
INTRODUCTION
Asthma is a kind of chronic lung disease characterized by airway inflammation, remodeling, and hyperresponsiveness [1-3]. In most cases, asthma is derived from sensitization via the airways to some common sensibiligen [4, 5]. They will cause airway inflammation through the release of inflammatory mediators, which is assumed to be the initiating event of airway remodeling. In allergic airway inflammation, including the innate and adaptive immune responses, eosinophils, macrophages, T helper type 2 (Th2) cells, and mast cells are involved in the inflammatory response of asthma [3, 6, 7]. Mast cells play key roles in the allergic inflammation response, airway remodeling and symptomatology [8]. Hence, the mechanism of function of mast cells in asthma is being actively studied now.
In the process of asthma, different inflammatory mediators, including cytokines, chemokines, and lipid mediators, will induce an inflammatory response and pulmonary structural changes [2, 9, 10]. In recent years, extracellular nucleotides (UDP, UTP, ATP, and ADP) have been considered as immunomodulatory mediators that have various functions in inflammatory diseases [11, 12]. Extracellular nucleotides exert their actions by activating ionotropic P2X receptors (P2X1-7) or transmembrane receptors of the P2 purinergic receptors family (P2Y1-14). Recently, P2Y2 and P2X7 have been found be involved in allergic airway inflammation [13, 14]. Purinergic receptor P2Y6, a G protein-coupled receptor activated by UDP or UTP, has also beeen shown to play an important role in inflammatory responses in some diseases [15-17]. P2Y6 was found to be overexpressed in some immune cells, including macrophages, neutrophils, and T cells during the inflammatory response [17-19]. Then, this receptor contributes to the mediation of proinflammatory effects, such as immune cell migration and release of cytokines and chemokines. It is suggested that P2Y6 also cause the pathogenesis of airway inflammation by activating some immune cells [16, 20]. However, the functions of P2Y6 in mediating inflammation were mostly focused on intestinal inflammation, bacterial infection, or vascular inflammation [15, 19, 21]. P2Y6 has been reported to have a potential role in pulmonary airway inflammation and remodeling in the development of asthma [16], but the regulatory mechanism of P2Y6 in the proinflammatory response in allergic asthma has not been reported and should be investigated in detail.
In the present study, we developed ovalbumin (OVA)-induced asthmatic mouse models and examined the alteration of the inflammatory response and airway morphologic alterations after P2Y6 deficiency. Cytokine release, immune cell invasion, and airway remodeling were measured in P2Y6-deficient mice following ovalbumin challenge and UDP treatment to clarify the potential function of P2Y6 in allergic asthma in mice. In our results, the release of UDP and overexpression of P2Y6 were demonstrated in the process of asthma in mice. Then, extracellular UDP induced more mast cell invasion into lung tissues and activated the functions of mast cells to deteriorate airway inflammation and remodeling in asthma by activating P2Y6 on mast cells. It indicated that P2Y6 enhanced functions of mast cells through the AKT signaling pathway in allergic asthma and deficiency of P2Y6 would limit the development of asthma in mice.
RESULTS
P2Y6 expression and UDP release in ovalbumin-induced asthmatic mice
To ensure the function of P2Y6 in airway inflammation, we developed ovalbumin-induced allergic asthmatic mice (Figure 1A). Firstly, the expression levels of cytokines and leukocyte invasion were detected to evaluate inflammation in ovalbumin-induced asthmatic mice. The ELISA results showed that the levels of IL-4 and IL-5 in bronchioalveolar lavage fluid (BALF) were increased on the 21st day after stimulation with 100 μg ovalbumin (Figure 1B). Then, the number of leukocytes in BALF was quantified using a blood cell counting plate. The results indicated that the invasion of leukocytes into the lung tissues showed time-dependence with ovalbumin induction in asthmatic mice (Figure 1C). To investigate whether P2Ys play roles in the development of asthma, we used real-time PCR to detect P2Ys expressions in mRNA levels in the lung tissues, and found that the expressions of P2Y1,2,6,12,13 were increased on the 10th day during ovalbumin treatment, especially that of P2Y2 and P2Y6. It was implied that these receptors were involved in airway inflammation. However, the expression of P2Y4 did not change markedly during ovalbumin treatment (Figure 1D). The expression of P2Y6 at the protein level was further confirmed by western blot. It was shown that P2Y6 expression was also increased at the protein level on the 21st day during asthma development in mice, but increased mRNA expression occurred later (Figure 1E). Fluorescence polarization was used in vivo to test the release level of UDP as the P2Y6 agonist during asthmatic development in mice. The results proved that the release of UDP was accompanied by alteration of expression of P2Y6 (Figure 1F). The peak of UDP release was on the 3th day of ovalbumin stimulation and after 10 days, its release level was decreased gently. It turned out that UDP activated the function of P2Y6 in the process of asthma in mice.
