
Introduction
Renal fibrosis is an inevitable outcome of all types of progressive chronic kidney disease. During the development and progression of renal fibrosis, alteration of the epithelial phenotype of renal tubular cells could impair the epithelial integrity and cellular function, which might subsequently trigger the fibrotic response in the kidney [1, 2]. The phenotypic conversion of epithelial cells involves the loss of epithelial proteins, such as E-cadherin, zonula occludens-1 (ZO-1) and cytokeratin, and the acquisition of new mesenchymal markers including vimentin, α-smooth muscle actin (α-SMA), fibroblast-specific protein-1 (FSP1), interstitial matrix components type I collagen, and fibronectin [3]. The loss of cell adhesion is accompanied by cytoskeletal remodeling and morphological changes, resulting in tubular basement membrane disruption. Consequently, these cells possess the ability to migrate from the tubule basement membrane into the interstitium [4]. Moreover, emerging evidence suggested that pathological insult-activated renal tubular cells might stimulate the proliferation of fibroblast cells located in the tubulointerstitial region via the transduction of fibrogenic signals [5].
PPAR-γ is a ligand-activated transcription factor and belongs to the nuclear hormone receptor superfamily. PPAR-γ is highly expressed in adipose tissue and plays a crucial role in adipocyte differentiation [6]. Moreover, PPAR-γ regulates a diverse spectrum of physiological and pathological processes including insulin sensitivity, lipid metabolism, inflammation, immune regulation, and extracellular matrix balance [7, 8]. In the kidney, PPAR-γ is expressed in all types of glomerular, tubular, and tubulointerstitial cells and is of importance in the pathogenesis of many types of kidney diseases [8, 9]. In the past decade, accumulating evidence from systemic activation of PPAR-γ showed an antifibrotic effect in CKD models [10-12]. However, the cell-specific role of PPAR-γ in modulating renal fibrosis remains uncertain.
In the present study, employing proximal tubule-specific PPAR-γ knockout (KO) mice and the PPAR-γ agonists rosiglitazone (RGZ) and 15-deoxyprostaglandin J2 (15d-PGJ2), we studied the role of renal tubular epithelium-targeted PPAR-γ in regulating the fibrotic response in vitro and in vivo and the potential mechanisms.
Results
TGF-β1 suppressed PPAR-γ expression and activity in HK-2 cells
HK-2 cells were used in all vitro experiment. First, we evaluated the influence of TGF-β1 on PPAR-γ expression and activity. After treatment with TGF-β1 (5 ng/ml) for 24 h, PPAR-γ mRNA expression was significantly decreased as determined by qRT-PCR (Figure 1A). Similarly, PPAR-γ protein expression was reduced in a dose-dependent manner (1.0, 2.5, 5.0 ng/ml) (Figure 1B & 1C). TGF-β1 strikingly inhibited PPAR-γ activity in time- and dose-dependent manners as detected by a PPAR-γ transcription factor assay kit (Figure 1D & 1E). These data indicate a potent role of TGF-β1 in suppressing PPAR-γ expression and activity.
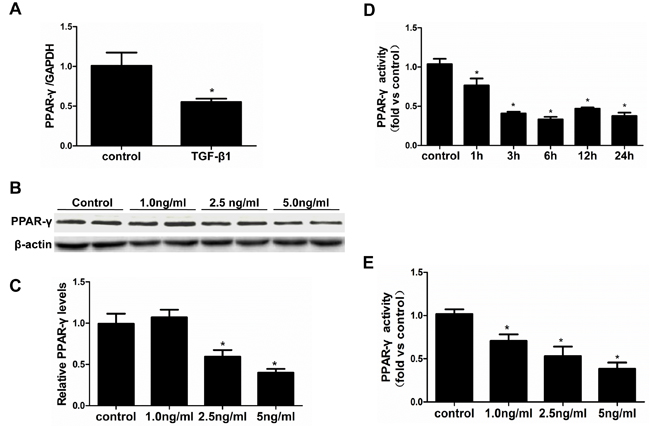
Figure 1: TGF-β1 suppressed PPAR-γ expression and activity in HK-2 cells. The cells were grown on 6-well plates until 80% confluence and then treated with vehicle or TGF-β1 for 24h. A. qRT-PCR analysis of PPAR-γ after treated with TGF-β1 (5ng/ml). B. Representative immunoblots. C. Densitometric analysis of PPAR-γ protein expression. D. & E. PPAR-γ transcription factor assay kit was used to detect PPAR-γ activity at different time points D. and different doses E.. Values are means ± SE, n = 6 in each group, * P < 0.05 vs. control.
Oxidative stress mediated the TGF-β1 effect on PPAR-γ reduction and cellular phenotype alteration
Oxidative stress has extensive actions in modulating physiological and pathological responses. We tested the potential of oxidative stress in mediating the TGF-β1 effect on PPAR-γ reduction and cellular phenotype alteration. First, we observed that TGF-β1 dose- and time-dependently elevated reactive oxygen species (ROS) production (Figure 2A & 2B). Next, the application of H2O2 similarly mimicked PPAR-γ expression (Figure 2C-2E).
