
INTRODUCTION
Colorectal cancer (CRC) is the fifth and fourth most commonly diagnosed cancer in males and females, respectively, and is the fifth most common cause of cancer-related death in China [1]. Colitis associated cancer (CAC) is a type of CRC that results from inflammatory bowel disease (IBD) [2, 3]. Traditional colorectal cancer therapies include surgical resection, chemotherapy, and radiotherapy [4]. In recent years, studies have investigated the influence of the tumor microenvironment on carcinogenesis and therapeutic efficacies [5, 6]. Tumor-associated macrophages (TAMs) that infiltrate tumors are abundant in the tumor microenvironment. TAMs play important roles in chronic inflammation and in the tumor microenvironment and not only promote tumor cell growth, but also affect the efficacy of different antineoplastic therapies [7–10].
Macrophages are generally classified as the classical ‘M1’ type activated by Th1 cytokines or the alternative ‘M2’ type activated by Th2 cytokines [11–14]. M1-like macrophages play a central role in killing invading pathogens and tumor cells, secrete pro-inflammatory cytokines such as IL-12, TNF-a, CXCL10, IFN-γ, and exhibit high nitric oxide synthase (iNOS) expression. In contrast, M2-like macrophages are important in tissue repair and tumor progression, secrete anti-inflammatory cytokines such as IL-10, IL-13, and IL-4, and exhibit high mannose receptor (MR, CD206) and Arg1 expression [13, 15, 16]. TAMs typically display the M2-like phenotype [17, 18].
The EGFR signaling pathway plays a crucial role in colon cancer survival, growth, and metastasis [19–21]. EGFR is overexpressed in 60-80% of colon cancers, and clinical studies of EGFR-targeted drugs have already been conducted [22]. Moreover, EGFR-targeted therapy might not only directly inhibit tumor cell growth, but also modulate the tumor microenvironment. For example, the EGF/EGFR pathway modulates cytokine profiles in breast cancer. Cannabidiol inhibits the EGF/EGFR pathway and alters cytokine production in tumor cells, leading to a reduction in the numbers of total and M2 macrophages at the primary and secondary tumor sites [23]. Inhibition of EGFR signals in human colon cancer also influences cytokine secretion [24]. However, whether the EGFR signaling pathway influences macrophage polarization in colorectal tumor microenvironment has not yet been investigated.
In this study, we found that the EGFR pathway modulated macrophage numbers and polarization in colon cancer, and consequently influenced tumor growth. Furthermore, IGF-1 secreted from colon cancer cells also influenced macrophage polarization.
RESULTS
Cetuximab modulates macrophage polarization in an AOM/DSS mouse model
The AOM/DSS mouse model was created by intraperitoneal injection of the pro-carcinogen AOM followed by two cycles of DSS (2AD) exposure (Figure 1A). As expected, colonic tumors were visible in all AOM/DSS mice (Figure 1B). After the second DSS cycle, mice were treated for four weeks with saline or cetuximab (1 mg/mouse, twice a week), a dose previously shown to prevent tumor formation [25]. Notably, cetuximab reduced the number of tumors, and most tumors were smaller (< 2 mm), in cetuximab-treated 2AD mice (2AD + cetu) compared to saline-treated 2AD (2AD) mice (Figure 1B and 1C). Histopathologically, 2AD mice developed adenoma tumors with high-grade dysplasia, while 2AD+cetu mice developed low-grade dysplasia (Supplementary Figure S1A). PCNA staining was stronger in 2AD mice than in normal mice and 2AD + cetu mice (Supplementary Figure S1B). These results suggest that cetuximab effectively protected against AOM/DSS-induced, colitis-associated tumorigenesis.
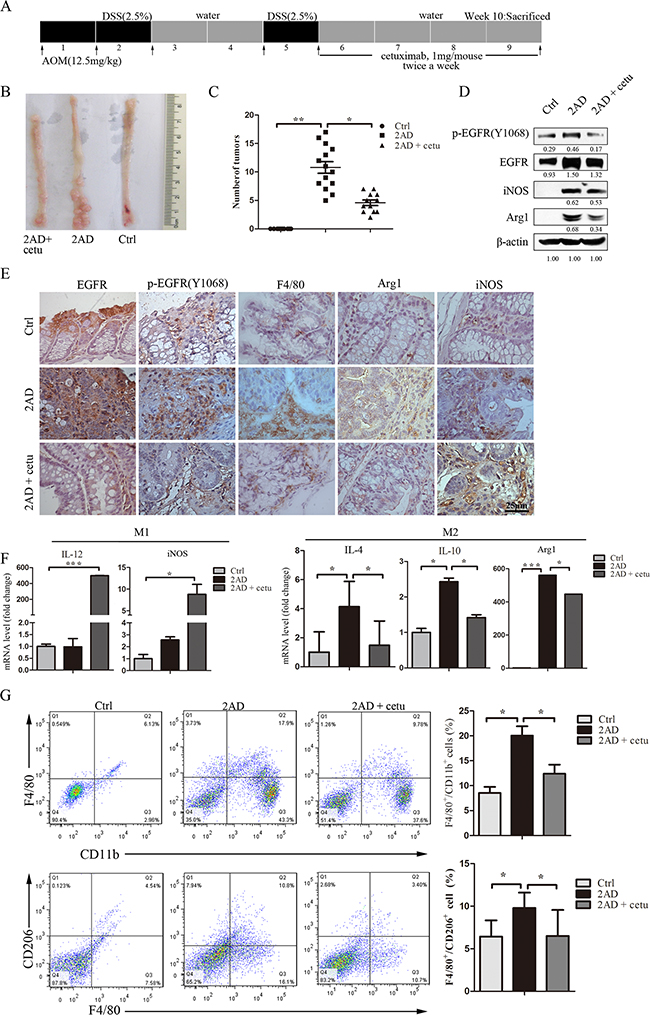
Figure 1: Cetuximab modulates macrophage polarization in an AOM/DSS mouse model. A. Establishment of the AOM/DSS mouse model. AOM was injected intraperitoneally at 12.5 mg/kg body weight. After one week, mice were given drinking water containing 2.5% DSS for 5 days followed by 16 days of regular drinking water. After two cycles of DSS treatment, cetuximab (1 mg/mouse, twice a week) was injected intraperitoneally for a month, and the mice were then sacrificed. B. Representative images of colon tumors in normal (right), AOM/DSS (2AD) (middle), and cetuximab-treated AOM/DSS mice (2AD+cetu) (left). C. Tumor quantification. Cetuximab treatment (2AD + cetu) reduced tumor numbers compared to 2AD mice. D. p-EGFR (Y1068), EGFR, Arg1, and iNOS protein levels in normal mice, 2AD and 2AD+cetu mouse tumors were detected by Western blot. All experiments were repeated three times. E. Representative photomicrographs of immunostaining for p-EGFR (Y1068), PCNA, F4/80, Arg1, and iNOS. Scale bars: 25 μm. F. M1 marker (IL-12, iNOS) and M2 marker (IL-4, IL-10, Arg1) mRNA levels in normal mouse colon tissues, 2AD and 2AD+cetu mouse tumor tissues were evaluated by q-PCR. G. Percentages of CD11b+/F4/80+ and F4/80+/CD206+ cells in normal, 2AD, and 2AD+cetu mice colon tissues were detected by flow cytometry. Colon tissues were cut into small pieces (1-2 mm) and incubated with collagenase D (1- mg/mL), dispase II (1 mg/mL), and DNase I (100 μg/mL) for 30-45 min in a shaking incubator at 37°C, and single-cell suspensions were then incubated with antibodies. Bars represent means ± SD (n = 3) for each treatment. *p < 0.05; **p < 0.01; ***p < 0.001.