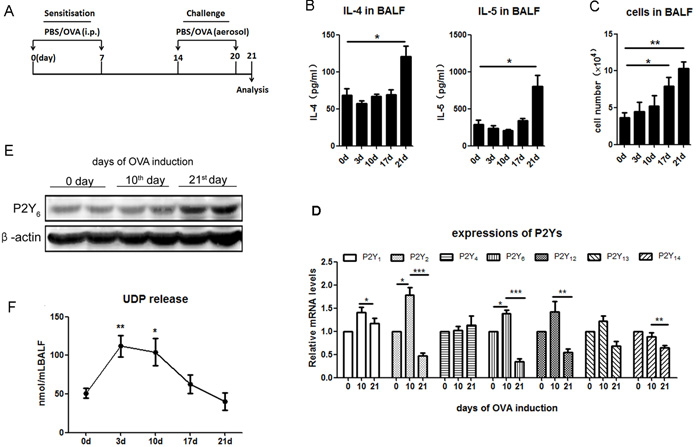
Figure 1: The expressions of P2Ys and UDP release in ovalbumin-induced asthmatic mice. A. The schematic treatment protocol of ovalbumin treatment for building asthmatic mouse model. B. Analysis of released levels of IL-4 and IL-5 in the BALF of ovalbumin-induced asthmatic mice using ELISA method (n = 5). C. Quantification of total cell invasion in the BALF of ovalbumin-induced asthmatic mice (n = 5). D. Real-time PCR was used to detect the expression of P2Ys at the mRNA level in ovalbumin-induced asthmatic mice. The relative mRNA levels of different P2Ys receptors were calculated as the method described in “Real-time PCR” of “Materials and Methods”. (n = 6) E. Detection of P2Y6 expression at the protein level in ovalbumin-induced asthmatic mice by western blot. F. UDP release in the ovalbumin-induced asthmatic mouse is checked by fluorescence polarization (n = 4). * P < 0.05, ** 0.01 < P < 0.05, *** P < 0.01. UDP is the abbreviation of uridine 5’-diphosphate; OVA is the abbreviation of ovalbumin.
P2Y6 was involved in immune cell invasion in ovalbumin-induced asthmatic mice
To study the role of P2Y6 in ovalbumin-induced airway conformation and inflammation, we used wild type and P2Y6-/- littermates to build asthmatic mouse models. Firstly, the goblet cell hyperplasia and subepithelial fibrosis in the lungs were observed under a light microscope with PAS and Masson’s trichrome staining in all asthmatic mice. In ovalbumin-induced asthmatic mice, there was a greater increase in hyperplasia of the goblet cells and the deposition of extracellular matrix in the lungs. However, these appearances were all relieved visibly in P2Y6-/- asthmatic mice (Figure 2A, 2B). P2Y6 deficiency relieved the airway conformation and mucus production in the development of asthma in mice.
Then we examined whether P2Y6 affected the airway construction through inflammatory reactions. We assessed the levels of IgE in serum and T helper type2 (Th2) relative cytokines IL-4, IL-5 and IL-13 in BALF. Although the level of them were increased in ovalbumin-treated mice, there were no striking difference between the wild type and P2Y6-/- asthmatic mice except IL-4. The level of IL-4 was decreased weakly after P2Y6 knockout in mice (Figure 2C, 2D, 2E, 2F). It indicated that P2Y6 influenced cytokine release slightly in the airway inflammatory reactions in asthma.
In association with airway remodeling in asthma are immune cell invasions, which are one of the major sources of released cytokines. Further, we detected the major type of immune cells including dendritic cells (DCs), mast cells and eosinophil invasion in the lungs of asthmatic mice to investigate whether P2Y6 has a role in recruiting inflammatory cells in the process of asthma. In ovalbumin-challenged mice, the total number of cells in BALF were much higher than those in the PBS-treated group. Meanwhile, in P2Y6-/- asthmatic mice, the invasion of cells in the lungs were markedly alleviated compared with wild type asthmatic mice (Figure 2G). Moreover, we analyzed the major types of immune cells in the lungs using flow cytometery. It was found that CD11c+/CD40+ DC cells, CD11c+/CD86+ DC cells, Siglec-F+/CCR3+ eosinophils, and CD117+/FcεRIα+ mast cells in the BALF were all increased significantly in ovalbumin-induced wild type asthmatic mice and no invasion of these immune cells occurred in the control group. After P2Y6-/- deficiency, the invasion of these cells in the lungs was decreased exceedingly in asthmatic mice, especially that of mast cells, which are important inflammatory cells in the development of asthma (Figure 2H, 2I, 2J, 2K). Furthermore, toluidine blue staining was used to evaluate mast cell invasion in the lung tissues and the same phenomenon was observed, in line with our previous result. Ovalbumin induced mast cell invasion in the lungs in wild type asthmatic mice, but this situation was relieved in P2Y6-deficient asthmatic mice (Figure 2L). The result revealed that P2Y6 enhanced inflammatory cell invasion, especially mast cells, in ovalbumin-induced airway inflammation in mice.
