
INTRODUCTION
The term “myeloid-derived suppressor cells” (MDSCs) was coined in 2007 to describe a non-lymphoid immune suppressor cell population of myeloid origin that was enriched in cancer patients [1]. We now know that MDSCs constitute a population of myeloid cells with heterogeneous morphology, surface phenotype, and function, but with strong immunosuppressive properties in common. These cells are enriched in different pathological conditions including cancer, trauma, and sepsis, with cancer being the predominant condition in which MDSCs have been described [2-5]. Indeed, the elimination of MDSCs dramatically improves immune response in cancer patients and tumor-bearing mice [2, 6, 7].
MDSCs are an important node in the cellular network that regulates immune responses. One of the hallmarks of MDSCs is their ability to suppress T-cell responses. MDSCs have also been described to regulate innate immune responses by modulating the cytokine production of macrophages [2, 4, 5]. Immunosuppressive myeloid cells have most likely been generated as a normal physiological response to acute and excessive inflammatory conditions during evolution. It is therefore not surprising that MDSCs are present at high numbers in tumors, since tumors show chronic inflammation normally controlled by regulatory immunosuppressive cells. In tumors, MDSCs also promote other non-immune functions such as tumor angiogenesis and eventually metastasis [8-10], perhaps reflecting the natural role of MDSCs during wound healing. Because of their suppression of anti-tumor immune responses, MDSCs are often described as “bad cells.” As such, MDSCs provide a favorable microenvironment in which transformed cells can proliferate, acquire new mutations, expand, and evade host immunosurveillance [2, 4]. Some typical MDSC functions are listed in Figure 1.
The accumulation of MDSCs in cancer patients is a generally accepted phenomenon [11, 12]. Its clinical relevance has also been reported for a substantial number of cancers, in which circulating MDSCs have been correlated with clinical cancer stage and tumor burden in patients with different tumors [13-19]. MDSCs have also been negatively correlated with immune responses to cancer therapy [20, 21]. The regulatory role of MDSCs is nonetheless crucial for limiting inflammation and for resolving immune responses in general, so that wound healing and recovery can take place, thereby restoring homeostasis [22, 23]. MDSCs are also thought to protect the host during severe infections through the regulation of inflammatory responses [24]. Indeed, MDSCs accumulate in acute life-threatening conditions such as sepsis, initially limiting the devastating effect of an excessive inflammatory response, and they even might promote bacterial clearance [25]. However, the high level of MDSCs generated probably also contribute to the potentially fatal immune paralysis observed during the later stages of sepsis [26]. Finally, their ability to suppress T-cells may also serve to prevent the development of autoimmune diseases by dampening inappropriate immune reactions [22, 23]. Although the functional importance of MDSCs as regulatory cells has emerged in recent years, there are still uncertainties about their generation and origins. In this review, we attempt to distinguish the different aspects of and theories on the origin of MDSCs with a focus on cancer.
MDSC CHARACTERIZATION
In terms of morphology, surface phenotype, and function, MDSCs are not a defined subset of myeloid cells, but rather a heterogeneous population. As such, they express a mixture of surface markers typical for myeloid cells, but lack lineage markers for lymphocytes, natural killer cells, macrophages, and dendritic cells [4, 27-29]. Two major groups of MDSCs have been characterized to date: cells with a morphology and surface phenotype typical for monocytes (Mo-MDSCs) and cells with a surface phenotype typical for granulocytes (G-MDSCs - also called polymorphonuclear [PMN]-MDSCs), but with a heterogeneous morphology including granulocytes, blasts, or cells with ring-shaped nuclei [27-29].
Historically, MDSCs have been regarded as immature cells. The main reasons for this would be their surface phenotype seen using flow-cytometric analysis, their morphology, and their ability to differentiate into macrophages, dendritic cells, or granulocytes [3, 4, 29]. The immature profile could possibly, at least in part, be opened for reevaluation. The surface phenotype does represent immature cells of myeloid origin, since they express myeloid cell lineage markers but lack activation and maturation markers. This view, however, could be criticized because mature cells of the myeloid lineage could also lose activation markers upon repeated toll-like receptor (TLR)-signaling, exposure to certain cytokines [30-34], or in response to hypoxia [35, 36]. Considerable evidence has emerged that Mo-MDSCs and G-MDSCs may even represent monocytes and neutrophils, respectively, that have been reprogrammed or activated into immunosuppressive populations [26, 29].
MDSCs were originally found in mice, and the surface phenotypes differ vastly between mice and humans. In mice, MDSCs are characterized as Gr-1+CD11b+ cells, and further Mo-MDSCs are described as CD11b+Ly6ChighLy6G- cells and G-MDSCs as CD11b+Ly6ClowLy6G+ expressing cells [4, 5, 28]. In humans, the phenotypic characterization of MDSCs has proven difficult. A great number of surface phenotypes have been described, with significant variations between different individuals, indicating that there may be distinct subpopulations of MDSCs besides G-MDSCs and Mo-MDSCs [4, 5, 28]. However, the increasingly accepted - although still debated - definitions of human MDSCs are CD11b+CD14+CD33+HLA-DR-/lowCo-receptor-/low (Mo-MDSCs) and CD11b+CD15+CD33+Lin-HLA-DR-/low (G-MDSCs) expressing cells, present in the mononuclear fraction of density gradients [5, 23, 37].
New potential candidates continue to be found to further characterize the human MDSCs such as CD66b, CD115 (CSF-1R; M-CSF receptor), CD124 (IL-4Rα), CD40, CD80, and S100A9 [4, 5, 23, 37-39]. Although these markers are undoubtedly expressed by MDSCs, they do not define a specific MDSC population with distinct suppressive functions. Given their heterogeneity, the definition and characterization of MDSCs are somewhat controversial and many studies concerning phenotypic characterization of MDSCs in humans have not studied their suppressive function, even though this activity is a mandatory criterion [40-44]. However, in recent years the suppressive function or markers specific for immune suppression (e.g., Arginase-1 [ARG1]) have been added to their phenotype definition [45].
MDSCs can employ a wide range of suppressive mechanisms that often includes more than one single mechanism (Figure 1). In humans, G-MDSCs are mostly known to inhibit T-cells via the production of reactive oxygen species (ROS) [46]. Mo-MDSCs, on the other hand, mediate T-cell suppression through the induction of high levels of NO/inducible nitric oxide synthase (iNOS), suppressive cytokines, and prostaglandin (PG)E2 [27, 28, 37, 47]. Both populations are able to express the immunosuppressive enzyme ARG1 [4]. The phenotypes and definitions of MDSCs comprise an important field discussed in several other excellent reviews [2, 4-6, 27, 28, 48, 49].
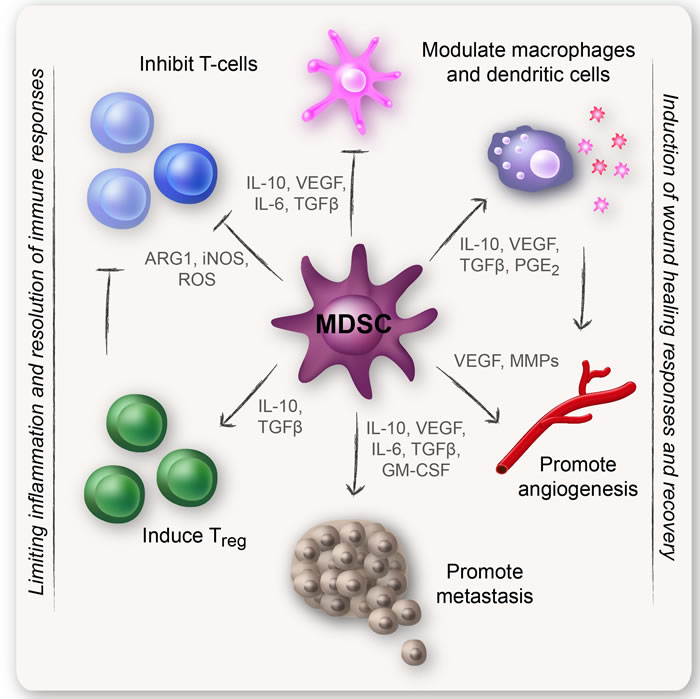
Figure 1: Classical MDSC functions. MDSCs have a strong immunosuppressive character that allows them to limit inflammation so that wound healing and recovery can take place. One of the hallmarks of MDSCs is their ability to suppress T-cell responses. MDSCs have also been described to induce regulatory T-cells (Tregs), modulate the cytokine production of macrophages, and promote other non-immune functions such as tumor angiogenesis and eventually metastasis.