Western blot results showed that levels of p-EGFR (Y1068), EGFR, Arg1, and iNOS proteins were higher in AOM/DSS mice than in normal mice. Treatment with cetuximab reduced levels of all of these proteins, except for iNOS, compared to 2AD mice (Figure 1D). Immunohistochemistry results were consistent with these findings. p-EGFR (Y1068) and EGFR levels were higher in 2AD mouse adenomas. F4/80-positive macrophage infiltration was present in 2AD and 2AD + cetu mice. Arg1 positive macrophages were abundant in 2AD mice, but rarely detected in normal mice and 2AD + cetu mice (Figure 1E). We then measured the expression of typical M1 and M2 macrophage marker mRNAs. Expression of iNOS and IL-12, which are typical M1 markers, did not differ between 2AD and normal mice, but were higher in 2AD + cetu mice (Figure 1F). In contrast, Arg1, IL-10, and IL-4, which are typical M2 markers, were higher in 2AD than in normal mice, and cetuximab treatment inhibited Arg1, IL-10, and IL-4 mRNA expression (Figure 1F).
Next, we analyzed macrophage populations in primary tumors using flow cytometry. 2AD mice had more total macrophages (F4/80+/CD11b+) and a higher percentage of M2 macrophages (F4/80+/CD206+) than normal mice, and cetuximab decreased both macrophage populations (Figure 1G). Taken together, these results suggest that cetuximab inhibits macrophage accumulation and M2 polarization in the AOM/DSS mouse model.
Inhibition of the EGFR signaling pathway in colon cancer cells reduces M2-like macrophage polarization
A previous study found that macrophages express EGFR [26], but we did not detect EGFR protein expression in macrophages (Figure 2A), and cetuximab alone had no effect on macrophage polarization (Supplementary Figure S2). It is possible that the EGFR monoclonal antibody cetuximab does not directly influence macrophage polarization in the AOM/DSS mouse model. Cetuximab might inhibit EGFR signaling in colon cancer cells and alter the secretion of other factors into the tumor microenvironment, consequently preventing macrophage polarization. To investigate this possibility, we overexpressed EGFR in HCT116 and CT26 cells, and knocked down EGFR expression in HCT116 cells (Figure 2B). Cancer cell conditioned media (CM) were then harvested and used to treat macrophage cells. CM from HCT116 cells induced the polarization of THP-1 cells into CD68+/CD11b+ macrophages (Figure 2C) and CD206-positive macrophages (Figure 2D). In addition, the expression of M1 and M2 macrophage marker mRNAs increased in HCT116 CM-treated THP-1 cells. In HCT116 siEGFR CM-treated THP-1 cells, M2-related markers IL-10, Arg1, CCL17, CCL22, and IL-4 were downregulated, but M1-related markers IL-12, CCR7, and TNF-α were upregulated, compared to HCT116-CM treated THP-1 cells (Figure 2E).
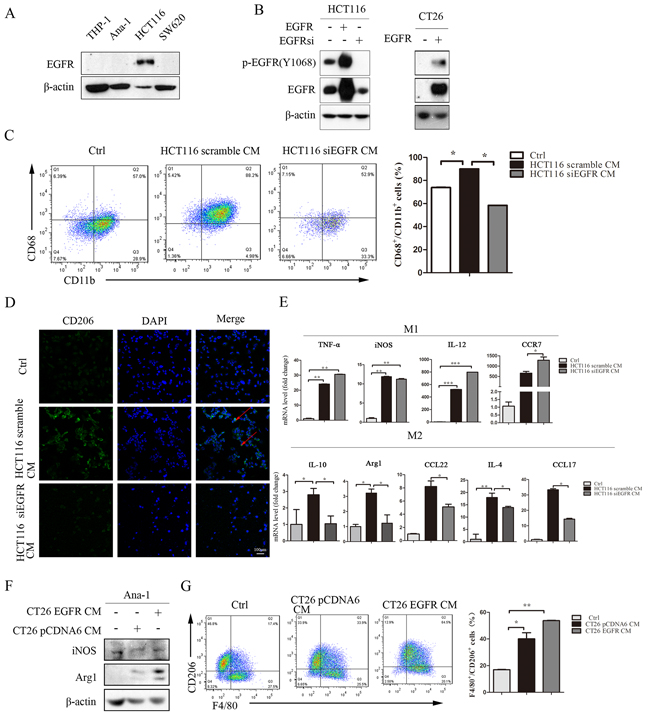
Figure 2: Inhibition of the EGFR signaling pathway in colon cancer cells prevents conditioned medium-induced M2-like macrophage polarization. A. EGFR protein levels in THP-1, Ana-1, HCT116, and SW620 cells were detected by Western blot. B. HCT116 cells were cultured to 50% confluence and then transfected with human scramble siRNA, pCDNA6-EGFR WT plasmid, or EGFR siRNA. CT26 cells were cultured to 50% confluence and then transfected with human pCDNA6 vector or pCDNA6-EGFR WT plasmid for 48 h; the cells were then harvested for Western blots for EFGR. C. Percentages of CD68+/CD11b+ in THP-1 cells after 48 h of treatment with normal RPMI1640, HC116 scramble CM, or HCT116 siEGFR CM were detected by flow-cytometry. D. Immunofluorescent staining for CD206+ was measured in THP-1 cells after incubation with normal RPMI1640, HCT116 scramble CM, or HCT116 siEGFR CM. E. M1-related marker (TNF-α, iNOS, IL-12 and CCR7) and M2-related marker (IL-4, CCL17, CCL22, IL-10 and Arg1) mRNA levels were detected by q-PCR in THP-1 cells after incubation with normal RPMI1640, HCT116 scramble CM, or HCT116 siEGFR CM. Scale bars: 100 μm. F. Arg1 and iNOS protein levels in Ana-1 cells were detected by Western blot after incubation with CT26 pCDNA6 CM or CT26 EGFR CM. G. Percentages of F4/80+/CD206+ in Ana-1 cells after incubation with CT26 pCDNA6 CM or CT26 EGFR CM were detected by flow cytometry. Red arrows indicate CD206 expression in the cell membrane. Nuclei were counterstained with DAPI. Bars represent means ± SD (n = 3) for each treatment.*p < 0.05; **p < 0.01; ***p < 0.001.
