
INTRODUCTION
DNA replication is a basic biochemical process during cell growth and division [1]. The initiation of DNA replication in eukaryotes occurs at specific genomic loci called “ORI” (origin of replication) or “RO” (replication origin) [2]. Timely duplication of the genome is an essential step in the reproduction of any cell [3]. There is only one ORI for most of bacterial genomes [4]. In contrast to that, eukaryotic genomes contain much more ORI sites [5]. Although eukaryotic replication mechanism is quite conservative, DNA replication initiator lacks obvious consensus sequence or structure between the different species [6].
The ORI in Saccharomyces cerevisiae (S. cerevisiae) is formed by domain A, domain B and domain C [7]. Each of the three domains has its special motif and function as elaborated in [8–9]. Interestingly, in the S. cerevisiae genome there are over 12,000 conservative sequences, of which, however, only 400 are of ORI [10].
For the detailed replication process in human DNA, see [11–13] as well as Figure 1.
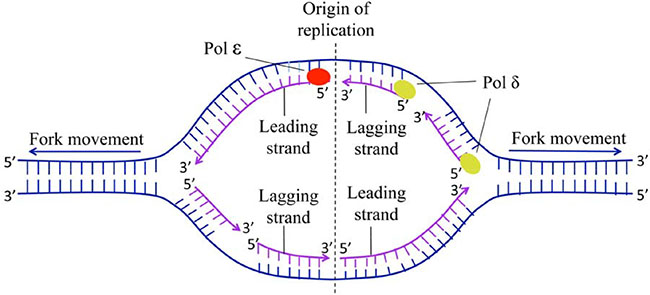
Figure 1: The schematic diagram of origin of replication of human. The process of DNA replication requires two DNA polymerase complexes traveling in opposite direction (i.e. two bidirectional replication forks) from the origin.
Although Chip (chromatin immunoprecipitation) is a very powerful technique to determine the ORI [14], it is time-consuming and costly. Therefore, it would be very helpful to develop bioinformatics tools in this regard.
Actually, considerable efforts [15–20] have been made for this purpose. Although these methods achieved encouraging results, the outcomes were often inconsistent and with limited accuracy. Particularly, none of these methods had taken into account the physicochemical properties of the DNA sequence concerned, one of the vitally important factors for conducting genome analysis, as indicated by a serious of recent studies (see, e.g., [21–27]).
The current study was devoted to establish a new computational method for predicting human ORIs based on the DNA’s physicochemical properties.
According to Chou’s 5-step rule [28], in developing a new statistical predictor we should make the following five procedures very clear as done in a series of recent publications [29–39]: (1) benchmark dataset; (2) sample formulation; (3) operation engine or algorithm; (4) accuracy evaluation; and (5) web-server. In the rest of this paper, we are to address these five steps one-by-one. However, to match the style of the Oncotarget journal, their order may be somewhat different.
RESULTS AND DISCUSSION
A new predictor as well as its web-server and user guide
A new and much more accurate sequence-based method, called iOri-Human, was developed for predicting the ORI sites in human DNA. In addition to the predictor’s high accuracy, it is also very important to make its web-server available to the public [40, 41]. Because only with this, can it be widely used by most experimental scientists. In view of this, the web-server for iOri-Human has also been established. Furthermore, a user’s guide is provided as follows.
(1) Click the web server at http://lin.uestc.edu.cn/server/iOri-Human.html, the top page of iOri-Human will be shown on your computer screen, as shown in Figure 2.
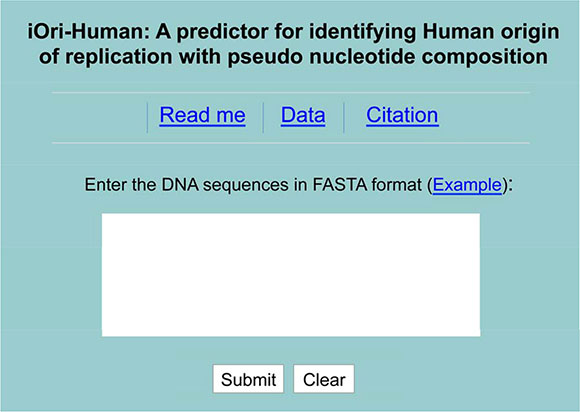
Figure 2: A semi-screenshot for the top-page of the iOri-Human web-server at http://lin.uestc.edu.cn/server/iOri-Human.html.
(2) In the input box at the center of Figure 2, type or copy/paste the query DNA sequences. The entered DNA sequences should be with the FASTA format. If not familiar with FASTA, click the button of Example.
(3) See the predicted results by clicking on the Submit button. If using the three query sequences in the Example window, you will see the following outcomes on your computer’s screen: the one for the first query sequence (with 300-bp long) is ‘Ori’; the one for the second query sequence (also with 300-bp long) is ‘non-Ori’; the one for the third query sequence (with 514-bp long) contains 514 – 300 + 1 = 215 sub-results, where the results for the segments from #1 to #134 are of ‘non-Ori’, those for the segments from #135 to #204 are of ‘Ori’, and those from #205 to #215 are of ‘non-Ori’, fully consistent with the experimental observations. The computational time is about a few seconds; of course, the more the number of query sequences, the longer the computational time will be.
(4) To get the benchmark dataset, click on the Data button.
(5) To find out the key relevant publications, click on the Citation button.
Caveats. The input query sequences should be 300 bp or longer, and expressed by DNA’s single-letter codes: ‘A’, ‘C’, ‘G’, and ‘T’.