GENERAL ORIGIN OF MDSCs
MDSCs are important for immune suppression in cancer patients and thereby also pose a major obstacle that needs to be overcome for successful anti-cancer immunotherapy treatments. Furthermore, MDSCs decrease after tumor resection and therefore the generation and maintenance of MDSCs appears to be an active process that is nourished by tumor cells [50, 51]. An important issue concerning MDSC generation to remember is that tumors do not invent new biology, they highjack existing mechanisms. To break this vicious cycle in which MDSCs are generated, and to target MDSCs to enhance the effects of cancer therapies, it is important to understand the origin of MDSCs. See Figure 2 for an overview of the theories on the origin of MDSCs.
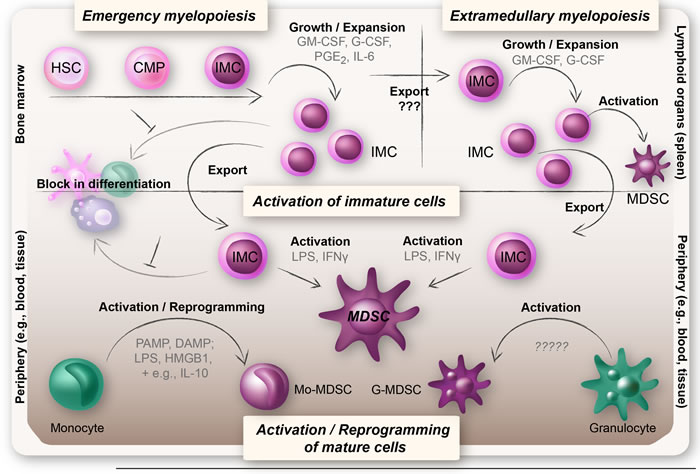
Figure 2: Overview of the theories on the origin of MDSCs. MDSCs arise under pathological conditions when there is an excessive inflammation. Upper left panel: Upon an increased demand for myeloid cells, IMCs expand in the bone marrow and migrate into the periphery, a process known as emergency myelopoiesis. The IMCs are believed to be blocked in their differentiation and to become functionally active MDSCs when exposed to inflammatory mediators (upper and lower panel). Upper right panel: IMCs may also expand and become functionally active MDSCs extramedullary (i.e. in organs outside of the bone marrow, foremost the spleen), a feature often seen in chronic inflammatory diseases such as cancer. Lower panel: More recent hypotheses suggest that Mo-MDSCs and G-MDSCs may represent reprogrammed or activated monocytes and granulocytes. Reprogramming of monocytes into Mo-MDSCs is believed to rely on a repeated TLR-signaling (triggered by PAMPs or DAMPs) in combination with certain cytokines and mediators (e.g., IL-10, Wnt5a, and PGE2), whereas G-MDSCs are thought to represent an activation state of neutrophils.
ABNORMAL MYELOPOIESIS
All MDSCs undoubtedly derive from common myeloid progenitors and their development is likely governed by the same growth factors that control normal myelopoiesis e.g., granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and macrophage colony-stimulating factor (M-CSF) [52-58]. MDSCs arise under pathological conditions, possibly as a result of a persistent signal of low strength coming from tumors or chronic inflammation [48, 59]. However, the strength-of-signal hypothesis might be difficult to defend for two reasons. First, the frequency of MDSCs positively correlates with clinical stage and tumor burden in different cancers [13-17] and second, MDSCs are also found in patients with overt, acute conditions such as trauma and sepsis [13, 60, 61]. Most likely, it is the persistent mixture or combination of signals that generate MDSCs. Among these signals, the CSFs seem to have a prominent role.
THE INVOLVEMENT OF CSFs IN MDSC GENERATION
It is widely debated whether the disease-dependent generation and expansion of MDSCs occurs in the bone marrow, periphery, or extramedullary predominantly in the spleen. MDSCs have been found in both the bone marrow and spleen of humans and mice [62-64]. The fact that MDSCs from the bone marrow, spleen, and blood as well as from tumors and metastases share a similar surface phenotype supports the notion that MDSCs have a common ancestor [49, 65]. There are several tumor-derived factors that could affect the myelopoiesis both in the bone marrow and extramedullary; the best described are GM-CSF, G-CSF, and M-CSF (Figure 3) [66].
GM-CSF, G-CSF, and M-CSF are hematopoietic growth factors that play an essential role in recruitment, proliferation, and maturation of granulocytes and macrophages. These growth factors are also crucial for the survival of myeloid cells [67-69]. Several in vitro studies have shown that bone marrow precursor cells treated with G-CSF or GM-CSF acquire a surface phenotype similar to MDSCs found in blood of cancer patients [70-73].
The administration of G-CSF and GM-CSF is a common therapy for a variety of disorders. In cancer, G-CSF and GM-CSF are often used as an adjuvant to ameliorate neutropenia and to limit the extent of infections due to neutropenia [74, 75]. In patients with colorectal carcinoma, rhGM-CSF was demonstrated to initiate wound healing through stimulation of angiogenesis after surgery [76]. There are, however, inconsistent results of the beneficial role of GM-CSF as adjuvant in cancer vaccines. For instance, one clinical study demonstrated that when GM-CSF was given as a low-dose adjuvant, an increase in peripheral blood MDSCs was observed in patients with stage IV metastatic melanoma [77], whereas no effect on MDSCs was seen in patients with advanced pancreatic cancer [78]. Still, GM-CSF and G-CSF are highly secreted by many cancer cells, and elevated serum levels of these cytokines have also been linked to an increase in circulating MDSCs [49, 53, 79]. In fact, tumor-derived GM-CSF affects both the generation, maintenance, and survival of MDSCs, and its constant production might be important for the large accumulation of MDSCs found in cancer patients. In contrast, the short administration of GM-CSF during clinical treatment is not believed to have the same effects on MDSC generation (Figure 3) [55]. It will be interesting to follow the ongoing clinical studies concerning the use of GM-CSF in combination with novel immune checkpoint inhibitors (e.g., anti-PD-1 [programmed cell death protein 1]), as it has been reported that myeloid cells lacking PD-1 evoke a stronger intra-cellular signal in response to GM-CSF [80]. Whether this response is beneficial for the generation of functionally mature myeloid cells, MDSCs, or both, remains to be seen.
GM-CSF, G-CSF, and M-CSF appear to affect the generation of Mo-MDSCs and G-MDSCs differently. It was shown early on that GM-CSF and M-CSF could generate suppressor cells from the bone marrow with different phenotypes. GM-CSF induced indomethacin-sensitive suppressor cells that release high levels of PGE2, whereas M-CSF gave rise to indomethacin-insensitive suppressor cells with low PGE2 release [81]. It is known that GM-CSF affects Mo-MDSC- and G-MDSC generation and mobilization from the bone marrow [54], whereas G-CSF induces the accumulation and suppressive functions of G-MDSCs [82] and M-CSF is important for the generation and immunosuppressive functions of Mo-MDSCs [83]. It is therefore not surprising that changes in expression levels of these mediators, either via infusion as a treatment option, peripherally in a primary tumor, or in the bone marrow as a result of bone metastases, would affect the generation of Mo-MDSCs and G-MDSCs differently [54].