Next, we incubated Ana-1 cells with CT26 or CT26 EGFR CM. Arg1 protein level and F4/80+/CD206+ positive cells increased after treatment with CT26 EGFR CM, while iNOS protein levels did not change after either CT26 CM or CT26 EGFR CM treatment (Figure 2F and 2G).
We also treated Ana-1 and bone marrow-derived macrophage (BMDM) cells with media conditioned by HCT116, SW480, and SW620 colon cancer cells and obtained similar results (Supplementary Figure S3-S5). Taken together, these results suggest that EGFR signaling activity in colon cancer cells might promote the polarization of M1-like macrophages into M2-like macrophages.
EGFR knockout in colon cancer cells inhibits macrophage-induced xenograft tumor growth
To investigate whether EGFR signaling activity in colon cancer cells stimulates Ana-1 macrophage polarization and how this polarization affects tumor growth, we subcutaneously injected HCT116 or HCT116 KO-EGFR cells with or without Ana-1 cells into Balb/c athymic nude mice. Injection of EGFR knockout HCT116 cells reduced tumor growth, and injection of Ana-1 cells with either HCT116 or HCT116 KO-EGFR cells increased tumor growth (Figure 3A, 3B). However, EGFR knockout in HCT116 cells dramatically reduced Ana-1-induced tumor growth; the tumor promotion efficiency of Ana-1 cells decreased from 2.7-fold with HCT116 cells to 1.3-fold with HCT116 KO-EGFR cells (Figure 3C).
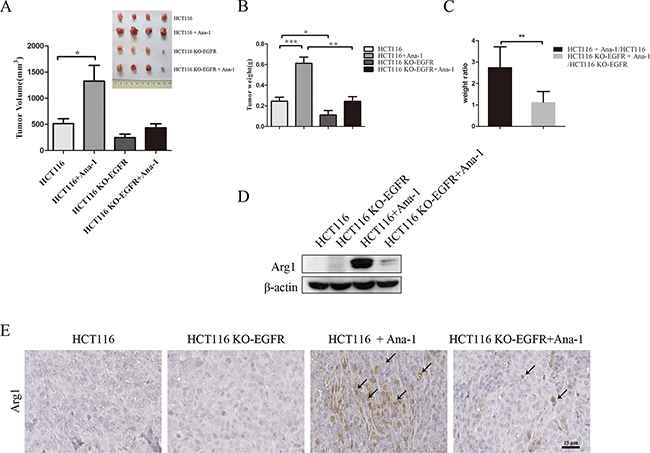
Figure 3: EGFR knockout in colon cancer cells inhibits macrophage-induced promotion of xenograft tumor growth. A. Mouse tumor volume quantification (bottom left). Images (top right) of tumors resulting from subcutaneous injection of HCT116 cells alone, HCT116 plus Ana-1 cells, HCT116 KO-EGFR cells alone, and HCT116 KO-EGFR plus Ana-1 cells (n=4) into Balb/c nude mice. After 18 days, the mice were sacrificed and tumors were excised. The experiment was repeated twice. B. Mouse tumor weights were measured. C. Weight ratio of mice receiving HCT116 plus Ana-1/HCT116 injections compared to HCT116 KO-EGFR plus Ana-1/HCT116 KO-EGFR injections. D. Arg1 protein levels were detected by Western blot in HCT116, HCT116 KO-EGFR, HCT116 plus Ana-1, and HCT116 KO-EGFR plus Ana-1 mouse tumors. E. Immunostaining for Arg1 in xenograft mouse tumor tissues. Scale bars: 50 μm. Bars represent means ± SD (n = 3) for each treatment. *p < 0.05; **p < 0.01.
Western blot showed that Arg1 protein was undetectable in HCT116 and HCT116 KO-EGFR tumors, but Arg1 levels increased in tumors induced by the injection of both HCT116 and Ana-1 cells compared to those induced by HCT116 KO-EGFR and Ana-1 cells (Figure 3D). IHC confirmed that HCT116 KO-EGFR plus Ana-1 tumors had fewer Arg1+ macrophages than HCT116 plus Ana-1 tumors (Figure 3E). HE staining of xenograft tissues is shown in Supplementary Figure S6A. Additionally, q-PCR showed that expression of the M1-related markers iNOS and CXCL10 was higher, while Arg1 expression was lower, in HCT116 KO-EGFR plus Ana-1 tumors than in HCT116 plus Ana-1 tumors (Supplementary Figure S6B). These results indicate that inhibition of the EGFR signaling pathway in HCT116 cells prevents M2 macrophage polarization and consequently inhibits tumor growth in vivo.
Colon cancer-derived IGF-1 promotes M2-like macrophage polarization
To investigate the mechanism by which conditioned medium promoted macrophage polarization, we performed a cytokine antibody array to compare differences between HCT116 siEGFR CM and HCT116 CM. We found that the IGF-1 concentration in HCT116 siEGFR CM was 53% of that in HCT116 CM (unpublished data). IGF-1 is a cytokine and a hormone factor, and might play a role in macrophage polarization [27]. Next, we examined whether IGF-1 levels in colon cancer CM were regulated by the EGFR pathway. Soluble IGF-1 in colon cancer CM was detected by ELISA. IGF-1 concentrations were higher in SW620 EGFR CM than in SW620 CM (Figure 4A). Overexpression of EGFR increased, and EGFR knockout decreased, IGF-1 secretion and mRNA expression in HCT116 cells (Figure 4B and 4C). We also investigated IGF-1 mRNA expression in AOM/DSS mouse colon tissues; IGF-1 expression was higher in 2AD mice than normal mice, and cetuximab treatment inhibited this increase in IGF-1 expression (Figure 4D).