The anticipated prediction accuracy
Listed in Table 1 are the success scores achieved by the predictor iOri-Human using the jackknife tests on the benchmark dataset (Supporting Information S1). Since it is the first predictor ever documented for identifying the ORI sites in human DNA sequences, it is not possible to demonstrate its power by a comparison with its published counterparts for exactly the same purpose. Nevertheless, it would be instructive to also list in Table 1 the corresponding optimal scores by the other machine-learning algorithms. As we can see from the table, the new iOri-Human achieved remarkably higher scores than its cohorts in almost all the four metrics, indicating clearly that the proposed new iOri-Human predictor is really very powerful. Note that, of the four metrics in Eq.8, the most important are the accuracy (Acc ) and Mathew’s correlation coefficient (MCC): the former reflects the overall accuracy of a predictor; while the latter, its stability in practical applications. The metrics sensitivity (Sn) and specificity (Sp) are used to measure a predictor from two different angles. When, and only when, both Sn and Sp of the predictor A are higher than those of the predictor B, can we say A is better than B. Actually, Sn and Sp are constrained with each other [42]. Therefore, it is meaningless to single out one from the two for making comparison. Accordingly, a really meaningful comparison in this regard should count the rates of both Sn and Sp, or even better the rate of their combination. That is exactly what MCC stand for.
Table 1: The success rates obtained by various machine-learning algorithms via jackknife tests on the benchmark dataset (Supporting Information S1)
Algorithm |
Sna |
Spa |
Acca |
MCCa |
AUCb |
|---|---|---|---|---|---|
iOri-Humanc |
0.762 |
0.739 |
0.75 |
0.501 |
0.835 |
SVMd |
0.688 |
0.544 |
0.616 |
0.400 |
0.651 |
Naive Bayes |
0.379 |
0.746 |
0.563 |
0.286 |
0.614 |
KNNe |
0.606 |
0.473 |
0.54 |
0.144 |
0.529 |
Decision Tree |
0.078 |
0.936 |
0.508 |
0.028 |
0.511 |
aSee Eq.8 for the definition of the metrics.
bAUC means the area under the ROC curves in Figure 3; the greater the AUC value is, the better the predictor will be [53, 54].
cThe proposed predictor in which the number of trees used was 100 with seed equal to 1.
dThe optimal parameters used for SVM were C= 0.5 and γ = 0.125.
eThe optimal parameters used for KNN (K nearest neighbor) was K = 1.
In studying complicated biological systems, graphical analysis is a very useful approach [43–52] due to its intuitivity. Here, let us use the ROC (receiver operating characteristic) graph [53, 54] to show the advantage of iOri-Human over its cohorts. The red graphic line in Figure 3 is the ROC curve for the iOri-Human predictor, while those of its cohorts are with different colors as directly marked on the figure. The area under the ROC curve is called AUC (area under the curve). The larger the area, the better the corresponding predictor [53, 54]. It can be seen from Figure 3, the iOri-Human has the largest AUC in comparison with its cohorts, once again indicating its power.
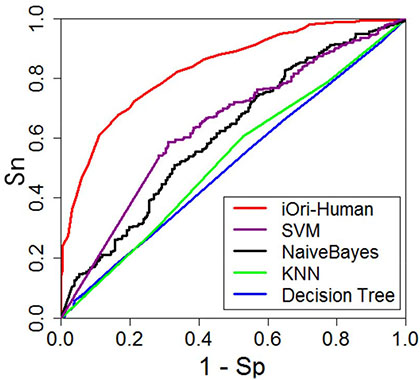
Figure 3: A graphical illustration to show the performances of iOri-Human and its cohorts via the ROC (receiver operating characteristic) curves [53, 54]. The area under the ROC curve is called AUC (area under the curve). The greater the AUC value is, the better the performance will be. See the text for further explanation.
MATERIALS AND METHODS
Benchmark dataset
The human ORIs data were collected from OriDB [55] (http://tubic.tju.edu.cn/deori/). These sequences were derived from Hela cell line. To construct a reliable benchmark dataset, the following steps were followed. (1) Collected were only experiment-confirmed data; thus we obtained 283 human ORIs with 300 bp in length. (2) For each of the 283 ORIs, extract a 300 bp segment from its upstream region at [-600 bp, -300 bp] as the corresponding non-ORI; a total 283 non-ORI samples were obtained. (3) Use the CD-HIT software [56] and set 0.75 as the threshold to remove redundant samples. Note that using 0.75 for the cutoff threshold was a compromise between reducing redundancy bias and keeping enough number of samples for statistical analysis. If further imposing more stringent cutoff, the number of DNA samples left would be too few to have statistical significance. Finally, we obtained 283 human OTI samples and 282 non-ORI samples.
In literature, the benchmark dataset usually consists of a training dataset and a testing dataset: the former is constructed for the purpose of training a proposed model, while the latter for the purpose of testing it. As pointed out by a comprehensive review [57], however, there is no need to separate a benchmark dataset into a training dataset and a testing dataset for validating a prediction method if it is tested by the jackknife or subsampling (K-fold) cross-validation since the outcome thus obtained is actually from a combination of many different independent dataset tests. Therefore, the benchmark dataset 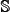 for the current study can be formulated as
for the current study can be formulated as

where the positive subset 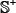 contains 283 human ORI samples, the negative subset
contains 283 human ORI samples, the negative subset  contains 282 non-ORI samples, and the symbol
contains 282 non-ORI samples, and the symbol  represents the union in the set theory. The 283 + 282 = 565 DNA samples are each consist of 300 bp, as can be generally formulated by
represents the union in the set theory. The 283 + 282 = 565 DNA samples are each consist of 300 bp, as can be generally formulated by
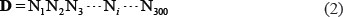
For readers’ convenience, their detained sequences are given in Supporting Information S1.