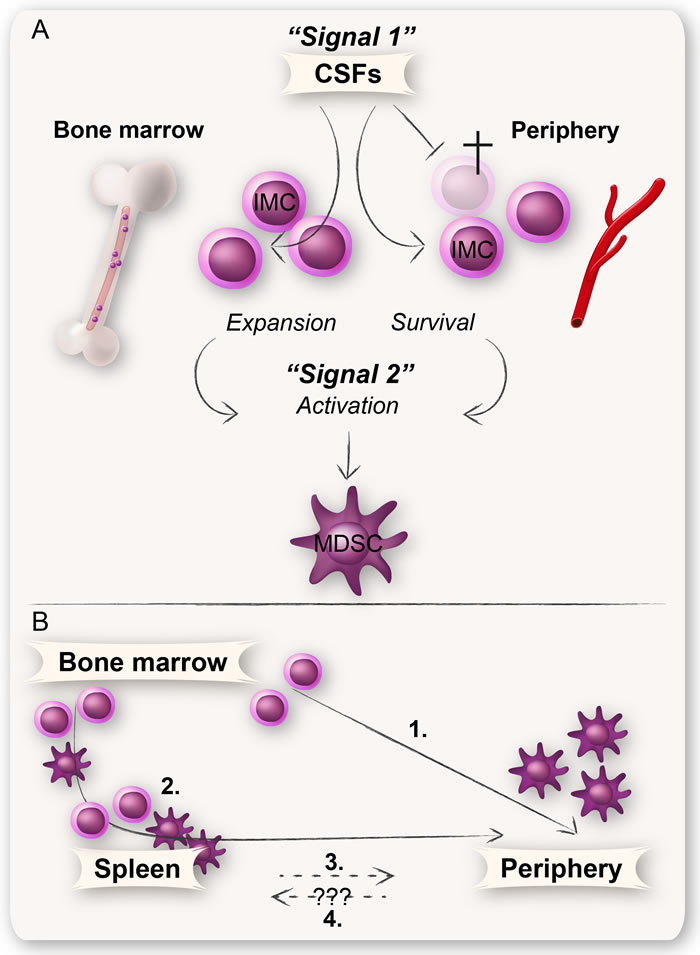
Figure 3: A schematic illustration of the involvement of CSFs in the generation of MDSCs. A: MDSC development is likely governed by the same growth factors that control normal myelopoiesis e.g., CSFs (GM-CSF, G-CSF, and M-CSF). CSFs are essential survival factors for myeloid cells, in the bone marrow as well as in the periphery. They are also involved in many aspects of MDSC generation such as expansion of IMCs in the bone marrow, migration of myeloid cells into the periphery, and in some instances also for activation of MDSCs. B: All MDSCs undoubtedly derive from common myeloid progenitors in the bone marrow. The IMCs either expand in and migrate from the bone marrow into the periphery where they attain their MDSC phenotype (1), or into the spleen where they expand and become activated MDSCs that further migrate into the periphery (2). IMCs or MDSCs derived from hematopoietic progenitor cells found in the spleen may also directly migrate into the periphery (3). Whether peripheral MDSCs also can migrate into the spleen is currently unknown (4).
EMERGENCY MYELOPOIESIS
In cancer and infections, elevated levels of CSFs induce emergency myelopoiesis to meet the increased demand for myeloid cells [55, 84-86]. Emergency myelopoiesis produces myeloid cells that migrate from the bone marrow, before they are fully mature, in response to inflammatory signals in order to renew or restore the peripheral populations that are consumed [67, 87]. Pathological conditions such as cancer and sepsis may invoke a prolonged and marked expansion of immature myeloid cells (IMCs) in the bone marrow, which eventually migrate into the blood stream where they become functionally active MDSCs with suppressive properties (Figure 2) [4, 5, 48, 88-91].
The classical hypothesis governing the molecular mechanism behind MDSC generation today is the “two-signal model” proposed by Gabrilovich et al. (Figure 4) [4]. This model states that an expansion signal 1, mediated mainly by STAT3 (induced by e.g., GM-CSF, G-CSF, and IL-6), mobilizes the IMCs from the bone marrow. This is followed by an activation signal 2, mediated mainly by the transcription factor NFκB (induced by pro-inflammatory stimuli e.g., TLR signaling and cytokines) (Figure 4) [4, 48]. However, Chalmin et al. demonstrated that the expansion of IMCs was induced by tumor-derived GM-CSF, but was not dependent on STAT3 activation. The activation of MDSCs, on the other hand, was induced by heat shock protein 72 on tumor-derived exosomes that triggered TLR2-NFκB signaling, with a subsequent production of IL-6 and activation of STAT3 [92]. This study highlights the fact that STAT3 might be implicated in several stages in the generation of MDSCs, and that many factors are involved and cooperates in the expansion and activation of MDSCs [92].
Recently, another critical transcription factor of downstream of GM-CSF, M-CSF, and G-CSF was shown to drastically affect the generation of both MDSCs and anti-inflammatory macrophages (M2), namely RORC1 (retinoic-acid-related orphan receptor C1). RORC1 was shown to drive cancer-induced emergency myelopoiesis by affecting other crucial transcription factors regulating myelopoiesis, e.g., C/EBPβ (CCAAT-enhancer-binding protein-β) and SOCS3 (suppressor of cytokine signaling 3), acting downstream of the CSFs. It is likely that both RORC1 and STAT3, together with NFκB, are important inducers of MDSC generation and expansion [48, 93].
Another study demontrated increased hematopoiesis in mice with IL-1β-secreting tumors, shown by splenomegaly, leukocytosis, and anemia [64]. The increased hematopoiesis was found in the bone marrow, where IL-1β stimulated the release of IMCs into the blood. The IMCs then migrated into the spleen where they proliferated and attained their suppressive phenotype [64]. This study indicated an increased emergency myelopoiesis in the bone marrow of mice, giving rise to increased IMCs in the blood. The IMCs then migrated into the spleen, where extramedullary myelopoiesis occurred, and received a second signal for activation [64]. Long before this article was published, Young et al. demonstrated a similar phenomenon with an increased amount of IMCs in blood, and an elevated hematopoiesis in the bone marrow and spleen in mice with metastatic Lewis lung carcinoma tumors [94]. These two studies demonstrate that the expansion of MDSCs may occur not only in the bone marrow, but also in peripheral organs or tumors.
EXTRAMEDULLARY MYELOPOIESIS
Extramedullary myelopoiesis is defined as myelopoiesis occurring in organs outside of the bone marrow and is frequently seen in chronic inflammatory diseases, cancer, trauma, and sepsis [95]. In mice, a fraction of hematopoietic stem and progenitor cells has been shown to migrate out of the bone marrow into the blood and further out into peripheral tissue and lymph vessels [96]. Tumor-derived factors might be responsible for the migration of precursor cells into the peripheral tissue in an emergency myelopoiesis manner [64]. The progenitor cells would then proliferate and become MDSCs through activation at extramedullary sites (Figure 2). In this model, the activation of TLRs appears to be central for the generation of MDSCs. For instance, LPS has been demonstrated to induce the accumulation of MDSCs at extramedullary sites, especially the spleen [96, 97]. Although LPS alone appears to have the ability to generate MDSCs, it seems there is an even more powerful generation of MDSCs with the right cytokine combination (e.g., IFNɣ) [60, 98].
It has been shown that c-kit+ hematopoietic precursors are increased in the spleen of tumor-bearing mice, a strong indication that increased extramedullary myelopoiesis does occur [52]. The generation of MDSCs from c-kit+ hematopoietic precursors in that study was dependent on GM-CSF [52]. Whether the increase of these hematopoietic precursors is directly connected to the simultaneous increase of MDSCs at the extramedullary sites in vivo is not fully proven. There are many indications that extramedullary myelopoiesis might be a consequence of an emergency myelopoiesis that induces the migration of IMCs out of the bone marrow into the periphery, where they then accumulate in the spleen and get their second signal to become MDSCs [64, 94]. However, during embryogenesis, before the hematopoiesis is established in the bone marrow, hematopoietic elements from the yolk sac are circulating in the embryo. These hematopoietic progenitor cells accumulate in the liver, but also in the spleen. Hematopoiesis can thus take place in these organs until birth [95, 99]. The persistence of progenitor cells in the spleen after birth might also be a source for MDSCs. Suppressive Gr1+CD11b+ cells in the spleen of healthy mice have been described, suggesting that MDSCs are not only induced upon infection or inflammation but also exist in steady state [98].
The finding of extramedullary hematopoiesis comes from experiments in mice; the equivalent in humans has not been fully explored. Nonetheless, it is well documented that treatment with G-CSF and GM-CSF causes an increase in spleen size in humans [100-102] that appears to be the result of extramedullary myelopoiesis [103]. This was confirmed with histology of patients receiving G-CSF, and indicates that G-CSF and GM-CSF not only induce the expansion of MDSCs in the bone marrow of humans, but also in the spleen [104].