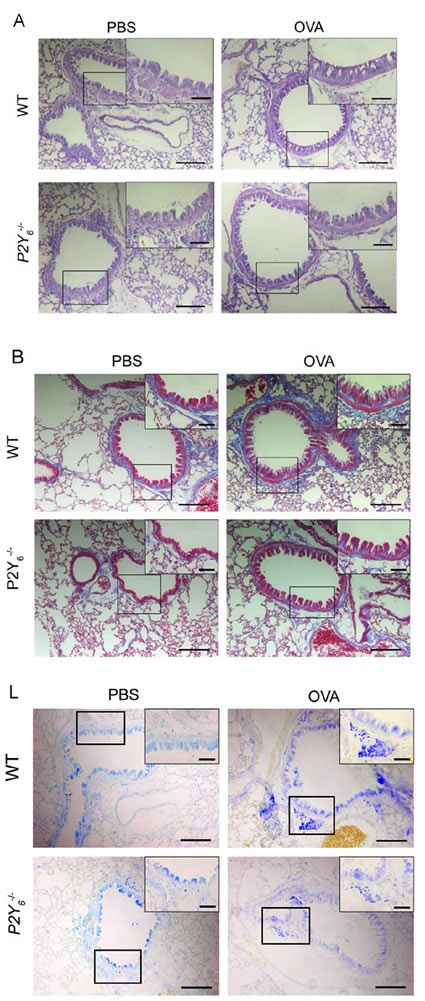
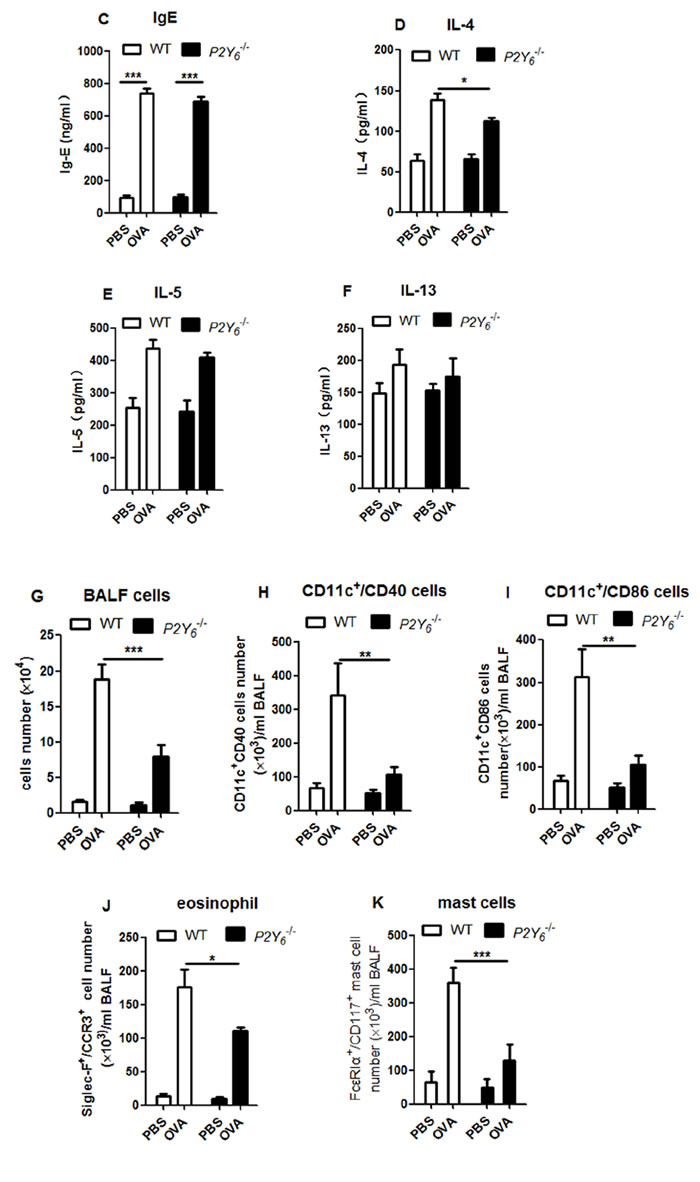
Figure 2: P2Y6 deficiency alleviates ovalbumin-induced inflammation in mice. A. PAS staining shows goblet cells in ovalbumin and saline-challenged mouse lung tissue. B. Masson’s trichrome staining is used to determine subepithelial fibrosis in the lung tissues of ovalbumin and saline-challenged mice. C.-F. ELISA method is used to detect the level of IgE expression in serum and IL-4, IL-5 and IL-13 levels in the BALF in ovalbumin and saline-challenged mice. G. The total number of cells in BALF was counted in a hemocytometer using an optical microscope. H.-K. Flow cytometry is used to quantify the dendritic cell, eosinophil and mast cell numbers in the BALF in ovalbumin and saline-challenged mice. L. The situation of mast cell invasion in the lung tissue was analyzed with toluidine blue stains. The lung sections were observed under a light microscopy at 10×magnification and in the upper right corner of each picture is at higher magnification of 40×. Scale bars: 100μm in the panels. * P < 0.05, ** 0.01 < P < 0.05, *** P < 0.01. WT is the abbreviation of wild type; OVA is the abbreviation of ovalbumin.
P2Y6 was activated by UDP to enhance ovalbumin-induced airway inflammation in mice
UDP, a P2Y6 agonist in vivo, was released during the process of ovalbumin stimulation in mice in our previous experiment. Here we intend to study whether UDP activates its purinergic receptor P2Y6 to affect ovalbumin-induced airway remodeling and inflammation in mice. We also used ovalbumin to induce allergic inflammation in littermates wild type and P2Y6-/- mice and meanwhile added UDP stimulation intratracheally after atomization for 30 min every three days during the process of model construction (Figure 3A). The results showed greater goblet cell hyperplasia in the airway epithelial layer in UDP-treated wild type asthmatic mice than in asthmatic mice, but not in P2Y6-/- asthmatic mice (Figure 3B). Unfortunately, a similar situation of deposition of extracellular matrix was found in UDP-treated asthmatic mice compared with single ovalbumin-induced mice, regardless of whether P2Y6 were deficiency (Figure 3C).