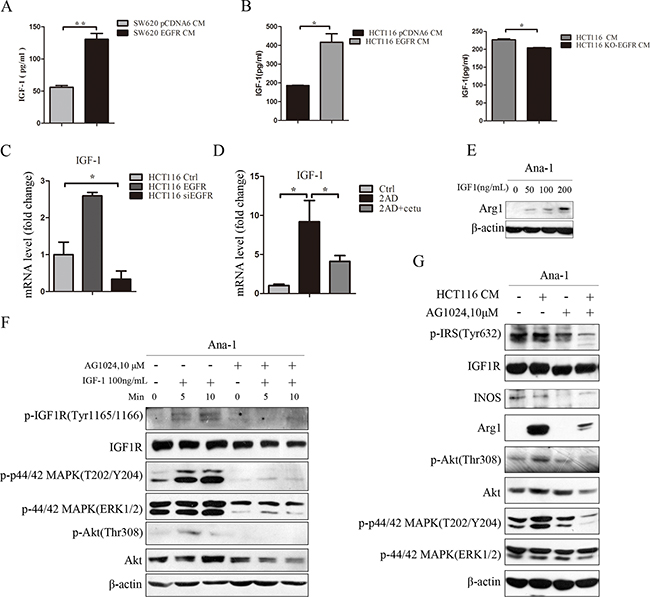
Figure 4: Colon cancer-derived IGF-1 promotes M2-like macrophage polarization. A. IGF-1 concentrations were determined by ELISA in SW620 and SW620 EGFR cell culture media. SW620 cells were cultured to 50% confluence and then transfected with human pCDNA6 vector or pCDNA6-EGFR WT plasmid. After 24 h, cells were transferred to normal medium and cultured for an additional 24 h. Conditioned medium from SW620 cells was collected, centrifuged at 3000 rpm for 10 minutes, and stored at -80°C until use. B. IGF-1 concentrations in HCT116 and HCT116 EGFR (left) and HCT116 and HCT116 KO-EGFR (right) cell culture media were measured by ELISA. C. IGF-1 mRNA levels in HCT116, HCT116 EGFR, and HCT116 siEGFR cells were measured by q-PCR. D. IGF-1 mRNA levels in normal, 2AD, and 2AD+cetu mouse tissues were measured by q-PCR. E. Arg1 protein levels were detected by Western blot in Ana-1 macrophages after treatment with mouse recombinant IGF1 (0, 50, 100, or 200 ng/mL) for 48 h. F. Levels of IGF1R signaling pathway-related proteins were detected by Western blot. Ana-1 cells were harvested after treatment with 100 ng/mL mouse recombinant IGF-1 for 5 or 10 min, or after pretreatment with AG1024 for 30 min followed by 100 ng/mL mouse recombinant IGF-1 for 5 or 10 min. G. Protein levels were detected by Western blot in Ana-1 cells. Ana-1 cells were pretreated with AG1024 for 30 min, and medium was then replaced with fresh RPMI 1640 with AG1024 (10 mM) or HCT116 CM with AG1024 (10 mM) followed by an additional 48 h of culture. Cells were harvested and levels of Arg1, iNOS, and IGF1R signaling pathway-related proteins p-IRS(Tyr632), IGF1R, p-p44/42 MAPK(T202/Y204), p-44/42 MAPK(Erk1/2), p-Akt(Thr308), and Akt were measured by Western blot. Bars represent means ± SD (n = 3) for each treatment. *p < 0.05; **p < 0.01.
To investigate the role of IGF-1 in macrophage polarization, we stimulated Ana-1 cells with recombinant IGF-1 (R&D) for 48 h. Western blot analysis showed that IGF-1 increased Arg1 protein levels in a dose-dependent manner (Figure 4E). IGF-1 also increased phosphorylation of the IGF1R, Akt, and MAPK signaling proteins (Figure 4F). Pretreatment with an Akt inhibitor abolished IGF-1-induced Arg1 expression in Ana-1 cells (Supplementary Figure S7A). Additionally, pretreatment with the IGF1R inhibitor AG1024 abolished IGF-1-induced IGF1R signaling pathway activation in Ana-1 cells (Figure 4F). HCT116 CM activated IGF1R signaling in Ana-1 cells and increased the expression of the M2 marker Arg1. After pretreatment with AG1024, HCT116 CM-induced IGF1R pathway activation was inhibited and Arg1 was downregulated in Ana-1 cells (Figure 4G). Human IGF-1 neutralizing antibody (R&D) also inhibited IGF-1 induced Arg1 expression (Supplementary Figure S7B). IGF-1 secretion was higher in EGF-stimulated HCT116 cells than in EGF-stimulated HCT116 KO-EGFR cells (Supplementary Figure S7C). Overall, these results demonstrate that IGF-1 from colon cancer cells induced M2 macrophage polarization, and inhibition of EGFR in HCT116 cells reduced both IGF-1 secretion and HCT116 CM-induced macrophage polarization.
DISCUSSION
The EGFR signaling pathway plays a critical role in colonic tumorigenesis [28] and is a target of many cancer therapies. The anti-EGFR monoclonal antibodies cetuximab (Erbitux™, Bristol Myers Squibb and Merck KGaA) and panitumumab (VectibixTM, Amgen) are approved for the treatment of metastatic colorectal cancer (mCRC) [29, 30]. In recent years, EGFR-targeted therapy has been reported to not only inhibit tumor cells, but also reduce the secretion of GM-CSF, CCL3, TGF-α, bFGF, and VEGF, further affecting the tumor microenvironment [23, 24]. In this study, we found cetuximab reduced both tumor growth in an AOM/DSS mouse model and F4/80+/CD206+ macrophage populations. Experimental colitis colonic and peritoneal macrophages and human CD14+ monocytes have been reported to express EGFR [26, 31], but we did not detect EGFR expression in Ana-1 and THP-1 cells here (Figure 2A). Cetuximab might therefore modulate the secretion of various factors from tumor cells and thus inhibit macrophage accumulation and polarization.
To investigate this possibility, we incubated macrophages with medium conditioned by colon cancer cells and found that M2 polarization increased. EGFR overexpression promoted M2 polarization, whereas inhibition of the EGFR pathway prevented M2 polarization. Lewis lung carcinoma tumor conditioned medium increases the production of immunosuppressive factors in macrophages and decreases phagocytosis activity [32]. Melanoma conditioned-media is very effective for differentiating macrophages in vitro [33]. Additionally, signal molecules produced by tumor cells, such as lactate, HRG/PIGF, chemokine ligand 2 (CCL2), soluble colony-stimulating factor 1 (sCSF1), and POSTN, play critical roles in macrophage polarization [34–39]. Our results suggest that inhibition of the EGFR pathway might alter the components of conditioned medium to contribute to macrophage polarization. We confirmed this possibility by analyzing the secreted cytokine profiles of HCT116 cells after EGFR knockdown. EGFR knockout inhibited the secretion of IGF-1, which plays a key role in cell growth, differentiation, survival, transformation, and metastasis. IGF-1 is overexpressed in pancreatic, colon, breast, and ovarian cancers [40–43]. Our results also indicated that colon cancer tumor cells secrete IGF-1. Cancer associated fibroblasts [44] and TAMs [45] also secrete IGF-1 and promote tumor progression. Additionally, IGF-1 alters macrophage numbers and activity in liver microenvironments in obese mice [27].