BLOCK IN DIFFERENTIATION
In pathological conditions such as cancer, emergency myelopoiesis creates a prolonged and marked expansion of bone marrow-derived IMCs that migrate out into the periphery [87]. The IMCs are believed to be arrested in their immature phase by inflammatory mediators such as S100A8, S100A9, VEGF, IL-10, and COX-2/PGE2 [89-91, 105-107]. It has also been suggested that systemically released inflammatory mediators are unable to induce MDSCs alone, but that a direct tumor cell contact or a close proximity to tumor cells would be required for MDSCs to be generated [107]. Either way, when MDSCs are taken from the tumor environment, the block is reversed and MDSCs can differentiate into mature myeloid cells, preferably monocytes/macrophages or dendritic cell [108, 109].
Both emergency myelopoiesis and block in differentiation are linked to an abnormal and persistent activation of STAT3, and many of the mediators involved in emergency myelopoiesis are also responsible for arresting IMCs in their immature phase [4]. The block in differentiation of IMCs might be an indirect effect of the tumor-derived mediators initially responsible for emergency myelopoiesis. For example, activating STAT3 in myeloid progenitor cells leads to the induction of S100A8 and S100A9 expression, which subsequently acts in an autocrine manner to arrest the cells in their immature phase (Figure 2) [4, 90].
As a treatment strategy, many studies have attempted to force the differentiation block of MDSCs to minimize the accumulation and immunosuppressive effects of these cells, in different diseases [41, 109-113]. One example is ATRA (all-trans-retinoic acid), a compound that is structurally similar to vitamin A and used to treat various malignancies. ATRA has been demonstrated to reverse the differentiation block of MDSCs and to enhance the maturation of these cells in humans [41, 109, 110]. Further studies have led to the identification of another vitamin, vitamin D3, with similar effects. In the presence of 1α,25-dihydroxyvitamin D3 the differentiation of CD34+ MDSCs into phenotypically and functionally DC-like cells in vitro was accelerated [111]. In addition, the number of circulating CD34+ MDSCs was reduced in patients with head and neck squamous cell carcinomas receiving 25 hydroxyvitamin D3 [112]. In a chronic inflammation model the use of a TNFα antagonist reversed the block in differentiation and augmented the maturation of dendritic cells and macrophages [113]. In addition, the blockage of the S100A8/S100A9 receptor on MDSCs with a carboxylated-N-glycan-specific antibody reduced the number of circulating MDSCs in tumor-bearing mice [65]. These studies demonstrate that the accumulation of MDSCs can be targeted by inducing differentiation.
ORIGIN OF MO-MDSCs
Although MDSCs have traditionally been viewed as immature cells, emerging evidence suggest that they are rather an intermediate or even alternative state of myeloid cell differentiation. Mo-MDSCs do not have an immature morphology, only a surface marker phenotype similar to myeloid cells of the monocytic lineage, lacking activation markers [4, 23, 114, 115]. However, Mo-MDSCs have been demonstrated to overexpress the co-receptors/activation markers CD80 and CD83 [13, 116] indicating that Mo-MDSCs are indeed not as immature as previously thought. Also, Mo-MDSCs are characterized, among other things, by their expression of CD14 [23, 37], which signifies lineage commitment. This contradicts the theory that Mo-MDSCs have their origin in emergency myelopoiesis, and there is no firm proof that Mo-MDSCs are generated in this way. Most of the studies confirming the existence of emergency myelopoiesis demonstrated results for MDSCs in general [64, 92] and the dominance in number of G-MDSCs relative to Mo-MDSCs may conceal the true nature of Mo-MDSC generation. Instead, Mo-MDSCs might originate from monocytes. This hypothesis is further strengthened by the fact that Mo-MDSCs have been shown to differentiate into tumor-associated macrophages in tumors [117], a finding that could simply represent the migration of the Mo-MDSCs into the tumor.
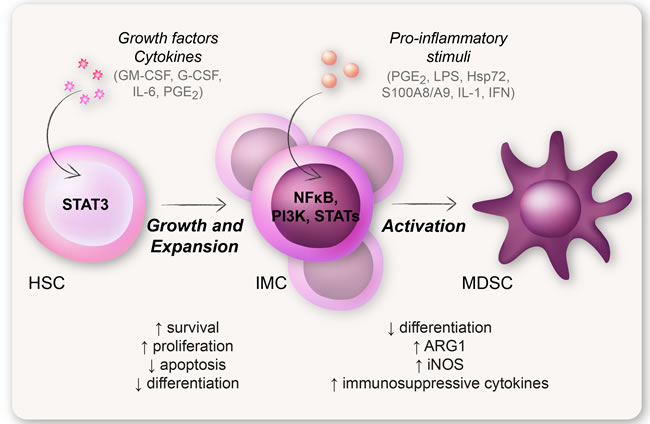
Figure 4: The two-signal model of MDSC generation. The classical hypothesis regarding the generation of MDSCs is the “two-signal model”. This model states that an expansion signal induced by e.g., CSFs (such as GM-CSF and G-CSF), IL-6, and PGE2, and mediated by STAT3, expands and mobilizes immature myeloid cells from the bone marrow. This is followed by an activation signal induced by pro-inflammatory stimuli e.g., LPS, PGE2 and S100A8/A9, and mediated by NFκB. According to this hypothesis it is not until the IMCs acquire an activation signal that they obtain a suppressive MDSC-phenotype.
REPROGRAMMING OF MONOCYTES
Myeloid cells with a Mo-MDSC phenotype have been demonstrated to originate from monocytes that acquire a suppressive phenotype under certain inflammatory conditions. One example of this is the endotoxin tolerance in sepsis patients where a subsequent dose of endotoxin, together with the right cytokines, results in a reprogramming of monocytes from a pro-inflammatory state to an anti-inflammatory state (Figure 5) [26, 118, 119]. These reprogrammed, anti-inflammatory monocytes (CARS-monocytes; compensatory anti-inflammatory response-monocytes) have the same surface phenotype (CD14+HLA-DR-/lowCo-receptor-/low) and function as Mo-MDSCs [26, 118, 119]. Such reprogramming can be regarded as an alternative differentiation, when the normal differentiation pathway is circumvented and the cell achieves another function. Under normal circumstances, monocytes differentiate into macrophages or dendritic cells. However, in conditions of excessive inflammation such as trauma and sepsis, where the proper cytokine milieu is created, monocytes can be reprogrammed and become Mo-MDSCs to limit the devastating effect of an inflammatory response [26, 118, 119]. This phenomenon was recently proposed to apply in cancer patients as well [13].
In a recent study, we showed that the gene expression profile of Mo-MDSCs from breast cancer patients was significantly more similar to that of reprogrammed anti-inflammatory monocytes from sepsis patients than to monocytes isolated from either healthy donors or tuberculosis patients [13]. The molecular mechanism behind monocyte reprogramming, in sepsis as well as in cancer, is suggested to rely on a repeated TLR-signal in combination with certain cytokines or mediators (e.g., IL-10, Wnt5a, and PGE2) thus leading to the formation of immunosuppressive NFκB p50:p50 homodimers instead of pro-inflammatory NFκB p65:p50 heterodimers (Figure 5) [13, 26, 120-122]. TLR-ligands (foremost LPS) are known to induce the expansion and activation of MDSCs [123]. In cancer, the TLR-ligands responsible for this are damage-associated molecular patterns (DAMPs; endogenous alarmin molecules), whereas in sepsis the TLR-ligands are mainly composed of the exogenous molecules pathogen-associated molecular patterns (PAMPs, such as LPS) [124]. In a way, monocyte reprogramming could be viewed as a “two-signal model,” again involving both STAT3 and NFκB, but with the difference that monocytes are affected.