Then we analyzed the alteration of airway inflammation caused by UDP in asthmatic mice, including the levels of IgE in serum, IL-4, IL-5 and IL-13 in BALF. As shown in Figure 3D, UDP did not affect the altering of IgE level in serum and there is no difference of that between wild type and P2Y6-/- mice. UDP enhanced the expression of IL-4, IL-5 and IL-13 in BALF in ovalbumin-treated wild type mice, and after P2Y6 deficiency, it caused reduction of the levels of IL-4 and IL-5 in BALF. As a proof of concept, more immune cells will influence cytokine release and allergic airway inflammation in the lungs. In this regard, the invasion of DCs, mast cells, and eosinophils in the lungs were measured after UDP treatment in asthmatic mice. We found that more immune cells invaded the lungs induced by UDP and ovalbumin together in mice, especially mast cells (Figure 3E). However, no more cells were observed in the lung in ovalbumin-sensitized P2Y6-/- mice, even those treated with UDP. In addition, UDP did not impact eosinophil invasion in the lungs of mice during the process of asthma. To verify the role of UDP in mast cell invasion, we further used toluidine blue staining to observe the mast cells in the lungs of asthmatic mice with and without P2Y6 deficiency (Figure 3F). According to the results, more mast cells were observed in the lung tissues of the UDP-treated asthmatic mice group and this appearance was reduced after P2Y6 deficiency. Therefore, P2Y6 activated by UDP enhanced mast cell invasion and IL-4 release to modulate mucus hypertrophy in the development of asthma in mice.
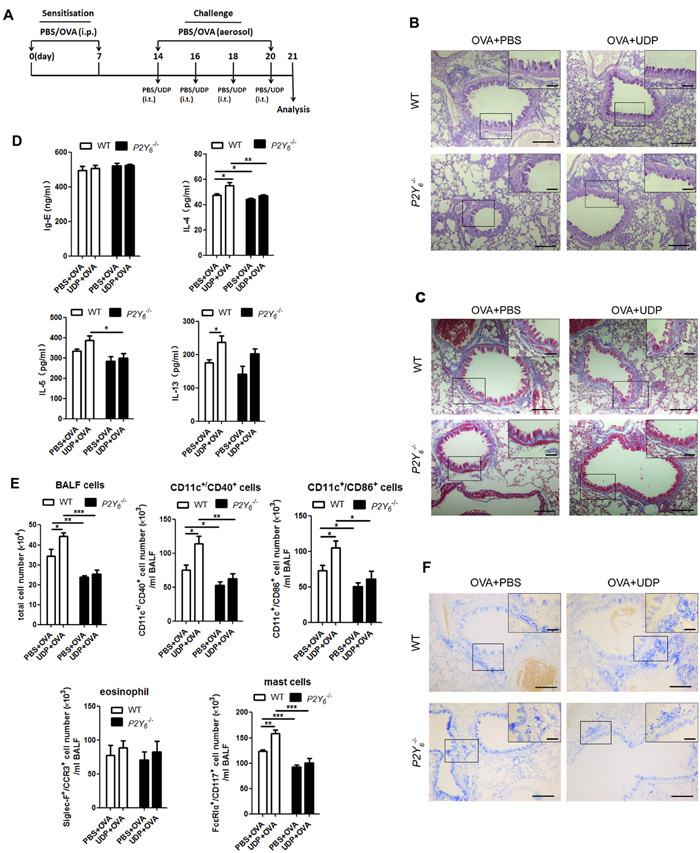
Figure 3: UDP enhance inflammation in ovalbumin-induced asthmatic mice. A. The schematic protocol of UDP treatment in ovalbumin-induced asthmatic mouse model. PAS staining B. and Masson’s trichrome staining C. results for lung tissues in ovalbumin-challenged mice with or without UDP treatment. D. The IgE level in serum and levels of IL-4, IL-5 and IL-13 in the BALF were analyzed using ELISA in ovalbumin-induced asthmatic mice with or without UDP treatment. E. The total numbers of cells in the BALF were quantified for ovalbumin and UDP-treated wild type or P2Y6-/- mice using a blood cell counting plate. Flow cytometry was used to quantify the numbers of dendritic cells, eosinophils, and mast cells in ovalbumin and UDP-treated wild type or P2Y6-/- mice. F. Toluidine blue stains was used to show the mast cell invasion in the lung tissues in mice. Scale bars: 100μm in the panels. * P < 0.05, ** 0.01 < P < 0.05, *** P < 0.01. WT is the abbreviation of wild type; UDP is the abbreviation of uridine 5’-diphosphate; OVA is the abbreviation of ovalbumin.
Activation of P2Y6 with UDP increased the function of mast cells in vitro
To investigate the regulatory function of P2Y6 in mast cells in asthma, mast cells were induced with IL-3 and SCF from primary separation of bone marrow cells, then stimulated with IgE overnight before treatment with UDP and ovalbumin in vitro. Firstly, alteration of the expression of P2Y6 at the mRNA level was detected in mast cells after stimulation with ovalbumin and UDP. The results indicated that this purinergic receptor on mast cells was overexpressed when treated with UDP and ovalbumin acting cooperatively (Figure 4A). Additionally, after activated mast cells were treated with UDP and ovalbumin together, the levels of inflammatory cytokines, including IL-4, IL-5, and IL-13, were increased compared with PBS treated, UDP-induced, and ovalbumin-induced groups (Figure 4A). However, in P2Y6-/- mast cells, no significant alteration of these cytokines was found when the cells were treated with UDP, ovalbumin or the two together. Therefore, we believe that in the process of ovalbumin stimulation, IgE-primed mast cells released more cytokines induced by UDP through the P2Y6 receptor on mast cells. Then we detected whether UDP stimulated the degranulation ability of mast cells during ovalbumin stimulation. The activity of β-hexosaminidase released from mast cells was analyzed to determine the function of degranulation in mast cells. Our evidence indicated that UDP also increased granule release from mast cells. Nonetheless, after P2Y6 knockout in mast cells, no enhancement of degranulation ability was observed when cells were induced with UDP or ovalbumin (Figure 4B). In our previous experiments, greater mast cell invasion in lung tissues was found in ovalbumin-induced mice additionally treated with UDP. Here, we detected whether UDP impacts the ability of chemotactic migration of mast cells in vitro. When 100 μm UDP was added to the lower compartment of transwell inserts, it induced a significant increase of mast cell migration to the lower chambers (Figure 4C). But this chemotactic response of mast cells was reduced after P2Y6 deficiency. In order to confirm the function of P2Y6 on UDP-induced migration of mast cells, the mast cells were treated with UDP for 30 min to activate the P2Y6 receptor before chemotactic assays. Then, the activated mast cells were added into the upper chambers of transwell inserts. According to the results of cell counting, an obviously increasing number of mast cells was also induced with UDP in the lower chamber for 3 h. In Figure 4C, it is shown that the migration of mast cells from upper to lower chambers was decreased after P2Y6-/- knockout, even without UDP induction. Here, our results indicated that P2Y6 on mast cells activated by extracellular UDP enhanced the functions of mast cells, including cytokine secretion, degranulation, and migration abilities, which affected the development of asthma.