Pseudo k-tuple nucleotide composition
With the explosive growth of biological sequences generated in the post-genomic age, one of the most challenging problems in computational biology is how to formulate a biological sequence with a discrete model or vector, yet still considerably keep its sequence pattern or key feature. This is because almost all the existing machine-learning algorithms were developed to handle vector but not sequence samples, as elaborated in [40]. But a vector defined in a discrete model may completely lose this kind of sequence-pattern information. To overcome this problem, the “pseudo amino acid composition” [58, 59] or Chou’s PseAAC [60–62] was developed to deal with protein/peptide sequences. Ever since PseAAC was proposed, it has penetrated into many biomedicine/drug development areas [63, 64] and nearly all the areas of computational proteomics (see, e.g., [65–70] as well as a long list of references cited in [71, 72]). Encouraged by its successes in computational proteomics, the idea of PseAAC was recently extended to dealing with DNA/RNA sequences in many important problems of genome analysis [23–27, 33, 35, 38, 73, 74] by introducing the pseudo nucleotide composition or PseKNC [75–79].
According to a recent review paper [41], the general form of PseKNC for a DNA sequence can be formulated as
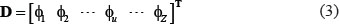
where T is the transpose operator, while Z an integer to reflect the vector’s dimension. The value of Z as well as the components 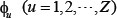 in Eq.3 will depend on how to extract the desired information from the DNA sequence. In the current study, we used the type-1 PseKNC [41], then the component in Eq.3 are given by
in Eq.3 will depend on how to extract the desired information from the DNA sequence. In the current study, we used the type-1 PseKNC [41], then the component in Eq.3 are given by
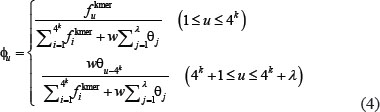
where 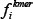 is the normalized occurrence frequency of the i-th kmer in the DNA sequence of Eq.2,
is the normalized occurrence frequency of the i-th kmer in the DNA sequence of Eq.2, 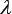 is the correlation tier used to reflect the long-range or global sequence pattern [41, 58], w is the factor used to adjust the weight between the local and global sequence coupling effects, and
is the correlation tier used to reflect the long-range or global sequence pattern [41, 58], w is the factor used to adjust the weight between the local and global sequence coupling effects, and 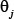 is the j-th structural correlation factor between all the j-th most contiguous dinucleotides as given by
is the j-th structural correlation factor between all the j-th most contiguous dinucleotides as given by
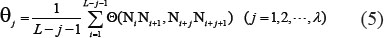
where the correlation factor 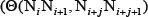 is given by
is given by
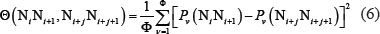
where 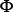 is the number of local DNA structural properties considered that is equal to 6 in the current study as will be explained below;
is the number of local DNA structural properties considered that is equal to 6 in the current study as will be explained below; 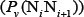 is the numerical value of the v-th physicochemical property for the dinucleotide at position i.
is the numerical value of the v-th physicochemical property for the dinucleotide at position i.
The spatial arrangements of any two successive base pairs could be characterized by six types of local structural parameters, of which three are local translational parameters (shift, slide and rise) and the other three are local angular parameters (twist, tilt and roll) [25, 26]. In recent years, more and more researches have demonstrated that the six DNA structural properties play important roles in many biological processes [80, 81]. There are 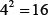 different dinucleotides, so the total number of local structural parameters is 6 × 16 = 96. Each of their parameter values can be found in Supplementary Table S1.
different dinucleotides, so the total number of local structural parameters is 6 × 16 = 96. Each of their parameter values can be found in Supplementary Table S1.
Before substituting these values into Eq.6, they were subjected to a standard conversion according to the following equation [82]
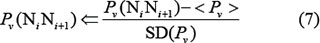
where the 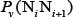 is the original value of the
is the original value of the  -th DNA physicochemical index for the dinucleotide
-th DNA physicochemical index for the dinucleotide 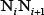 at position i; the symbol
at position i; the symbol  means the average value of the quantity therein for 16 different indices of dinucleotides, and SD denotes the corresponding standard deviation. The advantage to carry out the standard conversion is that the converted values obtained by Eq.7 will have a zero mean value over the 16 different indices, and will remain unchanged if they go through the same conversion procedure again. See Supplementary Table S2 for the corresponding values converted via Eq.7 from Supplementary Table S1.
means the average value of the quantity therein for 16 different indices of dinucleotides, and SD denotes the corresponding standard deviation. The advantage to carry out the standard conversion is that the converted values obtained by Eq.7 will have a zero mean value over the 16 different indices, and will remain unchanged if they go through the same conversion procedure again. See Supplementary Table S2 for the corresponding values converted via Eq.7 from Supplementary Table S1.
Random forest
The random forests (RF) algorithm is a very powerful algorithm, widely used in many areas of computational biology (see, e.g. [2, 31, 32, 34, 36, 39, 83–89]). The idea of RF is based on the ensemble of a large number of decision trees, with each giving a classification to choose the final outcome via a vote over all the trees in the forest. In this study, the number of trees is 100 and the seed is 1. The detailed procedures of RF and its formulation have been very clearly elaborated in [90], and hence there is no need to repeat here.
The predictor obtained via the aforementioned procedures is called iOri-Human, where “i” stands for “identify”, and “Ori-Human” for “human origin of replication”.
As stated in Introduction, how to objectively evaluate its anticipated success rates is an indispensable procedure for developing a useful predictor [28]. To realize this, we need to consider two issues: one is what metrics should be defined to measure the predictor’s quality; the other is what kind of test approach should be adopted to derive the metrics values. Below, let us address the two issues.