Other reports also confirm that monocytes can be reprogrammed into Mo-MDSCs by demonstrating that peripheral blood monocytes can serve as precursors to Mo-MDSCs under specific conditions in vitro [106, 107, 120, 125, 126]. This further indicates that monocytes can indeed be the source of Mo-MDSCs. For instance, tumor cell lines have been shown to convert peripheral blood monocytes into Mo-MDSC-like cells with T-cell suppressive functions [107, 125]. Furthermore, tumor-derived PGE2 was demonstrated to drive the differentiation of monocytes towards Mo-MDSCs instead of dendritic cells in the presence of GM-CSF and IL-4 in vitro [106]. These PGE2-induced Mo-MDSCs resembled patient-derived Mo-MDSCs in phenotype and suppressive functions [127].
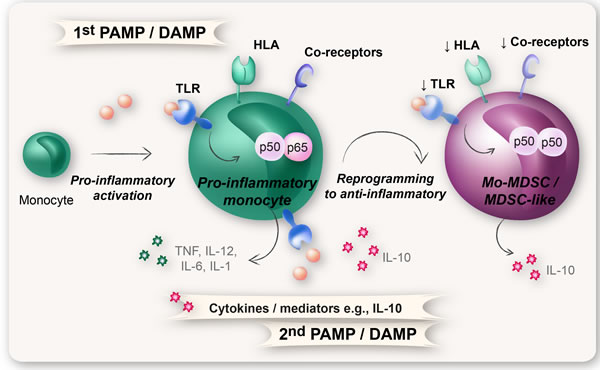
Figure 5: Reprogramming of monocytes into Mo-MDSCs. The molecular mechanism behind reprogramming of monocytes into Mo-MDSCs is suggested to rely on a repeated TLR-signaling in combination with the right cytokine milieu (e.g., IL-10). Upon a first encounter with PAMPs or DAMPs, the monocytes will attain an activated pro-inflammatory phenotype with formation of pro-inflammatory NFκB p65:p50 heterodimers. A subsequent exposure to PAMPs/DAMPs, in combination with certain cytokines (e.g., IL-10), results in the formation of immunosuppressive NFκB p50:p50 homodimers and a reprogramming of monocytes from a pro-inflammatory state to an anti-inflammatory state with down-regulation of TLR, HLA-DR, and co-receptors. These anti-inflammatory monocytes have the same surface phenotype and function as Mo-MDSCs.
ORIGIN OF G-MDSCs
G-MDSCs represent a heterogeneous population of cells with regards to nuclear morphology, ranging from blasts to PMNs [29, 128]. Whether the different cells within the G-MDSC-population all have a bona fide MDSCs function is unknown. Hence, it is not known whether the MDSCs with a PMN-morphology resemble neutrophils, or whether the blasts are just immature cells with no distinct function, or indeed functional MDSCs [29, 128-130]. The blast population might actually, functionally, represent the true G-MDSCs.
G-MDSCs are isolated from the mononuclear cell fraction of Ficoll density gradients and defined based on their granulocytic scatter profile (FSC/SSC) and surface phenotype using flow cytometry [29]. However, degranulated neutrophils, after activation, have also been seen to co-purify with mononuclear cells in density gradients [131]. Such degranulated neutrophils correlated with an increased level of serum ARG1 in patients with glioblastoma multiforme [131]. This implies that G-MDSC function, correlating with ARG1-mediated T-cell suppression in humans, might simply be mediated by activated neutrophils.
ACTIVATION OF NEUTROPHILS
The theory regarding whether G-MDSCs are activated granulocytes is controversial; as is the question of whether these cells arise in the periphery or in the bone marrow. The hypothesis that either immature or mature cells can attain a suppressive phenotype in the bone marrow was termed “suppressive granulopoiesis”, and this process was shown to be driven primarily by G-CSF [29]. Indeed, given the plasticity of neutrophils, G-MDSCs could be a functionally heterogeneous subsets of neutrophils [29].
Neutrophils were until recently thought to consist of one population, but accumulating evidence suggests the existence of distinct neutrophil subsets with diverse roles in infection, inflammation, and cancer [132-136]. Pillay et al. recently identified three distinct neutrophil subsets during acute systemic inflammation in humans based on the expression of CD16 and CD62L: CD16dim CD62Lhigh, CD16high CD62Lhigh, and CD16high CD62Ldim [134]. These subsets are thought to represent different stages of neutrophil activation. The same work demonstrated that the activated neutrophils (CD16high CD62Ldim) inhibited T-cell responses through cell-to-cell interactions via macrophage-1 antigen (Mac-1; consisting of CD11b and CD18) [134]. Neutrophils are known to interact with, and modulate, T-cell responses. Many of the molecules that inhibit T-cell responses such as ARG1 and ROS are present in both activated neutrophils and G-MDSCs [137], and at least ROS has also been shown to have a direct anti-tumor function by inducing tumor cell lysis [138, 139]. The activation of neutrophils in vitro by inflammatory cytokines such as GM-CSF, G-CSF, TNF, IL-1β, and IFN-ɣ has been shown to prolong the survival of neutrophils [140]. A longer lifespan could allow neutrophils to carry out more complex activities with regulatory functions.
Neutrophils have been ascribed both anti- and pro-tumorigenic functions (N1 and N2 neutrophils, respectively). Their different functions might depend on the microenvironment, whether it is an acute or chronic inflammation, the cells with which they co-operate, and their activation status per se [137, 141]. This illustrates the plasticity and heterogeneity of neutrophils, which also might explain the neutrophil “immunogenic switch” theory, where a switch from an anti-tumorigenic to a pro-tumorigenic immune phenotype is proposed to occur during tumor progression [135, 136, 142].
Neutrophils are very reactive cells that are easily activated in vitro. This, together with their relatively short half-life, makes them difficult to study in their native state and the findings may therefore be biased [29, 137]. Many of the studies of neutrophils have been performed in mice in vivo. The role and function of tumor-infiltrating myeloid cells, especially TANs, in humans may be different compared to mice [130, 136]. Indeed, mice have shorter life-spans and mouse tumor models are characterized by high tumor burden and rapid tumor growth. In contrast, human tumors evolve over years to decades indicating obvious differences in tumor characteristics. In addition, neutrophils are more abundant in humans compared to mice [130, 143]. Caution is therefore called for in the process of defining this heterogeneous cell population. Because neutrophils and G-MDSCs share similar granulocytic morphology and surface phenotype, and that there is no clear consensus on how to distinguish between them, it is difficult to make definite conclusions regarding the relationship between neutrophils and G-MDSCs. This issue thus merits further investigation.
CONCLUSIONS
MDSCs are regulatory cells with the ability to limit the extent of inflammation and to initiate wound healing and recovery. An excessive inflammatory response, such as in sepsis, will induce a signal to initiate an anti-inflammatory response and generate MDSCs. Similarly, cancer can be regarded as a site of chronic inflammation where the induction of anti-inflammatory MDSCs can be seen as a regulatory mechanism to dampen inflammation and to induce wound healing mechanisms. Indeed, cancer is not an inventor: it is an opportunist. As such, cancer exploits the simple homeostatic mechanism of MDSC generation to avoid immune surveillance and to promote tumor growth and metastasis.
A great number of MDSC-phenotypes have been described, but it is not only the surface phenotype that differs between different cancers. There are also variations in mechanisms of suppressions and in nuclear morphology. These dissimilarities in phenotype, morphology, suppressive capacity, and mechanisms might emerge from differences in origin or from different activations. Hence, Mo-MDSCs and G-MDSCs might be generated in distinct ways. Emerging evidence suggest that Mo-MDSCs are generated by a reprogramming of monocytes into Mo-MDSCs, whereas G-MDSCs might be a phenotype of neutrophils generated through the activation of immature or mature granulocytes and thereby merely represent different stages of activation. The ways in which Mo-MDSCs and G-MDSCs are generated may be distinct, but CSFs, STAT3, and NFκB seem to be central molecular players and as such may lead to new discoveries on how to target both MDSC-subtypes simultaneously. New factors that are important for the establishment of MDSCs are being unraveled, but there are still more to discover. It is very important to understand how MDSCs are generated and which factors are involved in the process to know how to design future MDSC-targeting therapy. To target MDSCs and the anti-inflammatory response might increase immunosurveillance and help improve overall survival in cancer patients.
ACKNOWLEDGMENTs
We would like to thank Daphne Sams for linguistic and editorial support.