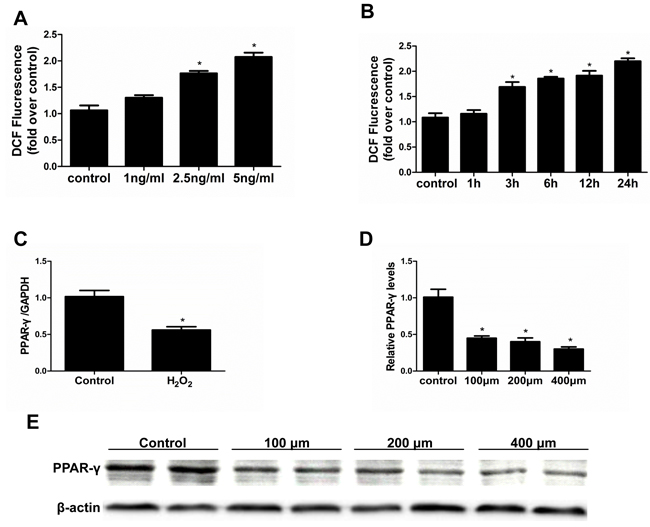
Figure 2: Effect of H2O2 administration on PPAR-γ expression. Cells were grown on 6-well plates until 80% confluence and then treated with TGF-β1 (0, 1, 2.5, 5 ng/ml) for 2 hours or treated with vehicle or H2O2 (200 µm) A. or cells were treated with TGF-β1 (5ng/ml) at different time points (0, 1, 3, 6, 12, 24h) B. then ROS levels were analyzed by flow cytometry. C. mRNA expression of PPAR-γ was detected by qRT-PCR. D. Densitometric analysis of PPAR-γ expression. E. Western-blot for PPAR-γ expression Values are means ± SE, n = 6 in each group, * P < 0.05 vs. control.
To define the contribution of different sources of ROS on the suppression of PPAR-γ, the mitochondrial inhibitor rotenone, NADPH oxidase inhibitor apocynin, and a nonspecific antioxidant N-acetyl-L-cysteine (NAC) were applied to treat the cells. As shown in Figure 3A-3C, NAC significantly blocked TGF-β1-induced PPAR-γ reduction, whereas the mitochondrial respiratory chain complex I inhibitor rotenone and the NADPH oxidase inhibitor apocynin partially prevented TGF-β1-induced PPAR-γ downregulation. The increase of α-SMA and decrease of E-cadherin were abolished by NAC, contrasting to a partial effect of rotenone or apocynin. indicating that mitochondria- and NADPH oxidase-derived ROS contributed to this pathological process.
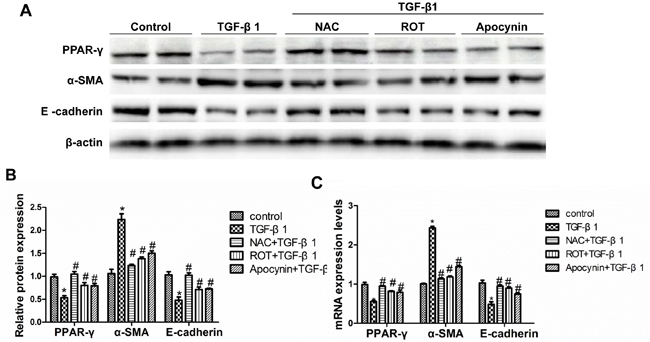
Figure 3: Inhibition of TGF-β1-induced oxidative stress restored PPAR-γ downregulation and the alteration of cellular phenotype. The cells were grown on 6-well plates until 80% confluence and then pretreated with NAC (5 mM), ROT (10 µmol/L), and Apocynin (500 µmol/L) for 30 min followed by TGF-β1 treatment for another 24h. A. Western-blot for PPAR-γ, α-SMA and E-cadherin expression. B. Densitometric analysis of the immunoblots for α-SMA and E-cadherin. C. qRT-PCR analysis of PPAR-γ, α-SMA and E-cadherin mRNA expression. Values are means ± SE, n = 6 in each group, * P < 0.05 vs. Control, # P < 0.05 vs. TGF-β1-treated group.
PPAR-γ activation prevented the cellular phenotype alteration induced by TGF-β1
To further study the role of PPAR-γ in antagonizing TGF-β1 in altering the epithelial cell phenotype, the PPAR-γ agonists RGZ and 15d-PGJ2 were administered to the cells with or without the PPAR-γ inhibitor T0070907. As shown in Figure 4A-4E, RGZ and 15d-PGJ2 restored the induction of α-SMA and the reduction of E-cadherin, which was abolished by a specific PPAR-γ inhibitor T0070907. To rule out a PPAR-γ-independent effect, we overexpressed PPAR-γ in HK-2 cells (Figure 5A-5B). As expected, PPAR-γ overexpression showed a similar effect as PPAR-γ agonists in protecting the epithelial phenotype in HK-2 cells (Figure 5C-5F).
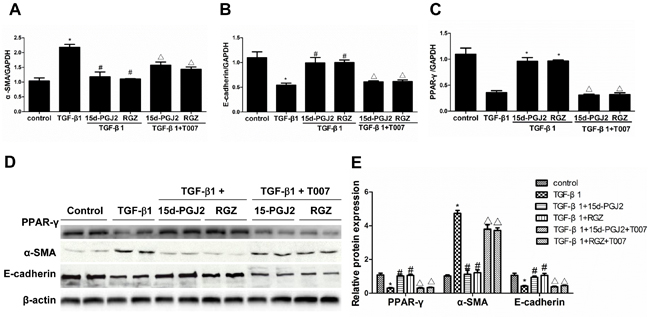
Figure 4: PPAR-γ activation inhibited TGF-β1-induced alteration of cellular phenotype. The cells were grown in 6-well plates until 80% confluence and then pretreated with RGZ or 15d-PGJ2 for 30 min followed by TGF-β1(10 ng/ml) treatment for 72h in the presence or absence of PPAR-γ inhibitor T0070907. A.-C. qRT-PCT analysis of α-SMA, E-cadherin, and PPAR-γ mRNA expression. D. Western blots of PPAR-γ, α-SMA, and E-cadherin. E. Densitometric analysis of PPAR-γ, α-SMA, and E-cadherin. Values are means ± SE. Values are means ± SE, n = 6 in each group, * P < 0.01 vs. control group, # P < 0.01 vs. TGF-β1-treated group, △P < 0.01 vs. PPAR-γ agonists (RGZ or 15d-PGJ2)-treated group.