A set of four intuitive metrics and their definitions
As stated in [91], to quantitatively evaluate the quality of a predictor in performing binary classification, four metrics are usually needed. They are: (1) Acc to measure the predictor’s overall accuracy; (2) MCC, the stability; (3) Sn, the sensitivity; and (4) Sp, the specificity. Unfortunately, the conventional formulations for the four metrics are not quite intuitive and most experimental scientists feel difficult to understand them, particularly the stability of MCC. Fortunately, as elaborated in [22, 92], by using the Chou’s symbols and derivation in studying signal peptides [93], the conventional metrics can be converted into a set of four intuitive equations, as given below:
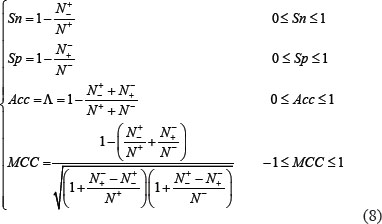
where 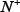 represents the total number of ORI samples investigated, while
represents the total number of ORI samples investigated, while 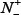 is the number of true ORIs incorrectly predicted to be of non-ORI;
is the number of true ORIs incorrectly predicted to be of non-ORI; 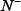 the total number of the non-ORI samples investigated, while
the total number of the non-ORI samples investigated, while 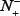 the number of the non-ORIs incorrectly predicted to be of ORI.
the number of the non-ORIs incorrectly predicted to be of ORI.
According to Eq.8, it is crystal clear to see the following. When 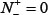 meaning none of the true ORI sequences are incorrectly predicted to be of non-ORI, we have the sensitivity
meaning none of the true ORI sequences are incorrectly predicted to be of non-ORI, we have the sensitivity 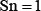 . When
. When 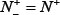 meaning that all the ORI samples are incorrectly predicted to be of non-ORI, we have the sensitivity
meaning that all the ORI samples are incorrectly predicted to be of non-ORI, we have the sensitivity 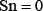 . Likewise, when
. Likewise, when 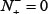 meaning none of the non-ORI samples are incorrectly predicted to be of ORI, we have the specificity
meaning none of the non-ORI samples are incorrectly predicted to be of ORI, we have the specificity 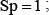 whereas
whereas 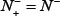 meaning that all the non-ORI sequences are incorrectly predicted to be of ORI, we have the specificity
meaning that all the non-ORI sequences are incorrectly predicted to be of ORI, we have the specificity 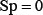 . When
. When 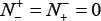 meaning that none of ORI samples in the positive dataset and none of the non-ORI samples in the negative dataset are incorrectly predicted, we have the overall accuracy
meaning that none of ORI samples in the positive dataset and none of the non-ORI samples in the negative dataset are incorrectly predicted, we have the overall accuracy 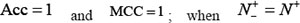 and
and 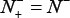 meaning that all the ORI samples in the positive dataset and all the non-ORI samples in the negative dataset are incorrectly predicted, we have the overall accuracy
meaning that all the ORI samples in the positive dataset and all the non-ORI samples in the negative dataset are incorrectly predicted, we have the overall accuracy 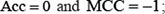 whereas when
whereas when 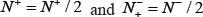 we have
we have 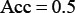 and
and 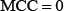 meaning no better than random guess. Therefore, Eq.8 has made the meanings of sensitivity, specificity, overall accuracy, and stability much more intuitive and easier-to-understand, particularly for the meaning of MCC, as concurred recently by many investigators (see, e.g., [24, 25, 27, 33, 38, 73, 86, 87, 89, 94–101]).
meaning no better than random guess. Therefore, Eq.8 has made the meanings of sensitivity, specificity, overall accuracy, and stability much more intuitive and easier-to-understand, particularly for the meaning of MCC, as concurred recently by many investigators (see, e.g., [24, 25, 27, 33, 38, 73, 86, 87, 89, 94–101]).
Note that, however, the set of equations defined in Eq.8 is valid only for the single-label systems. For the multi-label systems whose emergence has become more frequent in system biology [102–104] and system medicine [105] or biomedicine [39], a completely different set of metrics are needed as elaborated in [106].
Jackknife cross validation
With a set of intuitive metrics to measure the quality of a predictor, the next issue is what kind of validation method should be utilized to score these metrics. In statistics, the following three cross-validation methods are often used: (1) independent dataset test, (2) subsampling (or K-fold cross-validation) test, and (3) jackknife test [107]. Of these three, however, the jackknife test is deemed the least arbitrary that can always yield a unique outcome for a given benchmark dataset as elucidated in [28]. Accordingly, the jackknife test has been widely recognized and increasingly used by investigators to examine the quality of various predictors (see, e.g., [65–68, 70, 108–119]).
In view of this, here we also used the jackknife test to examine the quality of iOri-Human predictor. During the jackknifing process, both the training dataset and testing dataset are actually open, and each sample will be in turn moved between the two. The jackknife test can exclude the “memory” effect. Also, the arbitrariness problem as mentioned in [28] with the independent dataset and subsampling tests can be completely avoided because the outcome obtained by the jackknife cross-validation is always unique for a given benchmark dataset.