CONFLICTs OF INTEREST
The authors declare that there are no conflicts of interest.
GRANT SUPPORT
This work was generously supported by grants from the Vetenskapsrådet, Cancerfonden, Österlunds Foundation, Gunnar Nilsson Cancer Foundation, MAS Cancer Foundation, and Åke Wibergs Foundation.
REFERENCES
1. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007; 67: 425; author reply 6. doi: 10.1158/0008-5472.CAN-06-3037.
2. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009; 182: 4499-506. doi: 10.4049/jimmunol.0802740.
3. Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011; 17: 281-92. doi: 10.2119/molmed.2010.00178.
4. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9: 162-74. doi: 10.1038/nri2506.
5. Poschke I, Kiessling R. On the armament and appearances of human myeloid-derived suppressor cells. Clin Immunol. 2012; 144: 250-68. doi: 10.1016/j.clim.2012.06.003.
6. Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007; 13: 5243-8. doi: 10.1158/1078-0432.CCR-07-0182.
7. Ko JS, Bukowski RM, Fincke JH. Myeloid-derived suppressor cells: a novel therapeutic target. Curr Oncol Rep. 2009; 11: 87-93.
8. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007; 179: 977-83.
9. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008; 13: 23-35. doi: 10.1016/j.ccr.2007.12.004.
10. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008; 8: 618-31. doi: 10.1038/nrc2444.
11. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, Zanon A, Rossi CR, Nitti D, Bronte V, Zanovello P. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009; 182: 6562-8. doi: 10.4049/jimmunol.0803831.
12. Khaled YS, Ammori BJ, Elkord E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J Immunol Res. 2014; 2014: 879897. doi: 10.1155/2014/879897.
13. Bergenfelz C, Larsson AM, von Stedingk K, Gruvberger-Saal S, Aaltonen K, Jansson S, Jernstrom H, Janols H, Wullt M, Bredberg A, Ryden L, Leandersson K. Systemic Monocytic-MDSCs Are Generated from Monocytes and Correlate with Disease Progression in Breast Cancer Patients. PLoS One. 2015; 10: e0127028. doi: 10.1371/journal.pone.0127028.
14. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009; 58: 49-59. doi: 10.1007/s00262-008-0523-4.
15. Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol. 2013; 190: 794-804. doi: 10.4049/jimmunol.1202088.
16. Sun HL, Zhou X, Xue YF, Wang K, Shen YF, Mao JJ, Guo HF, Miao ZN. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012; 18: 3303-9. doi: 10.3748/wjg.v18.i25.3303.
17. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, Zhu J, Wei H, Zhao K. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One. 2013; 8: e57114. doi: 10.1371/journal.pone.0057114.
18. Huang A, Zhang B, Wang B, Zhang F, Fan KX, Guo YJ. Increased CD14(+)HLA-DR (-/low) myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother. 2013; 62: 1439-51. doi: 10.1007/s00262-013-1450-6.
19. Shen P, Wang A, He M, Wang Q, Zheng S. Increased circulating Lin(-/low) CD33(+) HLA-DR(-) myeloid-derived suppressor cells in hepatocellular carcinoma patients. Hepatol Res. 2014; 44: 639-50. doi: 10.1111/hepr.12167.
20. Antonia SJ, Mirza N, Fricke I, Chiappori A, Thompson P, Williams N, Bepler G, Simon G, Janssen W, Lee JH, Menander K, Chada S, Gabrilovich DI. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res. 2006; 12: 878-87. doi: 10.1158/1078-0432.CCR-05-2013.
21. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, Hilf N, Schoor O, Fritsche J, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012; 18: 1254-61. doi: 10.1038/nm.2883.
22. Kusmartsev S, Gabrilovich DI. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol Immunother. 2002; 51: 293-8. doi: 10.1007/s00262-002-0280-8.
23. Meirow Y, Kanterman J, Baniyash M. Paving the Road to Tumor Development and Spreading: Myeloid-Derived Suppressor Cells are Ruling the Fate. Front Immunol. 2015; 6: 523. doi: 10.3389/fimmu.2015.00523.
24. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M, Blander JM, Tacke F, Trautwein C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med. 2010; 207: 1453-64. doi: 10.1084/jem.20091474.
25. Derive M, Bouazza Y, Alauzet C, Gibot S. Myeloid-derived suppressor cells control microbial sepsis. Intensive Care Med. 2012; 38: 1040-9. doi: 10.1007/s00134-012-2574-4.
26. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009; 30: 475-87. doi: 10.1016/j.it.2009.07.009.
27. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010; 22: 238-44. doi: 10.1016/j.coi.2010.01.021.
28. Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010; 40: 2969-75. doi: 10.1002/eji.201040895.
29. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci. 2013; 70: 3813-27. doi: 10.1007/s00018-013-1286-4.
30. Fan HK, Cook JA. Molecular mechanisms of endotoxin tolerance. Journal of Endotoxin Research. 2004; 10: 71-84. doi: 10.1179/096805104225003997.
31. Ruppert J, Schutt C, Ostermeier D, Peters JH. Down-Regulation and Release of Cd14 on Human Monocytes by Il-4 Depends on the Presence of Serum or Gm-Csf. Dendritic Cells in Fundamental and Clinical Immunology. 1993; 329: 281-6.
32. Sinistro A, Ciaprini C, Natoli S, Sussarello E, Carducci FC, Almerighi C, Capozzi M, Bolacchi F, Rocchi G, Bergamini A. Lipopolysaccharide desensitizes monocytes-macrophages to CD40 ligand stimulation. Immunology. 2007; 122: 362-70. doi: 10.1111/j.1365-2567.2007.02648.x.
33. Xiu B, Lin Y, Grote DM, Ziesmer SC, Gustafson MP, Maas ML, Zhang Z, Dietz AB, Porrata LF, Novak AJ, Liang AB, Yang ZZ, Ansell SM. IL-10 induces the development of immunosuppressive CD14(+) HLA-DRlow/- monocytes in B-cell non-Hodgkin lymphoma. Blood Cancer Journal. 2015; 5. doi: ARTN e328 10.1038/bcj.2015.56.
34. Landmann R, Ludwig C, Obrist R, Obrecht JP. Effect of cytokines and lipopolysaccharide on CD14 antigen expression in human monocytes and macrophages. J Cell Biochem. 1991; 47: 317-29. doi: 10.1002/jcb.240470406.
35. Lahat N, Rahat MA, Ballan M, Weiss-Cerem L, Engelmayer M, Bitterman H. Hypoxia reduces CD80 expression on monocytes but enhances their LPS-stimulated TNF-alpha secretion. Journal of Leukocyte Biology. 2003; 74: 197-205. doi: 10.1189/jlb.0303105.
36. Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, Sozzani S, Austyn JM, Mantovani A, Sica A. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008; 112: 3723-34. doi: 10.1182/blood-2008-02-142091.
37. Filipazzi P, Huber V, Rivoltini L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother. 2012; 61: 255-63. doi: 10.1007/s00262-011-1161-9.
38. Feng PH, Lee KY, Chang YL, Chan YF, Kuo LW, Lin TY, Chung FT, Kuo CS, Yu CT, Lin SM, Wang CH, Chou CL, Huang CD, et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med. 2012; 186: 1025-36. doi: 10.1164/rccm.201204-0636OC.
39. Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, Greten TF, Korangy F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology. 2012; 136: 176-83. doi: 10.1111/j.1365-2567.2012.03566.x.
40. Mundy-Bosse BL, Young GS, Bauer T, Binkley E, Bloomston M, Bill MA, Bekaii-Saab T, Carson WE, 3rd, Lesinski GB. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4(+) T cells from patients with GI malignancy. Cancer Immunol Immunother. 2011; 60: 1269-79. doi: 10.1007/s00262-011-1029-z.
41. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006; 66: 9299-307. doi: 10.1158/0008-5472.CAN-06-1690.
42. Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011; 11: 856-61. doi: 10.1016/j.intimp.2011.01.030.
43. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011; 60: 1419-30. doi: 10.1007/s00262-011-1028-0.