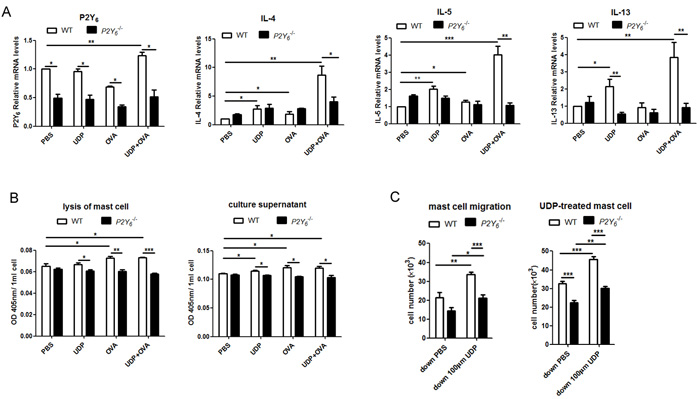
Figure 4: Analysis of the function of P2Y6 activated by UDP on mast cells in vitro. A. Detection of the expression of P2Y6, IL-4, IL-5 and IL-13 in mast cells after stimulation with ovalbumin and UDP by real-time PCR method. B. Mast cell degranulation ability was measured by assessing hexosaminidase activity in the culture supernatant or cell lysates. C. The chemotactic ability of mast cell was determined by the transwell method. * P < 0.05, ** 0.01 < P < 0.05, *** P < 0.01. WT is the abbreviation of wild type; UDP is the abbreviation of uridine 5’-diphosphate; OVA is the abbreviation of ovalbumin.
UDP regulates the function of mast cells through the AKT signal pathway
To explore the mechanism of UDP-induced enhancement of function of mast cells through its special receptor P2Y6, the phosphorylation of AKT, ERK, P38 and P65 was analyzed by western blot assay to assess the UDP/P2Y6-related signal pathway in mast cells. As shown in Figure 5A, the phosphorylation of AKT, ERK, and P38 was increased in mast cells induced by UDP, ovalbumin, and the two together. Meanwhile, in P2Y6-/- mast cells, the increase in phosphorylation of P38 was also induced by UDP and ovalbumin. The UDP and ovalbumin induced increased phosphorylation of AKT and ERK was decreased after P2Y6-/- deficiency in mast cells. These results suggested that AKT and ERK might be involved in the UDP/ P2Y6-mediated functions of mast cells in ovalbumin-induced asthma in vitro. In order to confirm the regulatory pathway of UDP/P2Y6 in its entirety, the levels of phosphorylated proteins were detected in the lung tissues of ovalbumin-induced asthmatic mice (Figure 5B). A sharp increase in the phosphorylation of AKT was found in ovalbumin-induced mice and this phenomenon was reduced observably in P2Y6-/- deficient mice. Moreover, the same alteration in phosphorylation of AKT occurred in the lung tissues of UDP-treated ovalbumin-induced asthmatic mice (Figure 5C). Thus, our results indicated that the AKT-related signaling pathway was more critical for regulatory function of UDP/P2Y6 on mast cells in the development of asthma. The molecular mechanism of how AKT signaling activates the function of mast cells still needs to be further explored.
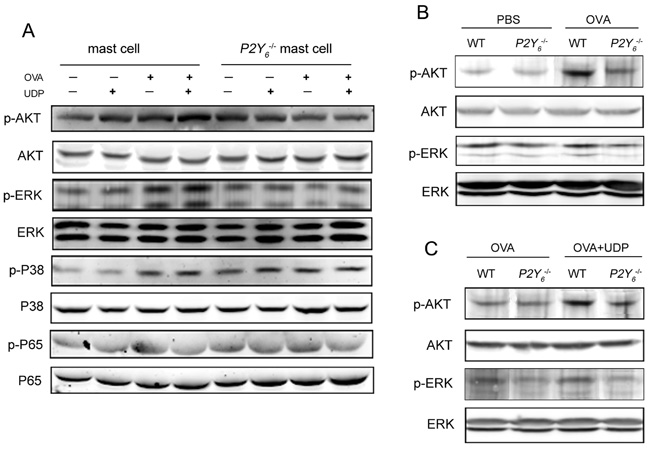
Figure 5: P2Y6-related signaling pathway detection by western blot in mast cells. A. The phosphorylation levels of P65, AKT, ERK, and P38 in mast cells were determined by western blot. B. The phosphorylation levels of AKT and ERK in the lung tissues in ovalbumin-induced wild type and P2Y6-/- mice. C. The phosphorylation levels of AKT and ERK in the lung tissues in UDP-treated wild type and P2Y6-/- asthmatic mice. WT is the abbreviation of wild type; UDP is the abbreviation of uridine 5’-diphosphate; OVA is the abbreviation of ovalbumin.