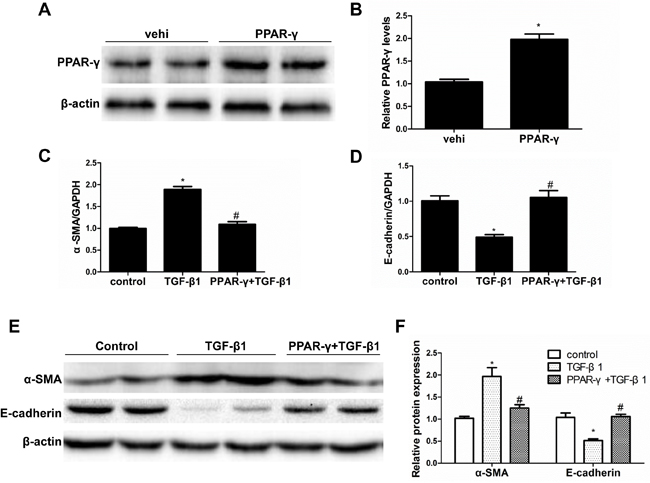
Figure 5: PPAR-γ overexpression blocked TGF-β1-induced phenotypic alteration. A. Western blotting analysis of PPAR-γ in HK-2 cells transfected with PPAR-γ or empty vector (vehicle). B. Densitometric analysis of PPAR-γ. C. qRT-PCR analysis of α-SMA. D. qRT-PCR analysis of E-cadherin. E. Western blots of E-cadherin and α-SMA. F. Densitometric analysis of E-cadherin and α-SMA. Values are means ± SE, n= 6 in each group, * P < 0.05 vs. Control, # P < 0.05 vs. TGF-β1-treated group.
Activation of PPAR-γ inhibited TGF-β1-induced ROS generation
In this experiment, we examined PPAR-γ action in modulating TGF-β1-indued ROS production. TGF-β1-induced ROS production was entirely abolished by treatment with RGZ or 15d-PGJ2. However, such an effect was remarkably blunted by a PPAR-γ antagonist (Figure 6A-6G).
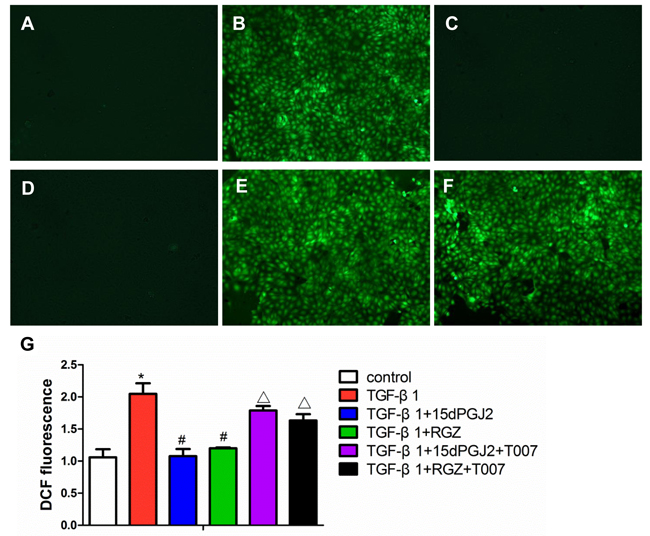
Figure 6: Activation of PPAR-γ inhibited TGF-β1-induced ROS production. Confluent HK-2 cells in chamber slides were pretreated with PPAR-γ agonists and inhibitors for 30 min and then exposed to TGF-β1 for another 2h in the presence of DCFDA. A. control. B. 10 ng/ml TGF-β1. C. 10 ng/ml TGF-β1 plus 15d-PGJ2. D. 10 ng/ml TGF-β1 plus RGZ. E. 10 ng/ml TGF-β1 plus 15d-PGJ2 and T0070907. F. 10 ng/ml TGF-β1 plus RGZ and T0070907. G. Quantitation of DCF fluorescence. Fluorescence was quantified using FLUOstar OPTIMA. Values are means ± SE, n = 6 in each group, * P < 0.05 vs. Control, # P < 0.05 vs. TGF-β1-treated group. ## P < 0.05 vs. TGF-β1plus RGZ or 15d-PGJ2 groups.
Proximal tubule epithelium-specific ablation of PPAR-γ aggravated renal fibrosis
To investigate the in vivo role of renal epithelial PPAR-γ in opposing fibrogenesis, we generated mice with specific PPAR-γ deletions in the proximal tubular epithelial cells (Figure 7A-7C). Following a 14-day UUO, the PT-PPAR-γ-CKO mice lost more of the epithelial phenotype, shown by a greater elevation of α-SMA, and more loss of E-cadherin (Figure 7D-7F). The PT-PPAR-γ-CKO mice developed more severe tubulointerstitial fibrosis than the WT controls, as evidenced by the greater deposition of extracellular matrix proteins (Figure 8A-8E). These data offered the first in vivo evidence showing that renal tubular epithelium-targeted PPAR-γ plays an important role in maintaining the epithelial phenotype and antagonizing the fibrogenic response.