We then investigated the role of IGF-1 in macrophage polarization. When the IGF-1 ligand binds to IGF1R, the immediate substrate insulin receptor substrate protein (IRS-1) is phosphorylated and recruits effectors containing SH2 or PTB domains to modulate the IGF1R pathway [46–48]. Here, IGF-1 directly promoted M2 macrophage polarization by activating the IGF1R signaling pathway, demonstrating for the first time that this pathway is important in macrophage polarization. Furthermore, we found that Akt signaling, which occurs downstream of IGF1R, was associated with macrophage polarization. Then we injected cancer cells and macrophages together to investigate the role of macrophage polarization in tumor growth. Ana-1 cells promoted tumor growth in the xenograft model, and most Ana-1 cells displayed the M2 phenotype. Moreover, EGFR knockout in HCT116 cells dramatically reduced the M2 macrophage population and tumor growth. Our results indicate that the tumor microenvironment induced M2 polarization in Ana-1 cells and that TAMs promote colon tumor growth. Inhibition of EGFR in HCT116 cells dramatically reduced TAM polarization and reduced tumor growth. Similar results have been reported for prostate cancer cells mixed with RAW264.7 cells. RAW264.7 cells promoted M2 polarization, which in turn promoted angiogenesis and tumor growth in vivo [36].
Colon cancer tissues contain large numbers of TAMs, which comprise the majority of immune cells within these tumors. Some studies have found that TAMs promote tumor progression in CRC patients; CD68+ macrophages are used as a marker of progression, and CD163+ macrophages are associated with early local recurrence and reduced survival times [49, 50]. Other studies indicate that macrophages inhibit tumor progression in CRC patients [51, 52]. However, the M1/M2 ratio in CRC patients is much higher than in prostate cancer patients, suggesting that M1 macrophages may be more important in CRC [52]. It is possible that the M1/M2 ratio rather than the total number of macrophages determines whether these cells promote or inhibit tumor growth. Moreover, TAMs that infiltrate the tumor invasive margin may be exposed to different tumor microenvironment signals than those that infiltrate the tumor stroma, possibly accounting for the different effects of TAMs on colon tumor growth [51, 53]. Because they are exposed to fewer signals produced by tumor cells, anti-tumor M1 polarization might predominate in peritumoral macrophages; in contrast, tumor microenvironment signals might increase pro-tumor M2 polarization in intratumoral macrophages. In our xenograft model, differences in the numbers of M2 macrophages rather than in total numbers of macrophages, which were similar among the groups, determined whether these cells inhibited or promoted tumor growth. Therefore, blocking tumor-induced M2 macrophage polarization might be a potential treatment strategy for inhibiting tumor growth.
In conclusion, our results suggest that inhibition of the EGFR signaling pathway in colon cancer cells alters cytokine secretion (e.g. IGF-1) and prevents M1- to M2-like polarization in macrophages, thus inhibiting cancer cell growth (Figure 5). Inhibiting such macrophage polarization might be a promising novel method for treating cancer.
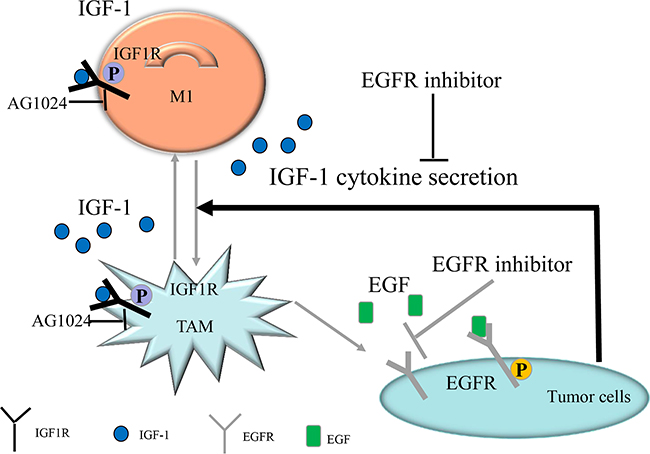
Figure 5: Diagrammatic model of EGFR pathway-modulated macrophage polarization in colon cancer. IGF-1-induced IGF1R pathway activation might promote TAM polarization. EGFR inhibition in colon cancer cells altered the secreted cytokine profile. Secreted IGF-1 concentrations decreased when EGFR was inhibited in colon cancer cells, and IGF-1-induced TAM polarization was inhibited; experiments using the IGF1R inhibitor AG1024 and IGF-1 neutralizing antibody confirmed this effect. In conclusion, EGFR signaling pathway activity promoted IGF-1 secretion, which in turn induced TAM polarization.
MATERIALS AND METHODS
Cell lines
HCT116, SW480, THP-1, SW620, CT26, and Ana-1 cells were maintained in RPMI1640 (Bioroc™, China) supplemented with 10% heated-inactivated fetal bovine serum, 100 UI/mL penicillin, and 100 μg/mL streptomycin. Bone marrow derived macrophage (BMDM) cells were maintained in DMEM (Bioroc™, China) supplemented with 10% heated-inactivated fetal bovine serum, 100 UI/mL penicillin, and 100 μg/mL streptomycin. Cells were cultured in a humidified environment at 37°C with 5% CO2. Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) was used for the transfection assay according to the manufacturer’s instructions. EGFR small interfering RNA (siRNA) (NM_005228, sequence: 5′AGCUAUGAGAUGGAGGAAGACGGCG3′) and scrambled negative control siRNA were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA). PCDNA6-EGFR WT plasmid was provided by Mien-Chie Hung (Addgene plasmid # 42665) [54]. Target sequences for CRISPR interference were designed using this website: http://tools.genome-engineering.org. The human EFGR target sequences (sense: CTCTAAAACACTCGCCGGGCAGAGCGCAG, anti-sense: AAACACCGTACTACTAGACGGGGATGT) were cloned into the Cas9/gRNA (Puro-GFP) Vector (Viewsolid Biotech, China). After transfection and selection with puromycin, knockout clones (HCT116 KO-EGFR cells) were identified by Western blot. Genomic mutations were identified by genome sequencing.
Isolation of bone marrow-derived macrophages (BMDMs)
BMDMs grown in macrophage colony-stimulating factor (M-CSF) were generated as previously described [35]. Briefly, BMDMs were isolated from the femurs of 4-5-week old C57 BL/6J mice, filtered through a 70 μm filter, and cultured in DMEM media supplemented with 10% heat-inactivated-FCS in the presence of M-CSF (10 ng/mL, R&D Systems). On day 3, non-adherent cells were discarded and adherent cells were cultured for an additional 3 days. On day 7, the culture medium was replaced with medium conditioned by tumor cells; 48 h later, the cells were collected and used in subsequent experiments.