Optimize parameters
As we can see from Eqs.4–5, the new predictor contains three parameters: one is 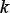 , the number of the nearest nucleotides considered to reflect the short-range or local pattern; one is λ, the number of the correlation tiers considered to reflect the long-range or global pattern; and one is
, the number of the nearest nucleotides considered to reflect the short-range or local pattern; one is λ, the number of the correlation tiers considered to reflect the long-range or global pattern; and one is 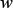 , the weight factor considered to adjust the effects between
, the weight factor considered to adjust the effects between 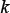 and λ. Their values will be determined via an optimization procedure according to various concrete problems. For the current study, the grid search for the optimal values of the three parameters was conducted within the scope given below
and λ. Their values will be determined via an optimization procedure according to various concrete problems. For the current study, the grid search for the optimal values of the three parameters was conducted within the scope given below

Where 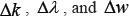 represent the step gaps for k,
represent the step gaps for k, 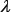 , and w, respectively. The reason why the search scope for
, and w, respectively. The reason why the search scope for 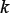 is limited under 4 is because the possible number of k-mers
is limited under 4 is because the possible number of k-mers 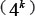 would be too large to be covered by the current benchmark dataset. As for the parameter
would be too large to be covered by the current benchmark dataset. As for the parameter 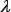 , generally speaking the greater it is, the more global sequence-order information the model will contain. However, if
, generally speaking the greater it is, the more global sequence-order information the model will contain. However, if 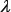 is too large, it would reduce the cluster-tolerant capacity [120] so as to lower down the cross-validation accuracy due to overfitting or “high dimension disaster” problem [121].
is too large, it would reduce the cluster-tolerant capacity [120] so as to lower down the cross-validation accuracy due to overfitting or “high dimension disaster” problem [121].
From Eq. 9, a total of 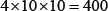 individual combinations were investigated for finding the optimal parameter combination. To reduce the computational time, the 10-fold cross-validation approach was used to assess the performances of the 400 combinations. Once the optimal values of the three parameters were determined, the rigorous jackknife test was adopted to calculate the scores for the four metrics defined in Eq.8 as well as the AUC in Figure 3. The final values thus obtained are given below
individual combinations were investigated for finding the optimal parameter combination. To reduce the computational time, the 10-fold cross-validation approach was used to assess the performances of the 400 combinations. Once the optimal values of the three parameters were determined, the rigorous jackknife test was adopted to calculate the scores for the four metrics defined in Eq.8 as well as the AUC in Figure 3. The final values thus obtained are given below
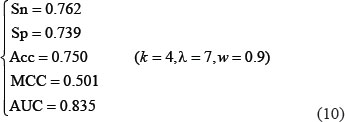
Also, see the results listed in Table 1, where the corresponding optimal parameters for various operation engines are also given.
CONCLUSIONS
One of the most important and fundamental processes in human cells is of the DNA replication. Knowledge of ORIs is crucial for in-depth understanding such a biological process, and hence computational method is highly demanded in this area. Unfortunately, it was very difficult to achieve decent success rates by computational approach. In the current model, both the local and global sequence patterns of DNA can be reflected via its physicochemical properties. That is why the iOri-Human predictor can yield remarkably high success rates. We anticipate that it will become a very useful high throughput tool for genome analysis.
ACKNOWLEDGMENTS AND FUNDING
The authors wish to thank the four anonymous reviewers for their constructive comments, which were very helpful for strengthening the presentation of this study. This work was supported by the Applied Basic Research Program of Sichuan Province (2015JY0100 and LZ-LY-45), the Scientific Research Foundation of the Education Department of Sichuan Province (11ZB122), the Nature Scientific Foundation of Hebei Province (C2013209105), the Fundamental Research Funds for the Central Universities of China (ZYGX2015J144 and ZYGX2015Z006), the Program for the Top Young Innovative Talents of Higher Learning Institutions of Hebei Province (BJ2014028), and the Outstanding Youth Foundation of North China University of Science and Technology (No. JP201502).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Halazonetis TD. Conservative DNA Replication. Nat Rev Mol Cell Bio. 2014; 15:300–300.
2. Xiao X, Ye HX, Liu Z. iROS-gPseKNC: predicting replication origin sites in DNA by incorporating dinucleotide position-specific propensity into general pseudo nucleotide composition. Oncotarget. 2016; 7:34180–34189. doi: 10.18632/oncotarget.9057.
3. Leonard AC, Mechali M. DNA replication origins. CSH Perspect Biol. 2013; 5:a010116.
4. Marczynski GT, Shapiro L. Bacterial chromosome origins of replication. Curr Opin Genet Dev. 1993; 3:775–782.
5. Schub O, Rohaly G, Smith RW, Schneider A, Dehde S, Dornreiter I, Nasheuer H-P. Multiple phosphorylation sites of DNA polymerase α-primase cooperate to regulate the initiation of DNA replication in vitro. J Biol Chem. 2001; 276:38076–38083.
6. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997; 61:212–238.
7. Foureau E, Courdavault V, Gallón SMN, Besseau S, Simkin AJ, Crèche J, Atehortùa L, Giglioli-Guivarc’h N, Clastre M, Papon N. Characterization of an autonomously replicating sequence in Candida guilliermondii. Microbiol Res. 2013; 168:580–588.
8. Dhar MK, Sehgal S, Kaul S. Structure, replication efficiency and fragility of yeast ARS elements. Res Microbiol. 2012; 163:243–253.
9. Crampton A, Chang F, Pappas DL, Frisch RL, Weinreich M. An ARS element inhibits DNA replication through a SIR2-dependent mechanism. Mol Cell. 2008; 30:156–166.
10. Méchali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Bio. 2010; 11:728–738.
11. Leman AR, Noguchi E. The replication fork: understanding the eukaryotic replication machinery and the challenges to genome duplication. Genes-Basel. 2013; 4:1–32.
12. Pursell ZF, Isoz I, Lundström E-B, Johansson E, Kunkel TA. Yeast DNA polymerase ε participates in leading-strand DNA replication. Science. 2007; 317:127–130.
13. Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem. 1994; 269:10923–10934.