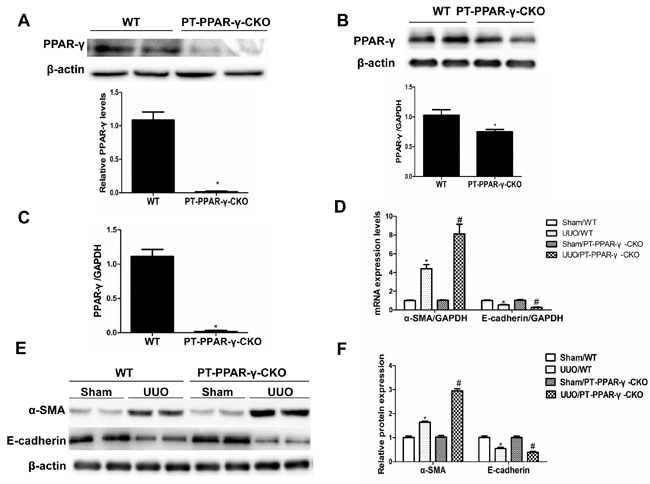
Figure 7: Proximal tubule epithelium-specific ablation of PPAR-γ aggravated the alteration of epithelial phenotype following UUO. A. Western blotting analysis of PPAR-γ protein expression in primary proximal tubule cells from WT and PT-PPAR-γ-CKO mice. B. Whole kidney PPAR-γ expression from WT and PT-PPAR-γ-CKO mice. C. PPAR-γ mRNA expression of primary proximal tubule cells from WT or PT-PPAR-γ-CKO mice. D. qRT-PCR analysis of E-cadherin and α-SMA. E. Western blotting analysis of E-cadherin and α-SMA. F. Densitometric analysis of E-cadherin and α-SMA. Values are means ± SE, n = 6 in each group, * P < 0.01 vs. Sham/WT, # P< 0.01 vs. UUO/WT.
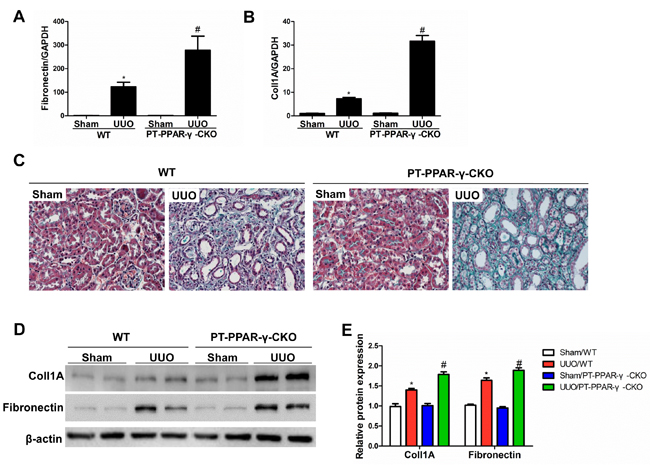
Figure 8: Proximal tubule epithelium-specific ablation of PPAR-γ aggravated renal fibrosis. A. qRT-PCR analysis of fibronectin. B. qRT-PCR analysis of collagen-1. C. Representative images of Masson’s trichrome staining. D. Western blotting analysis of TGF-β1, collagen-1 and fibronectin. E. Densitometric analysis of collagen-1 and fibronectin. Values are means ± SE, n = 6 in each group, * P < 0.01 vs. Sham/WT, # P < 0.01 vs. UUO/WT.
Rosiglitazone attenuated renal fibrosis in WT mice but not in PT-PPAR-γ-CKO mice
Along the whole nephron, PPAR-γ is expressed in glomeruli, proximal tubules, and distal nephron. To further figure out the contribution of proximal tubular PPAR-γ in antagonizing fibrotic response in UUO model, we treated UUO WT and UUO PT-PPAR-γ-CKO mice with a specific PPAR-γ agonist RGZ. As shown in Figure 9, RGZ could ameliorate fibrosis and epithelial phenotype transition in WT mice but not in KO mice, suggesting that PPAR-γ in proximal tubules but not in other nephron segments or cell types played a key role in opposing renal fibrotic response in this experimental setting.
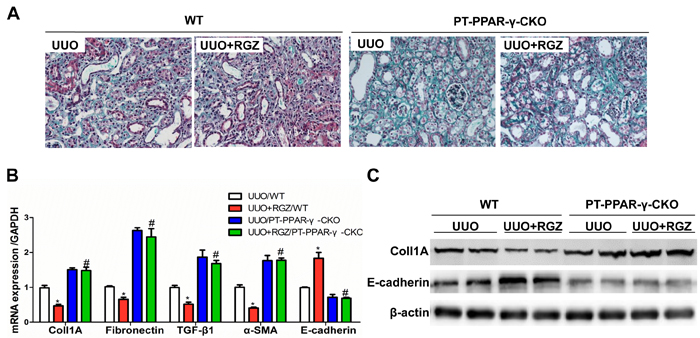
Figure 9: RGZ treatment attenuated renal fibrosis in WT mice but not in PT-PPAR-γ-CKO mice. A. Representative images of Masson’s trichrome staining. B. qRT-PCR analysis of fibronectin, collagen-1, TGF-β1, E-cadherin, and α-SMA. C.. Western blotting analysis of collagen-1 and E-cadherin. Values are means ± SE, n = 6 in each group, * P < 0.01 vs. Sham/WT, # P < 0.01 vs. UUO/WT.