Establishment of the AOM/DSS mouse model and the tumor xenograft model
The establishment of the AOM/DSS mouse model was conducted as previously described [55]. Briefly, C57BL/6J mice (7 weeks old, approximate body weight 18-20 g) were injected intraperitoneally with azoxymethane (AOM) (12.5 mg/kg; Sigma-Aldrich, St. Louis, MO, USA). Seven days after injection, mice were treated with 2.5% Dextran sodium sulfate (DSS) (NW36, 000-50,000Da, MP Biomedicals, USA) in drinking water for 5 consecutive days, followed by 16 days of normal drinking water. This DSS treatment was repeated for one additional cycle. After the second cycle, cetuximab (Merck KGaA, Germany) (1 mg/mouse, twice a week) was injected for one month.
For the tumor xenograft model, 5-week-old female Balb/c athymic nude mice received 0.1 mL subcutaneous injections of 1×107/mL HCT116 or HCT116KO-EGFR cells with or without 1×106/mL Ana-1 cells. After 18 days, the mice were sacrificed. Tumor tissues were weighed and tumor volumes were calculated according to the formula length×width2×0.5. All experimental procedures using animals were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Cancer Hospital of the Chinese Academy of Medical Science.
Treatment of macrophages with colon cancer cell conditioned medium
Colon cancer cells were cultured to 50% confluence and transfected with human EGFR siRNA or pCDNA6A-EGFR WT plasmid for 24 h. After replacement with fresh medium, cells were cultured for an additional 24 h. Tumor cell conditioned medium was collected and centrifuged at 3000 rpm for 10 minutes. THP-1 cells were pretreated with 320 nM phorbol 12-myrisatate13-acetate (PMA, Sigma-Aldrich) for 72 h and thoroughly washed five times with 0.9% saline, and Ana-1 and THP-1 cells were then cultured with 2 mL conditioned medium for 48 h. Ana-1 cells were then incubated with human and mouse recombinant IGF-1 (R&D Systems, Minneapolis, MN, USA) for 48 h. IGF1R signaling was blocked with IGF1R inhibitor AG1024 (Selleck Chemicals, Texas, USA) and IGF-1 neutralizing antibody (R&D Systems) to confirm the role of IGF-1 in conditioned medium-driven macrophage polarization.
Flow cytometry
Isolation of primary mouse macrophages from colon was performed as described previously [56]. Briefly, colon tissues were cut into small pieces (1-2 mm) and incubated in 10 mL PRMI 1640 medium with 10 mM HEPES and 5% fetal bovine serum containing 10 mg (1 mg/mL) collagenase D (Sigma-Aldrich), 10 mg (1 mg/mL) dispase II (Roche, Germany), and 100 μL 10 mg/ml DNase I (100 μg/mL) (Sigma-Aldrich) for 30-45 min in a shaking incubator at 37°C. For Ana-1 and BMDM cells, single-cell suspensions from mouse colon tissues were incubated with APC-anti-mouse CD11b and FITC-anti-mouse F4/80 (Affymetrix eBioscience, USA), or APC-anti-mouse F4/80 and FITC-anti-mouse CD206 (MMR) (Biolegend, San Diego, CA) antibodies. For THP-1 cells, single-cell suspensions were incubated with FITC anti-human CD68 intracellular marker before permeabilization with a BD Cytofix/Cytoooperm™ Fixation/Permeabilization Kit and incubation with PE-Cy™7 mouse anti-human CD11b/mac-1 (BD Biosciences, USA) antibody. Isotype control antibodies (Biolegend, eBioscience and BD Biosciences) were used as the negative control. All cells were incubated with antibodies for 30 min and washed with phosphate-buffered saline (PBS) before acquisition on a LSRII (BD, USA) and analysis with FlowJo software (Tree Stat, OR).
Western blot
Whole cell lysates were prepared using a lysis buffer (Cell Signaling Technology, CST, Danvers, MA, USA) and protein concentration was determined using the Pierce™ BCA Protein Assay kit (Thermo Scientific). Western blots were performed as previously described [57]. The antibodies included β-actin (Sigma-Aldrich), p-IGF1R (Tyr1165/1166), Arginase1 (H-52), NOS2 (N-20), and p-IRS (Tyr632) (Santa Crus Biotech., Santa Cruz, CA, USA), and p-EGFR (Tyr1068), p-p44/42 MAPK (T204/Y202), EGFR, Akt, p44/42 MAPK (Erk1/2), IGF1R (CST), and p-Akt (Thr308) (Affinity Biosciences Inc.)
Quantitative real-time polymerase chain reaction (q-PCR)
Total RNA was extracted with TRIZOL reagent (Invitrogen) according to the manufacturer’s protocol. 0.5 μg of total RNA were used as the template for synthesizing cDNA with PrimeScript™ RT Master Mix (Perfect real time) (Takara, Dalian, China). q-PCR was performed on the Step-One Plus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) with Power SYBR Green PCR Master Mix (Applied Biosystems) and analyzed using StepOne Software. GAPDH was used as an internal control. Sequences of the PCR primers [58–62] are showed in Table 1.
Table 1: Primers used for q-PCR
Species |
Oligo name |
Sense |
Anti-sense |
|---|---|---|---|
human |
GAPDH |
GTGAAGGTCGGAGTCAACGG |
CTCCTGGAAGATGGTGATGGG |
IL-10 |
TCCCTGTCAAAACAAGAGCA |
ATAGAGTCGCCACCCTGATG |
|
IL-4 |
CTGTGCTCCGGCAGTTCTA |
ACGTACTCTGGTTGGCTTCC |
|
IL-12 |
GCGGAGCTGCTACACTCTCT |
GGTGGGTCAGGTTTGATGAT |
|
TNF-α |
TGTAGCAAACCCTCAAGCTG |
TTGATGGCAGAGAGGAGGTT |
|
iNOS |
TCCAAGGTATCCTGGAGCGA |
CAGGGACGGGAACTCCTCTA |
|
Arg1 |
ACGGAAGAATCAGCCTGGTG |
GTCCACGTCTCTCAAGCCAA |
|
IGF-1 |
TGGATGCTCTTCAGTTCGTG |
TGGTAGATGGGGGCTGATAC |
|
CCL17 |
ACTTCAAGGGAGCCATTCCC |
CCTGCCCTGCACAGTTACAA |
|
CCL22 |
ATCGCCTACAGACTGCACTC |
GACGGTAACGGACGTAATCAC |
|
CCR7 |
TGTACGAGTCGGTGTGCTTC |
GGTAGGTATCCGTCATGGTCTTG |
|
mouse |
GAPDH |
AGGTCGGTGTGAACGGATTTG |
TGTAGACCATGTAGTTGAGGTCA |
INOS |
GTTCTCAGCCCAACAATACAAGA |
GTGGACGGGTCGATGTCAC |
|
CXCL10 |
CCAAGTGCTGCCGTCATTTTC |
GGCTCGCAGGGATGATTTCAA |
|
Arg1 |
TGGCTTGCGAGACGTAGAC |
GCTCAGGTGAATCGGCCTTTT |
|
IL-4 |
GGTCTCAACCCCCAGCTAGT |
GCCGATGATCTCTCTCAAGTGAT |
|
IL-10 |
GCTCTTACTGACTGGCATGAG |
CGCAGCTCTAGGAGCATGTG |
|
IL-12 |
ACTCTGCGCCAGAAACCTC |
CACCCTGTTGATGGTCACGAC |
|
IL-13 |
GGATATTGCATGGCCTCTGTAAC |
AACAGTTGCTTTGTGTAGCTGA |
|
IGF-1 |
CTGGACCAGAGACCCTTTGC |
GGTGCCCTCCGAATGCT |
|
CCL17 |
AGTGCTGCCTGGATTACTTCAAAG |
CTGGACAGTCAGAAACACGATGG |
|
CCL22 |
TAACATCATGGCTACCCTGCG |
TGTCTTCCACATTGGCACCA |
|
CCR7 |
TGAGGTCACGGACGATTACAT |
GTAGGCCCACGAAACAAATGAT |
Immunohistochemistry and Immunofluorescence
Sections (5 μm thick) of paraffin-embedded tissue were placed on glass slides, rehydrated, incubated with 3% hydrogen peroxide to quench endogenous peroxidase activity, and then blocked by incubation with 5% bovine serum albumin in PBS. Sections were then incubated with the arginase1 (N-20, Santa Cruz Biotech), PCNA (FL-261, Santa Cruz Biotech), F4/80 (MCA497GA, AbD Serotec), iNOS (Abcam, Cambridge, UK), EGFR, and p-EGFR (Y1068, CST) antibodies. Immunofluorescence was performed as previously described [63]. THP-1 cells were incubated with anti-mannose receptor antibody (Abcam).