14. Lubelsky Y, MacAlpine HK, MacAlpine DM. Genome-wide localization of replication factors. Methods. 2012; 57:187–195.
15. Ferris GR. Role of leadership in the employee withdrawal process: A constructive replication. J Appl Sport Psycho. 1985; 70:777.
16. Wold MS, Kelly T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. P Natl Acad Sci USA. 1988; 85:2523–2527.
17. Yin S, Deng W, Hu L, Kong X. The impact of nucleosome positioning on the organization of replication origins in eukaryotes. Biochem Biophy Res Com. 2009; 385:363–368.
18. Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. Conserved nucleosome positioning defines replication origins. Gene Dev. 2010; 24:748–753.
19. van Houten JV, Newlon CS. Mutational analysis of the consensus sequence of a replication origin from yeast chromosome III. Mol Cell Biol. 1990; 10:3917–3925.
20. Marsolier-Kergoat M-C. Asymmetry indices for analysis and prediction of replication origins in eukaryotic genomes. PLoS. 2012; 7:e45050.
21. Chen W, Lin H, Feng PM, Ding C. iNuc-PhysChem: A Sequence-Based Predictor for Identifying Nucleosomes via Physicochemical Properties. PLoS ONE. 2012; 7:e47843.
22. Chen W, Feng PM, Lin H, Chou KC. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition Ed: Insert a period Nucleic Acids Res. 2013; 41:e68.
23. Qiu WR, Xiao X, Chou KC. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int J Mol Sci (IJMS). 2014; 15:1746–1766.
24. Chen W, Feng PM, Deng EZ. iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal Biochem. 2014; 462:76–83.
25. Chen W, Feng PM, Lin H. iSS-PseDNC: identifying splicing sites using pseudo dinucleotide composition. Biomed Res Int. 2014; 2014:623149.
26. Guo SH, Deng EZ, Xu LQ, Ding H, Lin H. iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics. 2014; 30:1522–1529.
27. Lin H, Deng EZ, Ding H, Chen W. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014; 42:12961–12972.
28. Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J Theor Biol. 2011; 273:236–247.
29. Chen W, Ding H, Feng P, Lin H. iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget. 2016; 7:16895–16909. doi: 10.18632/oncotarget.7815.
30. Jia J, Liu Z, Xiao X, Liu B. iCar-PseCp: identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget. 2016; 7:34558–34570. doi: 10.18632/oncotarget.9148.
31. Qiu WR, Sun BQ, Xiao X, Xu ZC. iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget. 2016; 7:44310–44321. doi: 10.18632/oncotarget.10027.
32. Qiu WR, Xiao X, Xu ZH. iPhos-PseEn: identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget. 2016; 7:51270–51283. doi: 10.18632/oncotarget.9987.
33. Liu B, Fang L, Long R, Lan X. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics. 2016; 32:362–389.
34. Jia J, Liu Z, Xiao X, Liu B. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem. 2016; 497:48–56.
35. Liu Z, Xiao X, Yu DJ, Jia J. pRNAm-PC: Predicting N-methyladenosine sites in RNA sequences via physical-chemical properties. Anal Biochem. 2016; 497:60–67.
36. Jia J, Liu Z, Xiao X, Liu B. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol. 2016; 394:223–230.
37. Jia J, Zhang L, Liu Z, Xiao X. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics, doi: 101093/bioinformatics/btw387. 2016.
38. Liu B, Long R. iDHS-EL: Identifying DNase I hypersensi-tivesites by fusing three different modes of pseudo nucleotide composition into an en-semble learning framework. Bioinformatics. 2016; 32:2411–2418.
39. Qiu WR, Sun BQ, Xiao X, Xu ZC. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics; 2016. doi: 101093/bioinformatics/btw380. 2016.
40. Chou KC. Impacts of bioinformatics to medicinal chemistry. Med Chem. 2015; 11:218–234.
41. Chen W, Lin H, Chou KC. Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Mol BioSyst. 2015; 11:2620–2634.
42. Chou KC. A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. J Biol Chem. 1993; 268:16938–16948.
43. Jiang SP, Liu WM, Fee CH. Graph theory of enzyme kinetics: 1. Steady-state reaction system. Scientia Sinica. 1979; 22:341–358.
44. Forsen S. Graphical rules for enzyme-catalyzed rate laws. Biochem J. 1980; 187:829–835.
45. Zhou GP, Deng MH. An extension of Chou’s graphic rules for deriving enzyme kinetic equations to systems involving parallel reaction pathways. Biochem J. 1984; 222:169–176.
46. Chou KC. Graphic rules in steady and non-steady enzyme kinetics. J Biol Chem. 1989; 264:12074–12079.
47. Althaus IW, Gonzales AJ, Chou JJ, Kezdy FJ, Romero DL, Aristoff PA, Tarpley WG, Reusser F. The quinoline U-78036 is a potent inhibitor of HIV-1 reverse transcriptase. J Biol Chem. 1993; 268:14875–14880.
48. Althaus IW, Diebel MR, Romero DL, Aristoff PA, Tarpley WG, Reusser F. Steady-state kinetic studies with the non-nucleoside HIV-1 reverse transcriptase inhibitor U-87201E. J Biol Chem. 1993; 268:6119–6124.
49. Gonzales AJ, Diebel MR, Romero DL, Aristoff PA, Tarpley WG, Reusser F. Kinetic studies with the nonnucleoside HIV-1 reverse transcriptase inhibitor U-88204E. Biochemistry. 1993; 32:6548–6554.
50. Wu ZC, Xiao X. 2D-MH: A web-server for generating graphic representation of protein sequences based on the physicochemical properties of their constituent amino acids. J Theor Biol. 2010; 267:29–34.
