAT3. PloS one. 2012; 7:e42971.
35. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci. 2007; 104:1604–1609.
36. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015; 15:87–103.
37. Mediero A, Perez-Aso M, Cronstein BN. Activation of EPAC1/2 is essential for osteoclast formation by modulating NFkappaB nuclear translocation and actin cytoskeleton rearrangements. FASEB J. 2014; 28:4901–4913.
38. Zhu B, Ye J, Nie Y, Ashraf U, Zohaib A, Duan X, Fu ZF, Song Y, Chen H, Cao S. MicroRNA-15b Modulates Japanese Encephalitis Virus-Mediated Inflammation via Targeting RNF125. J Immunol (Baltimore, Md: 1950). 2015; 195:2251–2262.
39. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007; 28:138–145.
40. Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000; 49:171–186.
41. Bsibsi M, Ravid R, Gveric D and van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002; 61:1013–1021.
42. Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003; 43:281–291.
43. Sterka D, Jr., Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia. 2006; 53:322–330.
44. Alarcon R, Fuenzalida C, Santibanez M and von Bernhardi R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J Biol Chem. 2005; 280:30406–30415.
45. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012; 122:1164–1171.
46. Detje CN, Lienenklaus S, Chhatbar C, Spanier J, Prajeeth CK, Soldner C, Tovey MG, Schluter D, Weiss S, Stangel M, Kalinke U. Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon Beta producers that protect from lethal encephalitis. J Virol. 2015; 89:2731–2738.
47. Thounaojam MC, Kundu K, Kaushik DK, Swaroop S, Mahadevan A, Shankar SK, Basu A. MicroRNA 155 regulates Japanese encephalitis virus-induced inflammatory response by targeting Src homology 2-containing inositol phosphatase 1. J Virol. 2014; 88:4798–4810.
48. Thounaojam MC, Kaushik DK, Kundu K, Basu A. MicroRNA-29b modulates Japanese encephalitis virus-induced microglia activation by targeting tumor necrosis factor alpha-induced protein 3. J Neurochem. 2014; 129:143–154.
49. Ashraf U, Zhu B, Ye J, Wan S, Nie Y, Chen Z, Cui M, Wang C, Duan X, Zhang H, Chen H, Cao S. MicroRNA-19b-3p Modulates Japanese Encephalitis Virus-mediated Inflammation via Targeting RNF11. J Virol. 2016; pii: JVI.02586–15.
50. Chen B, Tang H, Liu X, Liu P, Yang L, Xie X, Ye F, Song C, Xie X, Wei W. miR-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett. 2015; 356:410–417.
51. Huang ZP, Wang DZ. miR-22 in cardiac remodeling and disease. Trends Cardiovasc Med. 2014; 24:267–272.
52. Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature. 2005; 434:338–345.
53. Qiu K, Huang Z, Huang Z, He Z, You S. miR-22 regulates cell invasion, migration and proliferation in vitro through inhibiting CD147 expression in tongue squamous cell carcinoma. Arch Oral Biol. 2016; 66:92–97.
54. Xiong F, Hu L, Zhang Y, Xiao X, Xiao J. miR-22 inhibits mouse ovarian granulosa cell apoptosis by targeting SIRT1. Biol. 2016; 5:367–371.
55. Chen H, Lu Q, Fei X, Shen L, Jiang D, Dai D. miR-22 inhibits the proliferation, motility, and invasion of human glioblastoma cells by directly targeting SIRT1. Tumour Biol. 2015; 1–8.
56. He Z, Zhu X, Wen W, Yuan J, Hu Y, Chen J, An S, Dong X, Lin C, Yu J, Wu J, Yang Y, Cai J, et al. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J Virol. 2016; pii: JVI.00221–16.
57. Lu LF, Li S, Lu XB, LaPatra SE, Zhang N, Zhang XJ, Chen DD, Nei P, Zhang YA. Spring viremia of carp virus N protein suppresses fish IFNφ1 production by targeting the mitochondrial antiviral signaling protein. J Immunol. 2016; 196: 3744–3753.
58. Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol (Baltimore, Md : 1950). 2009; 183:2150–2158.
59. Stewart CR, Marsh GA, Jenkins KA, Gantier MP, Tizard ML, Middleton D, Lowenthal JW, Haining J, Izzard L, Gough TJ, Deffrasnes C, Stambas J, Robinson R, et al. Promotion of Hendra virus replication by microRNA 146a. J Virol. 2013; 87:3782–3791.
60. Wu S, He L, Li Y, Wang T, Feng L, Jiang L, Zhang P, Huang X. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J Infect. 2013; 67:329–341.
61. Ho BC, Yu IS, Lu LF, Rudensky A, Chen HY, Tsai CW, Chang YL, Wu CT, Chang LY, Shih SR, Lin SW, Lee CN, Yang PC, et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat Commun. 2014; 5:3344.
62. Chen Y, Chen J, Wang H, Shi J, Wu K, Liu S, Liu Y, Wu J. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog. 2013; 9:e1003248.