ELISA analysis
Cytokine IGF-1 levels were measured by enzyme-linked immunosorbent assay (ELISA) using an ELISA kit (R&D Systems). The ELISA was performed according to the manufacturer’s protocol.
Statistical analysis
All results are expressed as means ± SD. Paired, two-tailed Student’s t-tests were used to identify significant differences between treatment and control groups. A p value of less than 0.05 was considered statistically significant. All analyses were conducted with GraphPad Prism5 software. Figures show the averages of three independent experiments.
ACKNOWLEDGMENTS
This work was supported by the National Basic Research Program of China (2011CB910704), National Key Research & Development (R&D) Plan(2016YFC0906002) and the National Natural Science Foundation (81321091, 31171322).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ and He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016.
2. Rustgi AK. The genetics of hereditary colon cancer. Genes & development. 2007; 21:2525-2538.
3. Cannon J. Colorectal Neoplasia and Inflammatory Bowel Disease. The Surgical clinics of North America. 2015; 95:1261-1269.
4. Markowitz SD, Dawson DM, Willis J and Willson JK. Focus on colon cancer. Cancer cell. 2002; 1:233-236.
5. Klemm F and Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends in cell biology. 2015; 25:198-213.
6. Johansson A, Hamzah J and Ganss R. License for destruction: tumor-specific cytokine targeting. Trends in molecular medicine. 2014; 20:16-24.
7. De Palma M and Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer cell. 2013; 23:277-286.
8. Mantovani A, Bottazzi B, Colotta F, Sozzani S and Ruco L. The origin and function of tumor-associated macrophages. Immunology today. 1992; 13:265-270.
9. Qian BZ and Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010; 141:39-51.
10. Mantovani A and Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Current opinion in immunology. 2010; 22:231-237.
11. Geissmann F, Gordon S, Hume DA, Mowat AM and Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nature reviews Immunology. 2010; 10:453-460.
12. Gordon S and Taylor PR. Monocyte and macrophage heterogeneity. Nature Reviews Immunology. 2005; 5:953-964.
13. Mantovani A, Sozzani S, Locati M, Allavena P and Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology. 2002; 23:549-555.
14. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA and Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. Journal of immunology. 1986; 136:2348-2357.
15. Martinez FO, Helming L and Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annual review of immunology. 2009; 27:451-483.
16. Murray PJ and Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews Immunology. 2011; 11:723-737.
17. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A and Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in immunology. 2004; 25:677-686.
18. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P and Mantovani A. Macrophage polarization in tumour progression. Seminars in cancer biology. 2008; 18:349-355.
19. Radinsky R, Risin S, Fan D, Dong Z, Bielenberg D, Bucana CD and Fidler IJ. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clinical cancer research. 1995; 1:19-31.
20. Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ and Threadgill DW. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proceedings of the National Academy of Sciences. 2002; 99:1521-1526.
21. Tong WM, Ellinger A, Sheinin Y and Cross HS. Epidermal growth factor receptor expression in primary cultured human colorectal carcinoma cells. British journal of cancer. 1998; 77:1792-1798.
22. Cohen RB. Epidermal Growth Factor Receptor as a Therapeutic Target in Colorectal Cancer. Clinical Colorectal Cancer. 2003; 2:246-251.
23. Elbaz M, Nasser MW, Ravi J, Wani NA, Ahirwar DK, Zhao H, Oghumu S, Satoskar AR, Shilo K, Carson WE, 3rd and Ganju RK. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of Cannabidiol in breast cancer. Molecular oncology. 2015; 9:906-919.
24. Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, De Placido S, Bianco AR and Tortora G. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clinical cancer research. 2001; 7:1459-1465.
25. Hotz B, Keilholz U, Fusi A, Buhr HJ and Hotz HG. In vitro and in vivo antitumor activity of cetuximab in human gastric cancer cell lines in relation to epidermal growth factor receptor (EGFR) expression and mutational phenotype. Gastric cancer. 2012; 15:252-264.
26. Lu N, Wang L, Cao H, Liu L, Van Kaer L, Washington MK, Rosen MJ, Dube PE, Wilson KT, Ren X, Hao X, Polk DB and Yan F. Activation of the epidermal growth factor receptor in macrophages regulates cytokine production and experimental colitis. Journal of immunology. 2014; 192:1013-1023.
27. Wu Y, Brodt P, Sun H, Mejia W, Novosyadlyy R, Nunez N, Chen X, Mendoza A, Hong SH, Khanna C and Yakar S. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer research. 2010; 70:57-67.
28. Salomon DS, Brandt R, Ciardiello F and Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Critical reviews in oncology/hematology. 1995; 19:183-232.
29. Gibson TB, Ranganathan A and Grothey A. Randomized phase III trial results of panitumumab, a fully human anti-epidermal growth factor receptor monoclonal antibody, in metastatic colorectal cancer. Clin Colorectal Cancer. 2006; 6:29-31.
30. Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C and Moore MJ. Cetuximab for the treatment of colorectal cancer. The New England journal of medicine. 2007; 357:2040-2048.