Discussion
Renal fibrosis is a common outcome of all types of CKDs. Although efforts have been made in the field, the pathogenic mechanisms remain uncertain. There is still no effective therapy against renal fibrosis. In recent years, PPAR-γ has drawn substantial attention for the treatment of kidney diseases including CKD-related renal fibrosis [7, 13]. However, the detailed molecular mechanisms and the role of cell-specific PPAR-γ in antagonizing fibrosis are unknown. In this study, we demonstrated that TGF-β1 suppressed PPAR-γ expression and activity via enhanced oxidative stress, and activated the PPAR-γ protected renal tubular cells via suppressing oxidative response. In agreement with the in vitro results, the conditional deletion of PPAR-γ in the tubular epithelium aggravated renal fibrosis and the tubular epithelial phenotype transition.
Accumulating evidence demonstrated that TGF-β1 serves as a principal mediator of fibrogenesis in the majority of organs [14, 15]. TGF-β1 alters the cellular phenotype and promotes renal fibrosis via its downstream components of Smads [2, 16]. TGF-β1 attenuates myocardial PPAR-γ expression in pressure overload-induced cardiac fibrosis and remodeling in mice [17]. Consistent with the previous findings, TGF-β1 treatment in renal epithelial cells caused typical changes including loss of E-cadherin, and de novo expression of α-SMA, indicating a fibrotic phenotype. In agreement with the alteration of the cellular phenotype, PPAR-γ was down-regulated by TGF-β1, suggesting a potential role of PPAR-γ in opposing the TGF-β1 effect.
PPAR-γ is a ligand-activated transcription factor and belongs to the nuclear hormone receptor superfamily. Ligands for PPAR-γ include a variety of natural and synthetic compounds. We demonstrated that TGF-β1 dose-dependently decreased PPAR-γ expression and increased oxidative stress. The formation of ROS is a natural consequence of aerobic metabolism and is integral for maintaining tissue oxygen homeostasis [18]. ROS are also important in the coordination and activation of numerous signal transduction pathways [19]. However, excessive ROS could cause cell damage and organ injury. In cultured HK-2 cells, TGF-β1-induced ROS was blocked by RGZ (a synthetic PPAR-γ agonist) and 15d-PGJ2 (a natural PPAR-γ agonist), demonstrating an anti-oxidative role of PPAR-γ. We determined that H2O2, a classical oxidant, mimicked the TGF-β1 effect in suppressing PPAR-γ and altering the epithelial phenotype. These data suggest a mutual antagonism between PPAR-γ and TGF-β1/ROS axis in renal epithelial cells. Moreover, using the mitochondrial inhibitor rotenone and the NADPH oxidase inhibitor apocynin, we showed that mitochondria- and NADPH oxidase-derived ROS participated in this pathological process. To rule out the nonspecific effect of PPAR-γ agonists, we performed another experiment by genetically overexpressing PPAR-γ in HK-2 cells. In this experiment, we observed a similar effect as RGZ and 15d-PGJ2 in the prevention of cellular phenotypic changes. PPAR-γ ligands including rosiglitazone, ciglitazone, and troglitazone and other members of TZD family, are popular used in gypoglycemic therapy for type 2 diabetes. PPAR-γ agonists could ameliorate renal fibrotic lesions in chronic kidney diseases including diabetic nephropathy. Studies in proximal epithelial cells, such as NRK52E and HK2 cell lines, shown PPAR-γ ligands play protective role on anti-fibrosis and TGF-β/Smads signaling is considered the most important pathway in EMT and fibrosis. In renal tubular epithelial cells, Li R et al shown curcumin blocked TGF-β1-induced EMT via ERK-dependent and then PPAR-γ-dependent pathway but not Smad2 or Smad3 [20]. Wei et al found CDDO, a synthetic PPAR-γ agonist, attenuates fibrogenesis by antagonistically targeting canonical TGF-β/Smad and Akt signaling in a PPAR-γ-independent manner [21]. In human lung fibroblasts, AA Kulkarni et al shown that PPAR-γ ligands play an anti-fibrotic role through inhibiting TGF-β1-induced Akt phosphorylation, the inhibition is independent of MAPK-p38 and PTEN but is dependent on TGF-β1-induced phosphorylation of FAK, a kinase that acts upstream of Akt [22].
PPAR-γ is expressed in multiple renal cell type including the cultured glomerular mesangial cells, podocytes, proximal epithelial cells [23, 24], and dominantly expressed in the collecting system of the mammalian urinary tract, including connective renal tubules and collecting ducts. The PPAR-γ ligand TZDs affect extracellular matrix and cytoskeleton remodeling and metabolic processes of the kidney collecting duct epithelia including carbohydrate, lipid metabolism [25, 26]. Studies shown activation of PPAR-γ in the distal collecting system causes severe water retention, a common side effect after thiazolidinedione treatment [27, 28]. Zhang H et al shown collecting duct-specific deletion of PPAR-γ blocks thiazolidinedione-induced fluid retention, rosiglitazone stimulated sodium transport in primary cultures of CD cells expressing PPAR-γ and not in cells lacking this receptor [29]. However, whether PPAR-γ in connective renal tubules and collecting ducts plays a similar role as proximal tubule in the pathogenesis of renal fibrosis have not be reported. In our study, rosiglitazone diminished renal fibrosis in WT mice, but not in mice with specific proximal epithelial cell PPAR-γ deletion.