DISCUSSION
As it is known, inflammatory mediators, such as Th2 cells, producing IL-4, IL-5 and IL-13 contribute to not only inflammation but also airway remodeling in asthma [22, 23]. IgE is considered a triggering antibody for the allergic response through activation of mast cells and basophils [24]. Recent years, extracellular nucleotides (UDP, UTP, ATP, and ADP) known as danger signals have been found to play important roles in inflammatory response as immunoregulatory mediators, including in pulmonary inflection [11, 20]. Some of these nucleotides take out their functions by activating P2Y purinergic receptors, which are expressed on different immune cells. In asthmatic airway inflammation, ATP was reported to mediate the P2Y2 receptor in triggering dendritic cell and eosinophil recruitment and reactive oxygen species production [14, 25]. However, the functions of P2Y receptors involved in asthma to other extracellular nucleotides remains insufficiently understood. In our ovalbumin-induced asthmatic mice, the expression of not only P2Y2 but also P2Y6 changed sharply during the development of asthma. The expression of P2Y6 was increased in the ovalbumin-challenged stage and was shown to be decreased in the later stimulated phase. Meanwhile, the same variable tendency to release UDP in lung tissues was found in the asthmatic mice (Figure 1). According to our results, UDP release induced more activation of its specific purinergic receptor P2Y6, which implied that P2Y6 was involved in asthmatic airway inflammatory development in mice.
The presence of P2Y6 transcripts was detected in some immune cells, including macrophages, neutrophils, dendritic cells, T cells, and mast cells, and it was verified to be involved in cytokine secretion and cell migration in proinflammatory reactions [17, 26]. Here, we found over-expression of P2Y6 in lung tissues in mice after challenge with ovalbumin, which suggested a possible role in allergic airway inflammation. In order to investigate the role of P2Y6 in asthma, P2Y6 -deficient mice were used to construct ovalbumin-induced asthmatic mice. The deficiency of P2Y6 would relieve the phenotype of airway remodeling, such as airway epithelial extensions, goblet cell formation, and subepithelial fibrosis in lung tissues in asthmatic mice. However, P2Y6 did not strongly affect the alteration of IgE in serum nor cytokine release, except that of IL-4 in BALF in mice. Further, the total number of immune cells in BALF was decreased markedly after P2Y6 knockout, and among them was mast cells (Figure 2). With the intrapulmonary application of the P2Y6 agonist UDP, more severe airway remodeling and inflammation, including immune cell invasion and high levels of cytokines occurred in lung tissues in ovalbumin-induced asthmatic mice (Figure 3). These symptoms were abrogated after P2Y6 deficiency. It was demonstrated that P2Y6 enhanced immune cell invasion in lung tissues by application of UDP. Here, mast cells invasion in lungs was shown to be reduced mostly in P2Y6-/- mice. Therefore, we focused on the function of P2Y6 in mast cells in our present research. As dendritic cells and eosinophils are also required in the airway inflammatory process [27, 28], the regulatory mechanism of P2Y6 in these should be further investigated.
Further, we examined function of P2Y6 on mast cells in vitro that purified and induced from bone marrow cells in wild type and P2Y6-/- mice. During a pulmonary allergic response, mast cell progenitors migrate from the blood into lung tissues. IgE released from B cells will activate mast cells to release immune mediators, such as histamine and cytokines, which will cause more serious airway remodeling [8, 29-31]. We found much higher levels of cytokine (IL-4, IL-5, and IL-13) mRNAs in mast cells with stimulation of UDP and ovalbumin together. In addition, the stronger degranulation and migration abilities of mast cells were shown when cells were treated with ovalbumin and UDP acting cooperatively. These proinflammatory capabilities of mast cells were blocked after P2Y6 deficiency. Taken together, these results indicate that P2Y6 enhanced the functions of mast cells in pro-inflammation reactions in the asthmatic process by triggering migration and cytokine and granule release. These mediators released from mast cells have been shown to cause tissue remodeling, especially in the airway [8, 32].
AKT and ERK signals were involved in degranulation and cytokine secretion in mast cells in allergic asthma [33-35]. Not surprisingly, the phosphorylation levels of AKT and ERK were increased after treatment with ovalbumin for IgE-stimulated mast cells, especially with application of UDP. The increased phosphorylation of AKT and ERK was reduced in P2Y6-/- mast cells. Next, in lung tissues of ovalbumin-induced asthmatic mice, only an increase in the phosphorylation level of AKT was indicated, including in those treated with UDP, and this increase was inhibited after P2Y6 knockout. Thus, P2Y6 appeared to be more important in mediating the AKT signaling pathway in asthmatic development in mice. Further study will be undertaken to reveal the regulatory mechanism of P2Y6 in AKT signaling in asthma in detail.
In conclusion, P2Y6 was evidenced to be involved in allergic airway inflammation and remodeling in asthmatic mice by enhancing the function of mast cells, such as migration into lung tissues, degranulation ability and cytokine secretion. Interestingly, in mast cells and lung tissue, P2Y6 deficiency will reduce IL-4 expression most obviously in ovalbumin-induced asthma. In asthma, IL-4 released from Th-2 cells contributes to stimulate proliferation and differentiation of B cells that release IgE to mast cells [27, 36]. Mast cells also secrete IL-4 to cause allergic inflammation and modulate pulmonary endothelial cell alteration [8]. According to our results, P2Y6 increased IL-4 release during the process of asthma and the function of P2Y6 on IL-4 should be actively studied. Further, UDP, the agonist of P2Y6, was found to aggravate asthmatic symptoms as an inflammatory mediator in lung tissues in mice by mediating the function of mast cells. Therefore, more research needs to investigate further to reveal the mechanism of P2Y6 regulating mast cells in the pathogenesis of asthma for finding innovate treatments of this disease.