31. Ma X, Wu D, Zhou S, Wan F, Liu H, Xu X, Xu X, Zhao Y and Tang M. The pancreatic cancer secreted REG4 promotes macrophage polarization to M2 through EGFR/AKT/CREB pathway. Oncology reports. 2016; 35:189-196.
32. Mulligan JK, Lathers DM and Young MR. Tumors skew endothelial cells to disrupt NK cell, T-cell and macrophage functions. Cancer immunology, immunotherapy : CII. 2008; 57:951-961.
33. Wang T, Ge Y, Xiao M, Lopez-Coral A, Azuma R, Somasundaram R, Zhang G, Wei Z, Xu X, Rauscher FJ, 3rd, Herlyn M and Kaufman RE. Melanoma-derived conditioned media efficiently induce the differentiation of monocytes to macrophages that display a highly invasive gene signature. Pigment cell & melanoma research. 2012; 25:493-505.
34. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, Squadrito ML, Segura I, Li X, Knevels E, Costa S, Vinckier S, Dresselaer T, Akerud P, De Mol M, Salomaki H, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer cell. 2011; 19:31-44.
35. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ and Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014; 513:559-563.
36. Chen PC, Cheng HC, Wang J, Wang SW, Tai HC, Lin CW and Tang CH. Prostate cancer-derived CCN3 induces M2 macrophage infiltration and contributes to angiogenesis in prostate cancer microenvironment. Oncotarget. 2014; 5:1595-1608. doi: 10.18632/oncotarget.1570.
37. Pyonteck SM, Gadea BB, Wang HW, Gocheva V, Hunter KE, Tang LH and Joyce JA. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene. 2012; 31:1459-1467.
38. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA and Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011; 475:222-225.
39. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Rich JN and Bao S. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nature cell biology. 2015; 17:170-182.
40. Huang CT, Chang MC, Chen YL, Chen TC, Chen CA and Cheng WF. Insulin-like growth factors inhibit dendritic cell-mediated anti-tumor immunity through regulating ERK1/2 phosphorylation and p38 dephosphorylation. Cancer letters. 2015; 359:117-126.
41. Samani AA, Yakar S, LeRoith D and Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocrine reviews. 2007; 28:20-47.
42. Fenton JI, Hord NG, Lavigne JA, Perkins SN and Hursting SD. Leptin, insulin-like growth factor-1, and insulin-like growth factor-2 are mitogens in ApcMin/+ but not Apc+/+ colonic epithelial cell lines. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2005; 14:1646-1652.
43. Singh P, Dai B, Yallampalli U, Lu X and Schroy PC. Proliferation and differentiation of a human colon cancer cell line (CaCo2) is associated with significant changes in the expression and secretion of insulin-like growth factor (IGF) IGF-II and IGF binding protein-4: role of IGF-II. Endocrinology. 1996; 137:1764-1774.
44. Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA and Massague J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013; 154:1060-1073.
45. Sprinzl MF, Puschnik A, Schlitter AM, Schad A, Ackermann K, Esposito I, Lang H, Galle PR, Weinmann A, Heikenwalder M and Protzer U. Sorafenib inhibits macrophage-induced growth of hepatoma cells by interference with insulin-like growth factor-1 secretion. Journal of hepatology. 2015; 62:863-870.
46. Kaburagi Y, Yamauchi T, Yamamoto-Honda R, Ueki K, Tobe K, Akanuma Y, Yazaki Y and Kadowaki T. The mechanism of insulin-induced signal transduction mediated by the insulin receptor substrate family. Endocrine journal. 1999; 46:S25-34.
47. Richards RG, Walker MP, Sebastian J and DiAugustine RP. Insulin-like growth factor-1 (IGF-1) receptor-insulin receptor substrate complexes in the uterus. Altered signaling response to estradiol in the IGF-1(m/m) mouse. The Journal of biological chemistry. 1998; 273:11962-11969.
48. Sun XJ, Miralpeix M, Myers MG, Jr., Glasheen EM, Backer JM, Kahn CR and White MF. Expression and function of IRS-1 in insulin signal transmission. The Journal of biological chemistry. 1992; 267:22662-22672.
49. Shabo I, Olsson H, Sun XF and Svanvik J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. International journal of cancer. 2009; 125:1826-1831.
50. Kang JC, Chen JS, Lee CH, Chang JJ and Shieh YS. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. Journal of surgical oncology. 2010; 102:242-248.
51. Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A and Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clinical cancer research. 2007; 13:1472-1479.
52. Lundholm M, Hagglof C, Wikberg ML, Stattin P, Egevad L, Bergh A, Wikstrom P, Palmqvist R and Edin S. Secreted Factors from Colorectal and Prostate Cancer Cells Skew the Immune Response in Opposite Directions. Scientific reports. 2015; 5:15651.
53. Erreni M, Mantovani A and Allavena P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer microenvironment. 2011; 4:141-154.
54. Hsu SC and Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. The Journal of biological chemistry. 2007; 282:10432-10440.
55. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF and Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004; 118:285-296.
56. Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, Malissen B, Osborne LC, Artis D and Mowat AM. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nature immunology. 2014; 15:929-937.
57. Zhou C, Liu S, Zhou X, Xue L, Quan L, Lu N, Zhang G, Bai J, Wang Y, Liu Z, Zhan Q, Zhu H and Xu N. Overexpression of human pituitary tumor transforming gene (hPTTG), is regulated by beta-catenin /TCF pathway in human esophageal squamous cell carcinoma. International journal of cancer. 2005; 113:891-898.
58. Fujimoto H, Sangai T, Ishii G, Ikehara A, Nagashima T, Miyazaki M and Ochiai A. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. International journal of cancer. 2009; 125:1276-1284.
59. Kang FB, Wang L, Li D, Zhang YG and Sun DX. Hepatocellular carcinomas promote tumor-associated macrophage M2-polarization via increased B7-H3 expression. Oncology reports. 2015; 33:274-282.
60. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K, Ueo T, Komekado H, Kawada M, Minami M and Chiba T. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011; 32:1333-1339.
61. Wu TH, Li YY, Wu TL, Chang JW, Chou WC, Hsieh LL, Chen JR and Yeh KY. Culture supernatants of different colon cancer cell lines induce specific phenotype switching and functional alteration of THP-1 cells. Cellular immunology. 2014; 290:107-115.
62. Hefetz-Sela S, Stein I, Klieger Y, Porat R, Sade-Feldman M, Zreik F, Nagler A, Pappo O, Quagliata L, Dazert E, Eferl R, Terracciano L, Wagner EF, Ben-Neriah Y, Baniyash M and Pikarsky E. Acquisition of an immunosuppressive protumorigenic macrophage phenotype depending on c-Jun phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:17582-17587.
63. Ma K, Xu Q, Wang S, Zhang W, Liu M, Liang S, Zhu H and Xu N. Nuclear accumulation of yes-associated protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer letters. 2016.