Recent studies identified that PPAR-γ agonists could inhibit renal fibroblast activation and the fibrosis of tubule cells [12, 30, 31]. To further evaluate if epithelium-targeted PPAR-γ plays a role in maintaining the normal cellular phenotype and antagonizing fibrosis, we specifically deleted PPAR-γ in the proximal tubular cells of mice. Following 14 days of UUO, the KO mice exhibited a remarkable deterioration in the tubulointerstitial fibrosis as evidenced by the greater deposition of extracellular matrix. The loss of E-cadherin and the gain of α-SMA were greater in the KO mice, suggesting a more serious change in the epithelial phenotype. These important data offered the first evidence showing an in vivo role of renal tubular PPAR-γ in opposing renal fibrosis and maintaining normal cellular phenotypes possibly via antagonizing TGF-β signaling in both epithelium and fibroblasts.
Employing genetic and pharmacological strategies, we demonstrated an essential role of renal tubular PPAR-γ in protecting kidneys against fibrosis and cellular phenotype alteration possibly via antagonizing TGF-β1 and TGF-β1-induced oxidative stress. The findings enriched our understanding of the PPAR-γ effect on modulating kidney function and the pathogenesis of renal fibrosis and also provided novel insights into CKD therapies by targeting renal tubular molecules and signaling pathways.
materials and Methods
Reagents and antibodies
Recombinant human transforming growth factor-β1 (TGF-β1, Cat#: 100-21) was from PeproTech (Rocky Hill, NJ), N-acetyl-L-cysteine (Cat#: A7250), apocynin (Cat#: A108109), rotenone (Cat#: R8875), RGZ (Cat#: R2408), T0070907 (Cat#: T8703, inhibitor of PPAR-γ), 2’,7’-dichlorofluoresceindiacetate (DCFDA, Cat#: D6883), and 15-deoxyprostaglandin J2 (Cat#: D8440), were from sigma (St. Louis, MO). Rat monoclonal anti-E-cadherin (Cat#: ab76055) and anti-α-SMA (Cat#: ab5694) antibodies, rabbit polyclonal anti-PPAR-γ antibody (Cat#: ab19481) were from abcam (Cambridge, MA). PPAR-γ transcription factor assay kit (Cat#: KA1358) was from abnova (Walnut, CA). PPAR-γ plasmid (Plasmid #8895) was from addgene (Cambridge, MA). Proteinase K (Cat#: P2308) was from sigma (St. Louis, MO).
Human kidney-2 (HK-2) cells
HK-2 cells, the immortalized human proximal tubular cell line, were grown in keratinocyte serum-free media (KSFM) supplemented with bovine pituitary extract and epidermal growth factor (Invitrogen). The cells were grown at 37°C in a humidified 5% CO2 incubator and subcultured at 60-80% confluence using 0.05% trypsin-0.02% EDTA (Invitrogen).
Transient transfection of HK-2 cells with PPAR-γ plasmid
Cells grown to 40% confluence in six-well plates were transiently transfected with PPAR-γ plasmid or vector with lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were transfected with PPAR-γ plasmid for 24 h before treatment with TGF-β1.
Animals
Mice with the floxed PPAR-γ gene and kap-cre mice were from Jackson Laboratory (Bar Harbor, ME) and had a genetic background of C57/B6J. In brief, 12-14-week-old male mice weighing 25-30 g were used in the experiment. All mice were maintained on a 12-h light-dark cycle in a temperature-controlled room and were fed standard rodent chow. The animal study protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Nanjing Medical University, China (No. 20090053).
Isolation of DNA from mouse tails
Cut off 0.2cm tail from 2- to 3-week old mice and put it into the tube with 300μl tail lysate including Proteinase K (10mg/ml). After incubation overnight at 55 degrees in water bath, 600μl 100% EtOH was added and vortexed vigorously. Then the sample was centrifuged by 12,000g for 5 min at room temperature. After removing the supernatant, the DNA pellet was rinsed with 70% EtOH followed by 12,000g centrifugation for 5 min at room temperature. Then the supernatant was removed and the DNA pellet was allowed to air-dry for 5-10 min.
Proximal tubule-specific deletion of PPAR-γ in mice and genotyping
To generate the proximal tubule-specific deletion of PPAR-γ in mice, homozygous mice with PPAR-γ flox/flox were crossed with kap-cre mice to obtain PPAR-γflox/+ kap-cre(+) mice. Then, the PPAR-γflox/+ kap-cre(+) mice were crossed with PPAR-γ flox/flox mice to obtain PPAR-γflox/flox kap-cre(+) male mice (PT-PPAR-γ-CKO). All male Cre transgene negative littermates served as controls (wild type, WT). A routine PCR protocol was used for genotyping tail DNA with the following primer pairs: PPAR-γ genotyping, forward: 5’-TGTAATGGAAGGGCAAAAGG-3’ and reverse: 5’-TGGCTTCCAGTG CATAAGTT-3’, which yielded 250- and 200-bp bands, respectively, for the floxed and wild-type alleles; Cre transgene genotyping, forward: 5’-AGATGCCAGGACATCAG GAACCTG-3’ and reverse: 5’-ATCAGCCACACCAGACACAGAGATC-3’, which generated a 200-bp fragment.