51. Lin WZ, Xiao X. Wenxiang: a web-server for drawing wenxiang diagrams. Natural Science. 2011; 3:862–865.
52. Zhou GP. The disposition of the LZCC protein residues in wenxiang diagram provides new insights into the protein-protein interaction mechanism. J Theor Biol. 2011; 284:142–148.
53. Fawcett JA. An Introduction to ROC Analysis. Pattern Recogn Let. 2005; 27:861–874.
54. Davis J, Goadrich M. The relationship between Precision-Recall and ROC curves. Proceedings of the 23rd international conference on Machine learning. 2006; pp. 233–240.
55. Nieduszynski CA, Hiraga S-i, Ak P, Benham CJ, Donaldson AD. OriDB: a DNA replication origin database. Nucleic Acids Res. 2007; 35:D40–D46.
56. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659.
57. Chou KC, Shen HB. Review: Recent progresses in protein subcellular location prediction. Anal Biochem. 2007; 370:1–16.
58. Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS: (Erratum: ibid, 2001, Vol44, 60). 2001; 43:246–255.
59. Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005; 21:10–19.
60. Cao DS, Xu QS, Liang YZ. propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics. 2013; 29: 960–962.
61. Du P, Gu S, Jiao Y. PseAAC-General: Fast building various modes of general form of Chou’s pseudo amino acid composition for large-scale protein datasets. Int J Mol Sci. 2014; 15:3495–3506.
62. Lin SX, Lapointe J. Theoretical and experimental biology in one —A symposium in honour of Professor Kuo-Chen Chou’s 50th anniversary and Professor Richard Giegé’s 40th anniversary of their scientific careers. J Biomed Sci Eng (JBiSE). 2013; 6:435–442.
63. Zhong WZ, Zhou SF. Molecular science for drug development and biomedicine. Int J Mol Sci. 2014; 15:20072–20078.
64. Zhou GP, Zhong WZ. Perspectives in Medicinal Chemistry. Curr Top Med Chem. 2016; 16:381–382.
65. Kabir M, Hayat M. iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou’s PseAAC to formulate DNA samples. Mol Genet Genomics. 2016; 291:285–296.
66. Dehzangi A, Heffernan R, Sharma A, Lyons J, Paliwal K, Sattar A. Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou’s general PseAAC. J Theor Biol. 2015; 364:284–294.
67. Kumar R, Srivastava A, Kumari B, Kumar M. Prediction of beta-lactamase and its class by Chou’s pseudo amino acid composition and support vector machine. J Theor Biol. 2015; 365:96–103.
68. Mondal S, Pai PP. Chou’s pseudo amino acid composition improves sequence-based antifreeze protein prediction. J Theor Biol. 2014; 356:30–35.
69. Wang X, Zhang W, Zhang Q, Li GZ. MultiP-SChlo: multi-label protein subchloroplast localization prediction with Chou’s pseudo amino acid composition and a novel multi-label classifier. Bioinformatics. 2015; 31:2639–2645.
70. Tang H, Chen W, Lin H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Mol Biosyst. 2016; 12:1269–1275.
71. Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteomics. 2009; 6: 262–274.
72. Chou KC. An unprecedented revolution in medicinal science (doi:10.3390/MOL2NET-1-b040). Proceedings of the MOL2NET (International Conference on Multidisciplinary Sciences) 2015; 1:1–10.
73. Chen W, Feng P, Ding H, Lin H. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal Biochem (also, Data in Brief, 2015, 5: 376–378). 2015; 490:26–33.
74. Liu B, Fang L, Liu F, Wang X. iMiRNA-PseDPC: microRNA precursor identification with a pseudo distance-pair composition approach. J Biomol Struct Dyn. 2016; 34:223–235.
75. Chen W, Lei TY, Jin DC, Lin H. PseKNC: a flexible web-server for generating pseudo K-tuple nucleotide composition. Anal Biochem. 2014; 456:53–60.
76. Chen W, Zhang X, Brooker J, Lin H. PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics. 2015; 31:119–120.
77. Liu B, Liu F, Fang L, Wang X. repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics. 2015; 31:1307–1309.
78. Liu B, Liu F, Fang L, Wang X. repRNA: a web server for generating various feature vectors of RNA sequences. Mol Genet Genomics. 2016; 291:473–481.
79. Liu B, Liu F, Wang X, Chen J, Fang L. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015; 43(W1):W65-W71.
80. Zuo Y-C, Li Q-Z. The hidden physical codes for modulating the prokaryotic transcription initiation. Physica A. 2010; 389:4217–4223.
81. Soltani S, Askari H, Ejlali N, Aghdam R. The structural properties of DNA regulate gene expression. Mol BioSyst. 2014; 10:273–280.
82. Chou KC, Shen HB. Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-nearest neighbor classifiers. J Proteome Res. 2006; 5:1888–1897.
83. Kandaswamy KK, Moller S, Suganthan PN, Sridharan S, Pugalenthi G. AFP-Pred: A random forest approach for predicting antifreeze proteins from sequence-derived properties. J Theor Biol. 2011; 270:56–62.
84. Lin WZ, Fang JA, Xiao X. iDNA-Prot: Identification of DNA Binding Proteins Using Random Forest with Grey Model. PLoS ONE. 2011; 6:e24756.
85. Pugalenthi G, Kandaswamy KK, Kolatkar P. RSARF: Prediction of Residue Solvent Accessibility from Protein Sequence Using Random Forest Method. Protein Pept Lett. 2012; 19:50–56.