MATERIALS AND METHODS
Animals
Wild-type C57BL/6 mice were purchased from the Shanghai Laboratory Animal Company (Shanghai, China) and P2Y6-/- C57BL/6 mice were obtained from the Laboratory Animal Center of East China Normal University (Shanghai, China). All mice were bred and housed under pathogen-free conditions and maintained according to institutional guidelines. The mice were 6-8 weeks old at the time of the experiments and they were sacrificed using the method of cervical dislocation before experiments. All experimental protocols were approved by the Animal Investigation Committee of East China Normal University. Age-matched littermates (6-8 weeks old) with different genotypes were used for ovalbumin-induced asthmatic mouse models. Before the experiments, P2Y6 deficiency in the mice was confirmed by PCR.
Ovalbumin sensitization and challenge in mice
For the construction of a suitable asthma mouse model, a sensitization concentration of 100 μg ovalbumin (grade V, Sigma-Aldrich, St. Louis, USA) plusing 1% Al(OH)3 in a volume of 200 μL sterile PBS was intraperitoneally injected into different groups of wild type and P2Y6-deficient mice on day 0 and 7(Figure 1A) [37]. On days 14 to 20, sensitive mice were challenged by atomization with 1% ovalbumin in PBS or PBS only as control for 30 min each time. The UDP-stimulated group were injected intratracheally (i.t.) with 1 mM UDP (Sigma-Aldrich, St. Louis, USA) or PBS as a control at 30 min after atomization on days 14, 16, 18, and 20 (Figure 3A). At 24 h after the last challenge, serum, bronchoalveolar lavage fluid and lung tissues were collected after all mice were euthanized.
Bronchoalveolar lavage fluid leukocyte count
Lungs were flushed twice with cold 0.5% fetal bovine serum in 1 mL PBS. BALF was obtained after lavage and centrifuged at 2000 g at 4 °C for 5 min. The depositions were resuspended in 50 μL PBS, and the total number of cells was counted in a hemocytometer.
IgE and cytokine analysis
The serum samples were obtain by centrifugation of blood samples at 3000 g at 4 °C for 10 min. Then the levels of total IgE in serum were analyzed with a commercial enzyme-linked immunosorbent assay (ELISA) kit (Biolegend, California, USA). The levels of interleukin IL-4, IL-5 and IL-13 in the BALF were detected using the corresponding ELISA kit (Biolegend, California, USA) following manufacturer’s instructions.
Histology and immunohistochemistry
To analyze goblet cell hyperplasia, the left upper lung from each mouse was fixed in 4% paraformaldehyde overnight (24 h), embedded in paraffin and cut into 5-µm sections. Dewax the slices in dimethylbenzene and then rehydrate them in decreasing concentrations of ethanol. The slices were incubated in periodic acid alcohol before stained with periodic acid-Schiff (PAS) reagent [38]. Then, they were washed with sulfurous acid, followed by nucleolus staining using hematin and mounting in glycerol. PAS-positive cells and mucus secretion in the lungs were observed under a light microscope.
The lung tissue slices were incubated in periodic acid alcohol before observing subepithelial fibrosis in the lungs. The slices were washed with deionized water, and nucleolus were stained using hematoxylin. Then the subepithelial fibrosis in lung tissues were determined by Masson’s trichrome-stained reagent staining (Zhongshanjinqiao, Beijing, China) [39]. The slices were observed under a light microscope after mounting in glycerol.
To observing the mast cells in the lung tissue, the lung tissue slices were dewaxed in dimethylbenzene and rehydrated in decreasing concentrations of ethanol. Then stained with toluidine blue and wash with glacial acetic acid [40]. The slices were observed under a light microscope after mounting in glycerol.
Ovalbumin sensitization and cell challenge
After the primary separation of bone marrow cells from femora and tibiae of wild-type mice, 10 ng/ml IL-3 and 10 ng/mL SCF (Biolegend, California, USA) were added into RPMI-1640 cell culture medium to induce bone marrow cell differentiation to mast cells. After 4-6 weeks culture, flow cytometry analysis was used to separate CD117 and FcεRIα-positive mast cells (APC anti-CD117 and FITC anti-FcεRIα, Biolegend, California, USA) [41]. The purified mast cells were activated using human IgE full-length protein (Abcam, Massachusetts, USA) overnight and then challenged with 200 µg ovalbumin for 1 h [42]. Real-time PCR was used to detect the mRNA expression levels of P2Y6, IL-4, IL-5 and IL-13 on mast cells.
Flow cytometry analysis and cell sorting
The BALF were incubated with different antibodies for 1 h: FITC anti-CCR3, PE anti-Siglec-F for eosinophil [43], FITC anti-FcεRIα, and APC anti-CD117 for mast cells, FITC anti-CD11c, and APC anti-CD40 and APC anti-CD86 for DCs (BD Biosciences, New Jersey, USA or Biolegend, California, USA). After the BALF were washed by PBS three times, the different types of cells were analyzed and sorted using a flow cytometer (BD FACSAria, USA) and data were analyzed using CellQuest software (BD Biosciences, USA).