44. van Cruijsen H, Hoekman K, Stam AG, van den Eertwegh AJ, Kuenen BC, Scheper RJ, Giaccone G, de Gruijl TD. Defective differentiation of myeloid and plasmacytoid dendritic cells in advanced cancer patients is not normalized by tyrosine kinase inhibition of the vascular endothelial growth factor receptor. Clin Dev Immunol. 2007; 2007: 17315. doi: 10.1155/2007/17315.
45. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005; 65: 3044-8. doi: 10.1158/0008-5472.CAN-04-4505.
46. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007; 13: 828-35. doi: 10.1038/nm1609.
47. Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, van der Veen RC. Mycobacteria-induced Gr-1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. J Leukoc Biol. 2007; 81: 1205-12. doi: 10.1189/jlb.1006640.
48. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 2015; 98: 913-22. doi: 10.1189/jlb.4RI0515-204R.
49. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013; 13: 739-52. doi: 10.1038/nrc3581.
50. Rashid OM, Nagahashi M, Ramachandran S, Graham L, Yamada A, Spiegel S, Bear HD, Takabe K. Resection of the primary tumor improves survival in metastatic breast cancer by reducing overall tumor burden. Surgery. 2013; 153: 771-8. doi: 10.1016/j.surg.2013.02.002.
51. Salvadori S, Martinelli G, Zier K. Resection of solid tumors reverses T cell defects and restores protective immunity. J Immunol. 2000; 164: 2214-20.
52. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell. 2012; 21: 822-35. doi: 10.1016/j.ccr.2012.04.025.
53. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999; 162: 5728-37.
54. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, Grassi F, Bronte V. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010; 40: 22-35. doi: 10.1002/eji.200939903.
55. Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010; 123: 39-49. doi: 10.1007/s10549-009-0622-8.
56. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, Yu L, Ross J, Korsisaari N, Cao T, Bou-Reslan H, Kallop D, Weimer R, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A. 2010; 107: 21248-55. doi: 10.1073/pnas.1015855107.
57. Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One. 2011; 6: e27690. doi: 10.1371/journal.pone.0027690.
58. Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, Blay JY. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood. 1998; 92: 4778-91.
59. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016; 37: 208-20. doi: 10.1016/j.it.2016.01.004.
60. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007; 204: 1463-74. doi: 10.1084/jem.20062602.
61. Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006; 176: 2085-94.
62. Kusmartsev SA, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000; 165: 779-85.
63. Schmidt-Wolf IG, Dejbakhsh-Jones S, Ginzton N, Greenberg P, Strober S. T-cell subsets and suppressor cells in human bone marrow. Blood. 1992; 80: 3242-50.
64. Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol. 2005; 175: 8200-8.
65. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008; 181: 4666-75.
66. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012; 12: 253-68. doi: 10.1038/nri3175.
67. Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ’emergency’ hematopoiesis. Cytokine. 2008; 42: 277-88. doi: 10.1016/j.cyto.2008.03.002.
68. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. 2016; 12: 37-48. doi: 10.1038/nrrheum.2015.161.
69. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008; 8: 533-44. doi: 10.1038/nri2356.
70. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010; 32: 790-802. doi: 10.1016/j.immuni.2010.05.010.
71. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, Francescato S, Basso G, Zanovello P, Onicescu G, Garrett-Mayer E, Montero AJ, Bronte V, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011; 118: 2254-65. doi: 10.1182/blood-2010-12-325753.
72. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010; 116: 5738-47. doi: 10.1182/blood-2010-06-287839.
73. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007; 67: 4507-13. doi: 10.1158/0008-5472.CAN-06-4174.
74. American Society of Clinical Oncology. Recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. J Clin Oncol. 1994; 12: 2471-508.
75. Ozer H, Armitage JO, Bennett CL, Crawford J, Demetri GD, Pizzo PA, Schiffer CA, Smith TJ, Somlo G, Wade JC, Wade JL, 3rd, Winn RJ, Wozniak AJ, et al. 2000 update of recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. American Society of Clinical Oncology Growth Factors Expert Panel. J Clin Oncol. 2000; 18: 3558-85. doi:
76. Wu FP, Westphal JR, Hoekman K, Mels AK, Statius Muller MG, de Waal RW, Beelen RH, van Leeuwen PA, Meijer S, Cuesta MA. The effects of surgery, with or without rhGM-CSF, on the angiogenic profile of patients treated for colorectal carcinoma. Cytokine. 2004; 25: 68-72.
77. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007; 25: 2546-53. doi: 10.1200/JCO.2006.08.5829.
78. Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, Valle J, Smith D, Wadsley J, Cunningham D, Pandha H, Neoptolemos JP, Middleton G. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother. 2014; 63: 175-83. doi: 10.1007/s00262-013-1502-y.
79. Tsuchiya Y, Igarashi M, Suzuki R, Kumagai K. Production of colony-stimulating factor by tumor cells and the factor-mediated induction of suppressor cells. J Immunol. 1988; 141: 699-708.
80. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999; 11: 141-51.
81. Oghiso Y, Yamada Y, Ando K, Ishihara H, Shibata Y. Differential induction of prostaglandin E2-dependent and -independent immune suppressor cells by tumor-derived GM-CSF and M-CSF. J Leukoc Biol. 1993; 53: 86-92.
82. Abrams SI, Waight JD. Identification of a G-CSF-Granulocytic MDSC axis that promotes tumor progression. Oncoimmunology. 2012; 1: 550-1.
83. Stromnes IM, Greenberg PD, Hingorani SR. Molecular pathways: myeloid complicity in cancer. Clin Cancer Res. 2014; 20: 5157-70. doi: 10.1158/1078-0432.CCR-13-0866.
84. Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young MR. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res. 1995; 1: 95-103.
85. Cheers C, Haigh AM, Kelso A, Metcalf D, Stanley ER, Young AM. Production of Colony-Stimulating Factors (Csfs) during Infection - Separate Determinations of Macrophage-Csf, Granulocyte-Csf, Granulocyte-Macrophage-Csf, and Multi-Csf. Infection and Immunity. 1988; 56: 247-51.
86. Kawakami M, Tsutsumi H, Kumakawa T, Abe H, Hirai M, Kurosawa S, Mori M, Fukushima M. Levels of Serum Granulocyte Colony-Stimulating Factor in Patients with Infections. Blood. 1990; 76: 1962-4.
87. Ueha S, Shand FH, Matsushima K. Myeloid cell population dynamics in healthy and tumor-bearing mice. Int Immunopharmacol. 2011; 11: 783-8. doi: 10.1016/j.intimp.2011.03.003.
88. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001; 166: 678-89.
89. Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, Mantovani A. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur J Immunol. 1998; 28: 359-69.
90. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008; 205: 2235-49. doi: 10.1084/jem.20080132.
91. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998; 92: 4150-66.
92. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010; 120: 457-71. doi: 10.1172/JCI40483.
93. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, Porta C, Anselmo A, Tartari S, Doni A, Zitelli F, Tripodo C, Colombo MP, et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell. 2015; 28: 253-69. doi: 10.1016/j.ccell.2015.07.006.
94. Young MR, Newby M, Wepsic HT. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res. 1987; 47: 100-5.
95. Kim CH. Homeostatic and pathogenic extramedullary hematopoiesis. J Blood Med. 2010; 1: 13-9. doi: 10.2147/JBM.S7224.
96. Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007; 131: 994-1008. doi: 10.1016/j.cell.2007.09.047.
97. Skabytska Y, Wolbing F, Gunther C, Koberle M, Kaesler S, Chen KM, Guenova E, Demircioglu D, Kempf WE, Volz T, Rammensee HG, Schaller M, Rocken M, et al. Cutaneous innate immune sensing of Toll-like receptor 2-6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity. 2014; 41: 762-75. doi: 10.1016/j.immuni.2014.10.009.
98. Greifenberg V, Ribechini E, Rossner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. 2009; 39: 2865-76. doi: 10.1002/eji.200939486.
99. O’Malley DP. Benign extramedullary myeloid proliferations. Mod Pathol. 2007; 20: 405-15. doi: 10.1038/modpathol.3800768.
100. Platzbecker U, Prange-Krex G, Bornhauser M, Koch R, Soucek S, Aikele P, Haack A, Haag C, Schuler U, Berndt A, Rutt C, Ehninger G, Holig K. Spleen enlargement in healthy donors during G-CSF mobilization of PBPCs. Transfusion. 2001; 41: 184-9.