Primary culture of proximal tubule cells
Primary proximal tubular epithelial cells were isolated and cultured as previous described [13]. WT and PT-PPAR-γ-CKO mice (20-30 days) were sacrificed, and the renal cortices were sliced into pieces ~1 mm wide in ice-cold dissection solution (DS) (HBSS with in mmol/l: 10 glucose, 5 glycine, 1 alanine, 15 HEPES, pH 7.4 and osmolality 325 mosmol/kgH2O), followed by collagenase solution [DS with 0.1% (wt/vol) type-2 collagenase and 96 μg/ml soybean trypsin inhibitor] digestion for 30 min at 37°C. The supernatant was then sieved through two nylon sieves (pore size 250-μm and 80-μm). The longer PT fragments remained in the 80-μm sieve and were resuspended by flushing the sieve in the reverse direction with warm DS (37°C) containing BSA 1% (wt/vol). The PTs in the BSA solution were centrifuged for 5 min at 170 g, washed, and then resuspended in the appropriate amount of culture medium: 1:1 DMEM/F12 without phenol red and supplemented with heat-inactivated FCS 1%, HEPES 15 mmol/l, L-glutamine 2 mmol/l, hydrocortisone 50 nmol/l, insulin 5 g/ml, transferrin 5 μg/ml, selenium 50 nmol/l, sodium pyruvate 0.55 mmol/l, 100× Nonessential amino acids 10 ml/l, penicillin 100 IU/ml and streptomycin 100 μg/ml buffered to pH 7.4 and osmolality of 325 mOsmol/kgH2O. The PT fragments were seeded onto collagen-coated permeable PTFE-filter supports and left unstirred for 48 h at 37°C and 95% air/5% CO2 in a standard humidified incubator (Jouan, Winchester, VA). The culture medium was then changed for the first time. The medium was replaced every 2 days. After 7 days, the cell cultures were organized as a confluent monolayer.
Animal model of unilateral ureteral obstruction (UUO)
Male PT-PPAR-γ-CKO and WT mice (24-28 g) were used in these studies. The animals were provided with ample food and water and maintained on a 12 hr-12 hr light-dark cycle. UUO was performed using an established protocol as previously described [14]. Briefly, all mice received preoperative analgesia (subcutaneous injection of 50 mg/kg buprenorphine), and the right ureter was subsequently ligated with 4.0 surgical silk sutures through a small abdominal incision under 2.0% isoflurane-induced anesthesia. The abdomen was closed in two layers, and the mice were allowed to recover from surgery for 12 hours in a 28°C room. To evaluate the effect of PPAR-γ on renal fibrosis, the mice were randomized into the following four groups (n = 6 in each group): (1) sham operated mice of WT mice, (2) UUO model of WT mice, (3) sham operated PT-PPAR-γ-CKO mice, and (4) UUO model of PT-PPAR-γ-CKO mice. All of the mice were euthanized after 14 days of UUO.
Histological analysis
Two-micrometer sections of paraffin-embedded kidney tissue were subjected to Masson’s trichrome staining using commercial kits (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s protocol.
Western blotting
Cells were grown and stimulated with TGF-β1. After treatment, the cells were washed with PBS and lysed in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride). SDS-PAGE gel was used to separate the proteins. The gel was transferred to a PVDF membrane, which was blocked with 5% non-fat milk in TBST. The blots were probed with a primary antibody followed by a HRP-conjugated secondary antibody. The signal was detected using a Chemiluminescent ECL reagent kit (Millipore).
Real-time quantitative reverse transcription (qRT-PCR)
The total RNA was isolated from HK-2 cells and the kidney cortex using a Trizol Total RNA Isolation kit (Invitrogen) according to the manufacturer’s protocol. The RNA was eluted with RNase-free water. Reverse transcription was performed using the Superscript III RT kit (Invitrogen) according to the manufacturer’s protocols. Briefly, the reactions were incubated at 65°C for 5 min and then at 50°C for 60 min. Oligo nucleotides were designed by Primer3 software (available at http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and synthesized by Invitrogen. Real-time PCR amplification was performed using the SYBR Green master mix (Roche company) and the Prism 7500 Real-time PCR Detection System (Applied Biosystems). The cycling conditions were 95°C for 10 min followed by 40 repeats of 95°C for 15 s and 60°C for 1 min. The relative amounts of mRNA were normalized by GAPDH and calculated using the delta-delta method from threshold cycle numbers.
DCFDA fluorescence measurement of ROS
The fluorogenic substrate 2’,7’-dichlorofluorescein diacetate (DCFDA) is one of the most widely used techniques for directly measuring the redox state of a cell and could be used to evaluate the intracellular generation of ROS. For measurement of ROS, cells were grown onto glass cover slides. Then, the cells were washed twice with PBS and incubated for 30 min with DCFDA 50 µM when they reached 80% confluence. The cells were then treated with TGF-β1 for 30 min. At the end of the incubation period, the cells were again washed twice with PBS and imaged by confocal laser microscopy. To monitor the ROS levels, cells were seeded into 96-well plates and treated as mentioned above. The relative fluorescence was measured by a fluorescence plate reader (FLUO star OPTIMA) at excitation and emission wavelengths of 485 and 528 nm, respectively, three times at 90-s intervals. The relative fluorescence unit (RFU) was expressed as the fold increase over untreated cells.
Statistical analysis
All data are shown as the means ±SE. Analysis of variance (ANOVA) was followed by a Bonferroni post-test. A P value < 0.05 was considered significant.
Acknowledgments
We thank Yan Guo, Shuang Chen for Pathology/Histotechnology. We also thank Dr. Jianming Li in Experimental Animal Center of Nanjing Medical University for technical assistance.
Conflicts of interest
The authors have declared that no conflict of interest exists.