86. Jia J, Liu Z, Xiao X. iPPI-Esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J Theor Biol. 2015; 377:47–56.
87. Jia J, Liu Z, Xiao X, Liu B. Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition (iPPBS-PseAAC). J Biomol Struct Dyn. 2016; 34:1946–1961.
88. Jia J, Liu Z, Xiao X, Liu B. iPPBS-Opt: A Sequence-Based Ensemble Classifier for Identifying Protein-Protein Binding Sites by Optimizing Imbalanced Training Datasets. Molecules. 2016; 21:95.
89. Qiu WR, Sun BQ, Xiao X. iPhos-PseEvo: Identifying human phosphorylated proteins by incorporating evolutionary information into general PseAAC via grey system theory. Mol Inform. 2016; doi:10.1002/minf.201600010.
90. Breiman L. Random forests. Machine learning. 2001; 45:5–32.
91. Chen J, Liu H, Yang J. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids. 2007; 33: 423–428.
92. Xu Y, Ding J, Wu LY. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS ONE. 2013; 8:e55844.
93. Chou KC. Prediction of protein signal sequences and their cleavage sites. Proteins. 2001; 42:136–139.
94. Xu Y, Shao XJ, Wu LY. iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013; 1:e171.
95. Ding H, Deng EZ, Yuan LF, Liu L, Lin H, Chen W. iCTX-Type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed Res Int. 2014; 2014:286419.
96. Liu B, Fang L, Liu F, Wang X. Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS ONE. 2015; 10:e0121501.
97. Liu B, Fang L, Wang S, Wang X, Li H. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J Theor Biol. 2015; 385:153–159.
98. Xiao X, Min JL, Lin WZ, Liu Z, Cheng X. iDrug-Target: predicting the interactions between drug compounds and target proteins in cellular networking via the benchmark dataset optimization approach. J Biomol Struct Dyn . 2015; 33:2221–2233.
99. Liu Z, Xiao X, Qiu WR. iDNA-Methyl: Identifying DNA methylation sites via pseudo trinucleotide composition. Anal Biochem (also, Data in Brief, 2015, 4: 87–89). 2015; 474:69–77.
100. Chen W, Feng P, Ding H, Lin H. Using deformation energy to analyze nucleosome positioning in genomes. Genomics. 2016; 107:69–75.
101. Chen W, Tang H, Ye J, Lin H. iRNA-PseU: Identifying RNA pseudouridine sites. Molecular Therapy - Nucleic Acids 2016; 5:e332.
102. Chou KC, Wu ZC, Xiao X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol BioSyst. 2012; 8:629–641.
103. Lin WZ, Fang JA, Xiao X. iLoc-Animal: A multi-label learning classifier for predicting subcellular localization of animal proteins. Mol BioSyst. 2013; 9:634–644.
104. Xiao X, Wu ZC. iLoc-Virus: A multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites. J Theor Biol. 2011; 284:42–51.
105. Xiao X, Wang P, Lin WZ, Jia JH. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem. 2013; 436:168–177.
106. Chou KC. Some Remarks on Predicting Multi-Label Attributes in Molecular Biosystems. Mol BioSyst. 2013; 9:1092–1100.
107. Chou KC, Zhang CT. Review: Prediction of protein structural classes. Crit Rev Biochem Mol Biol. 1995; 30:275–349.
108. Zhou GP. An intriguing controversy over protein structural class prediction. J Protein Chem. 1998; 17:729–738.
109. Zhou GP, Assa-Munt N. Some insights into protein structural class prediction. Proteins. 2001; 44:57–59.
110. Cai YD, Zhou GP. Support vector machines for predicting membrane protein types by using functional domain composition. Biophys J. 2003; 84:3257–3263.
111. Zhou GP, Doctor K. Subcellular location prediction of apoptosis proteins. Proteins. 2003; 50:44–48.
112. Shen HB, Yang J. Euk-PLoc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids. 2007; 33:57–67.
113. Chou KC, Cai YD. Prediction and classification of protein subcellular location: sequence-order effect and pseudo amino acid composition. J Cell Biochem (Addendum, ibid 2004, 91, 1085). 2003; 90:1250–1260.
114. Chou KC, Cai YD. Prediction of membrane protein types by incorporating amphipathic effects. J Chem Inf Model. 2005; 45:407–413.
115. Fan GL, Zhang XY, Liu YL, Nang Y, Wang H. DSPMP: Discriminating secretory proteins of malaria parasite by hybridizing different descriptors of Chou’s pseudo amino acid patterns. J Comput Chem. 2015; 36:2317–2327.
116. Khan ZU, Hayat M, Khan MA. Discrimination of acidic and alkaline enzyme using Chou’s pseudo amino acid composition in conjunction with probabilistic neural network model. J Theor Biol. 2015; 365:197–203.
117. Ali F, Hayat M. Classification of membrane protein types using Voting Feature Interval in combination with Chou’s Pseudo Amino Acid Composition. J Theor Biol. 2015; 384:78–83.
118. Ahmad K, Waris M, Hayat M. Prediction of Protein Submitochondrial Locations by Incorporating Dipeptide Composition into Chou’s General Pseudo Amino Acid Composition. J Membr Biol. 2016; 249:293–304.
119. Ju Z, Cao JZ, Gu H. Predicting lysine phosphoglycerylation with fuzzy SVM by incorporating k-spaced amino acid pairs into Chou’s general PseAAC. J Theor Biol. 2016; 397:145–150.
120. Chou KC. A key driving force in determination of protein structural classes. Biochem Biophy Res Com. 1999; 264:216–224.
121. Wang T, Yang J, Shen HB. Predicting membrane protein types by the LLDA algorithm. Protein Pept Lett. 2008; 15:915–921.