101. Stroncek D, Shawker T, Follmann D, Leitman SF. G-CSF-induced spleen size changes in peripheral blood progenitor cell donors. Transfusion. 2003; 43: 609-13.
102. Picardi M, De Rosa G, Selleri C, Scarpato N, Soscia E, Martinelli V, Ciancia R, Rotoli B. Spleen enlargement following recombinant human granulocyte colony-stimulating factor administration for peripheral blood stem cell mobilization. Haematologica. 2003; 88: 794-800. doi:
103. Molineux G, Pojda Z, Dexter TM. A comparison of hematopoiesis in normal and splenectomized mice treated with granulocyte colony-stimulating factor. Blood. 1990; 75: 563-9.
104. Falzetti F, Aversa F, Minelli O, Tabilio A. Spontaneous rupture of spleen during peripheral blood stem-cell mobilisation in a healthy donor. Lancet. 1999; 353: 555. doi: 10.1016/S0140-6736(99)00268-8.
105. Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997; 159: 28-35.
106. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011; 118: 5498-505. doi: 10.1182/blood-2011-07-365825.
107. Mao Y, Poschke I, Wennerberg E, Pico de Coana Y, Egyhazi Brage S, Schultz I, Hansson J, Masucci G, Lundqvist A, Kiessling R. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013; 73: 3877-87. doi: 10.1158/0008-5472.CAN-12-4115.
108. Gabrilovich DI, Corak J, Ciernik IF, Kavanaugh D, Carbone DP. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin Cancer Res. 1997; 3: 483-90.
109. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003; 63: 4441-9.
110. Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kubler H, Yancey D, Dahm P, Vieweg J. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008; 14: 8270-8. doi: 10.1158/1078-0432.CCR-08-0165.
111. Garrity T, Pandit R, Wright MA, Benefield J, Keni S, Young MRI. Increased presence of CD34(+) cells in the peripheral blood of head and neck cancer patients and their differentiation into dendritic cells. International Journal of Cancer. 1997; 73: 663-9. doi: Doi 10.1002/(Sici)1097-0215(19971127)73:5<663::Aid-Ijc9>3.0.Co;2-V.
112. Lathers DMR, Clark JI, Achille NJ, Young MRI. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D-3. Cancer Immunology Immunotherapy. 2004; 53: 422-30. doi: 10.1007/s00262-003-0459-7.
113. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor Necrosis Factor-alpha Blocks Differentiation and Enhances Suppressive Activity of Immature Myeloid Cells during Chronic Inflammation. Immunity. 2013; 38: 541-54. doi: 10.1016/j.immuni.2013.02.007.
114. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008; 111: 4233-44. doi: 10.1182/blood-2007-07-099226.
115. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T, Takami T. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008; 83: 1136-44. doi: 10.1189/jlb.0907611.
116. Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010; 70: 4335-45. doi: 10.1158/0008-5472.CAN-09-3767.
117. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010; 207: 2439-53. doi: 10.1084/jem.20100587.
118. Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. 2011; 186: 7243-54. doi: 10.4049/jimmunol.1001952.
119. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P, Mantovani A, Sica A. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A. 2009; 106: 14978-83. doi: 10.1073/pnas.0809784106.
120. Bergenfelz C, Medrek C, Ekstrom E, Jirstrom K, Janols H, Wullt M, Bredberg A, Leandersson K. Wnt5a induces a tolerogenic phenotype of macrophages in sepsis and breast cancer patients. J Immunol. 2012; 188: 5448-58. doi: 10.4049/jimmunol.1103378.
121. Goldsmith M, Avni D, Ernst O, Glucksam Y, Levy-Rimler G, Meijler MM, Zor T. Synergistic IL-10 induction by LPS and the ceramide-1-phosphate analog PCERA-1 is mediated by the cAMP and p38 MAP kinase pathways. Mol Immunol. 2009; 46: 1979-87. doi: 10.1016/j.molimm.2009.03.009.
122. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009; 15: 42-9. doi: 10.1038/nm.1905.
123. Ray A, Chakraborty K, Ray P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front Cell Infect Microbiol. 2013; 3: 52. doi: 10.3389/fcimb.2013.00052.
124. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007; 81: 1-5. doi: 10.1189/jlb.0306164.
125. Rodrigues JC, Gonzalez GC, Zhang L, Ibrahim G, Kelly JJ, Gustafson MP, Lin Y, Dietz AB, Forsyth PA, Yong VW, Parney IF. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010; 12: 351-65. doi: 10.1093/neuonc/nop023.
126. Fang Z, Li J, Yu X, Zhang D, Ren G, Shi B, Wang C, Kosinska AD, Wang S, Zhou X, Kozlowski M, Hu Y, Yuan Z. Polarization of Monocytic Myeloid-Derived Suppressor Cells by Hepatitis B Surface Antigen Is Mediated via ERK/IL-6/STAT3 Signaling Feedback and Restrains the Activation of T Cells in Chronic Hepatitis B Virus Infection. J Immunol. 2015; 195: 4873-83. doi: 10.4049/jimmunol.1501362.
127. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. 2014; 20: 4096-106. doi: 10.1158/1078-0432.CCR-14-0635.
128. Janols H, Bergenfelz C, Allaoui R, Larsson AM, Ryden L, Bjornsson S, Janciauskiene S, Wullt M, Bredberg A, Leandersson K. A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J Leukoc Biol. 2014; 96: 685-93. doi: 10.1189/jlb.5HI0214-074R.
129. Brandau S, Moses K, Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol. 2013; 23: 171-82. doi: 10.1016/j.semcancer.2013.02.007.
130. Hagerling C, Werb Z. Neutrophils: Critical components in experimental animal models of cancer. Semin Immunol. 2016. doi: 10.1016/j.smim.2016.02.003.
131. Sippel TR, White J, Nag K, Tsvankin V, Klaassen M, Kleinschmidt-DeMasters BK, Waziri A. Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res. 2011; 17: 6992-7002. doi: 10.1158/1078-0432.CCR-11-1107.
132. Arnardottir HH, Freysdottir J, Hardardottir I. Two circulating neutrophil populations in acute inflammation in mice. Inflamm Res. 2012; 61: 931-9. doi: 10.1007/s00011-012-0484-0.
133. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: ”N1” versus ”N2” TAN. Cancer Cell. 2009; 16: 183-94. doi: 10.1016/j.ccr.2009.06.017.
134. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, Ulfman LH, Leenen LP, Pickkers P, Koenderman L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012; 122: 327-36. doi: 10.1172/JCI57990.
135. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov RV, Ariel A, Hovav AH, Henke E, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015; 10: 562-73. doi: 10.1016/j.celrep.2014.12.039.
136. Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, Quatromoni JG, Stephen TL, Litzky L, Deshpande C, Feldman MD, Hancock WW, Conejo-Garcia JR, et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell. 2016; 30: 120-35. doi: 10.1016/j.ccell.2016.06.001.
137. Brandau S, Dumitru CA, Lang S. Protumor and antitumor functions of neutrophil granulocytes. Semin Immunopathol. 2013; 35: 163-76. doi: 10.1007/s00281-012-0344-6.
138. Lichtenstein A, Seelig M, Berek J, Zighelboim J. Human neutrophil-mediated lysis of ovarian cancer cells. Blood. 1989; 74: 805-9.
139. Zivkovic M, Poljak-Blazi M, Zarkovic K, Mihaljevic D, Schaur RJ, Zarkovic N. Oxidative burst of neutrophils against melanoma B16-F10. Cancer Lett. 2007; 246: 100-8. doi: 10.1016/j.canlet.2006.02.002.
140. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of Granulocyte Survival and Programmed Cell-Death by Cytokines and Bacterial Products. Blood. 1992; 80: 2012-20.
141. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012; 33: 949-55. doi: 10.1093/carcin/bgs123.
142. Granot Z, Fridlender ZG. Plasticity beyond cancer cells and the “immunosuppressive switch”. Cancer Res. 2015; 75: 4441-5. doi: 10.1158/0008-5472.CAN-15-1502.
143. Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003; 188: 49-71.