Grant support
This work was supported by grants from National Natural Science Foundation of China (Nos. 81530023, 81370802, 81300591, 81325004, and 81270797), and the Natural Science Foundation of Jiangsu Province (Nos. BK20141079, and BL2014007).
References
1. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008; 214(2):199-210.
2. Zeisberg M and Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010; 21(11):1819-1834.
3. Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010; 21(2):212-222.
4. Hills CE and Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol. 2010; 31(1):68-74.
5. Abbate M, Zoja C, Rottoli D, Corna D, Tomasoni S and Remuzzi G. Proximal tubular cells promote fibrogenesis by TGF-beta1-mediated induction of peritubular myofibroblasts. Kidney international. 2002; 61(6):2066-2077.
6. Janani C and Ranjitha Kumari BD. PPAR gamma gene—a review. Diabetes & metabolic syndrome. 2015; 9(1):46-50.
7. Fogo AB. PPARgamma and chronic kidney disease. Pediatr Nephrol. 2011; 26(3):347-351.
8. Kiss-Toth E and Roszer T. PPARgamma in Kidney Physiology and Pathophysiology. PPAR research. 2008; 2008:183108.
9. Zhou TB, Drummen GP, Jiang ZP, Long YB and Qin YH. Association of peroxisome proliferator-activated receptors/retinoic acid receptors with renal diseases. Journal of receptor and signal transduction research. 2013; 33(6):349-352.
10. Panchapakesan U, Sumual S, Pollock CA and Chen X. PPARgamma agonists exert antifibrotic effects in renal tubular cells exposed to high glucose. American journal of physiology Renal physiology. 2005; 289(5):F1153-1158.
11. Zafiriou S, Stanners SR, Saad S, Polhill TS, Poronnik P and Pollock CA. Pioglitazone inhibits cell growth and reduces matrix production in human kidney fibroblasts. Journal of the American Society of Nephrology. 2005; 16(3):638-645.
12. Wang W, Liu F and Chen N. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists attenuate the profibrotic response induced by TGF-beta1 in renal interstitial fibroblasts. Mediators Inflamm. 2007; 2007:62641.
13. Lepenies J, Hewison M, Stewart PM and Quinkler M. Renal PPARgamma mRNA expression increases with impairment of renal function in patients with chronic kidney disease. Nephrology (Carlton). 2010; 15(7):683-691.
14. Hinz B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix biology. 2015; 47:54-65.
15. Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R and Kietzmann T. Redox-fibrosis: Impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox biology. 2015; 6:344-352.
16. Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011; 7(7):1056-1067.
17. Gong K, Chen YF, Li P, Lucas JA, Hage FG, Yang Q, Nozell SE, Oparil S and Xing D. Transforming growth factor-beta inhibits myocardial PPARgamma expression in pressure overload-induced cardiac fibrosis and remodeling in mice. J Hypertens. 2011; 29(9):1810-1819.
18. Castro L and Freeman BA. Reactive oxygen species in human health and disease. Nutrition. 2001; 17(2):161, 163-165.
19. Seifried HE, Anderson DE, Fisher EI and Milner JA. A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem. 2007; 18(9):567-579.
20. Li R, Wang Y, Liu Y, Chen Q, Fu W, Wang H, Cai H, Peng W and Zhang X. Curcumin inhibits transforming growth factor-beta1-induced EMT via PPARgamma pathway, not Smad pathway in renal tubular epithelial cells. PloS one. 2013; 8(3):e58848.
21. Wei J, Zhu H, Komura K, Lord G, Tomcik M, Wang W, Doniparthi S, Tamaki Z, Hinchcliff M, Distler JH and Varga J. A synthetic PPAR-gamma agonist triterpenoid ameliorates experimental fibrosis: PPAR-gamma-independent suppression of fibrotic responses. Annals of the rheumatic diseases. 2014; 73(2):446-454.
22. Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP and Sime PJ. PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PloS one. 2011; 6(1):e15909.
23. Saeki T, Morioka T, Arakawa M, Shimizu F and Oite T. Modulation of mesangial cell proliferation by endothelial cells in coculture. The American journal of pathology. 1991; 139(4):949-957.
24. Sugawara A, Uruno A, Kudo M, Matsuda K, Yang CW and Ito S. Effects of PPARgamma on hypertension, atherosclerosis, and chronic kidney disease. Endocrine journal. 2010; 57(10):847-852.
25. Corinaldi J, Nasrallah R, Clark J, Paris G, Miura P, Jasmin BJ and Hebert RL. Troglitazone induces extracellular matrix and cytoskeleton remodeling in mouse collecting duct cells. Journal of biomedicine & biotechnology. 2012; 2012:507057.
26. Beltowski J, Rachanczyk J and Wlodarczyk M. Thiazolidinedione-induced fluid retention: recent insights into the molecular mechanisms. PPAR research. 2013; 2013:628628.
27. Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y and Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nature medicine. 2005; 11(8):861-866.
28. Fu Y, Gerasimova M, Batz F, Kuczkowski A, Alam Y, Sanders PW, Ronzaud C, Hummler E and Vallon V. PPARgamma agonist-induced fluid retention depends on alphaENaC expression in connecting tubules. Nephron. 2015; 129(1):68-74.
29. Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ and Yang T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(26):9406-9411.
30. Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N and Yorioka N. PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab Invest. 2009; 89(1):47-58.
31. Liu Y, Dai B, Xu C, Fu L, Hua Z and Mei C. Rosiglitazone inhibits transforming growth factor-beta1 mediated fibrogenesis in ADPKD cyst-lining epithelial cells. PloS one. 2011; 6(12):e28915.