Western blot
After treatment or stimulation, the lung tissues were lysed by RIPA buffer (Cell Signaling Technology, USA). The concentration of total protein was measured by BCA assay (Pierce) and adjusted to the same concentration with extraction reagent. Samples were heated at 100 °C for 5 min and loaded onto 10% SDS-PAGE. After electrophoresis, the gel was transferred onto polyvinylidene fluoride (PVDF) membrane and blocked with 5% BSA. The membrane was incubated with primary Abs and then with the appropriate fluorescent secondary Abs. Subsequently, the immunoreactive proteins were detected using the Odyssey laser digital imaging system (Gene Company, Hongkong, China). The primary antibodies including anti-AKT, anti-phospho-AKT, anti-ERK, anti-phospho-ERK, anti-P38, anti-phospho-P38, anti-P65 and anti-phospho-P65 are all rabbit monoclonal antibodies (Cell Signaling Technology, USA). The P2Y6 antibody is a rabbit polyclonal IgG (Santa Cruz Biotechnology, USA). The secondary Antibody is IRDye® 680LT Goat anti-Rabbit IgG (LI-COR Bioscience, USA).
Real-time PCR
The sorted cells were stored in the TRIzol (TaKaRa Clontech, Japan). Total RNAs were extracted according to the manufacturer’s protocol of TakaRa. Template cDNAs were obtained by reverse transcription of total RNA using PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa Clontech, Japan). Amplification was carried out by using SYBR® Premix Ex Taq™ (Tli RNaseH Plus) (TaKaRa Clontech, Japan). The mRNA expression level of β-actin was used as internal control. The relative mRNA levels of P2Ys were calculated as follows [44]. P2Ys genes were taken for example and the average CT for β-actin was subtracted from the average value for P2Ys to generate Δ for each sample. CT is the cycle number of PCR amplification. Δ = CT× (P2Ys) - CT× (β-actin). Therefore, ΔΔ = Δ(P2Ys value for the samples at day10 or day21)- Δ(P2Ys value for the samples at day0). Finally, the formula 2-ΔΔ was taken to calculate the relative mRNA level compared with the control. The sequences of the primers for real-time PCR are listed in Table 1.
Table 1: Sequences of the primers for real-time PCR
Gene name |
Primers (5'- 3') |
Il-4 |
forward: GGTCTCAACCCCCAGCTAGT |
reverse: GCCGATGATCTCTCTCAAGTGAT |
|
Il-5 |
forward: AGGCTTCCTGTCCCTACTCA |
reverse: AATCCAGGAACTGCCTCGTC |
|
Il-13 |
forward: CCTGGCTCTTGCTTGCCTT |
reverse: GGTCTTGTGTGATGTTGCTCA |
|
P2Y1 |
forward: CGCACACAGGTACAGTGGCGT |
reverse: TTCCGAGTCCCAGTGCCAGAGT |
|
P2Y2 |
forward: GTGCGGGGAACCCGGATCAC |
reverse: AGCCGCCTGGCCATAAGCAC |
|
P2Y4 |
forward: GTGTGCACCGCTACATGGGCA |
reverse: TCAGGCAGAGTCGTGTCATGGCA |
|
P2Y6 |
forward: AGGGGACCACTGGCCCTTCG |
reverse: TACCCAAGCAGCACGGCGAC |
|
P2Y12 |
forward: AACTCTATCGGTCTTATGTCA |
reverse: AGAATACAGCAATGATGATGAA |
|
P2Y13 |
forward: GGGCCTCATCGCTTTCGACAGG |
reverse: TCACGGATGATGGCGTTGCCT |
|
P2Y14 |
forward: GGGGCGGAAGTGGCACAAGG |
reverse: GCGGCTGGACTTCCTCTTGACG |
|
β-actin |
forward: GTACGCCAACACAGTGCTG |
reverse: CGTCATACTCCTGCTTGCTG |
Degranulation
Bone marrow mast cells were incubated with IgE (Sigma-Aldrich, St. Louis, USA) to activate overnight, then washed by Tyrode’s buffer [45]. The mast cells were resuspended in Tyrode’s buffer at 2 × 105 per well in a 96-well plate and stimulated with UDP, ovalbumin, or UDP + ovalbumin for 1 h. After that, the mast cells were treated with RIPA cell lysis buffer and supernatants were collected after centrifugation. These supernatants and cell culture medium supernatants were mixed with 0.1 M sodium citrate buffer (pH 4.5) including p-nitrophenyl N-acetyl-β-D-glucosamide (Sigma-Aldrich, St. Louis, USA) for 90 min at 37 °C. The reaction was stopped by addition of 0.2 M glycine (pH 10.7). The absorbance of β-hexosaminidase released from mast cells was read at a wavelength of 405 nm.
Chemotaxis assays
Chemotactic responses of mast cells were examined in 24-well Transwell plates (BD Falcon, USA) with 8-μm-pore-size polycarbonate filters. Cells (2 × 106 cells/mL) in 200 μL RPMI 1640 were added to the top wells with or without UDP. The migration buffer containing PBS or 100 μm UDP was added to the lower wells. After 3 h at 37 °C in 5% CO2, the mast cells in the lower chambers were harvested by centrifugation and resuspended with 100 μL PBS. The cells were counted on a blood cell counting plate.
Statistical analysis
Significant differences were assessed with the t-test for comparisons between two groups and ANOVA test for comparisons among more than two groups. The data are presented as the mean ± SEM. All statistical values were measured using GraphPad Prism version 5.0 (GraphPad Prism Software, USA). Results were considered significant at P < 0.05.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
GRANT SUPPORT
This work was supported by National Basic Research Program of China (2012CB910404), National Natural Science Foundation of China (81272369, 81172816, 31470040 and 31000346), Doctoral Fund of Ministry of Education of China (20130076110013) and the Shanghai Committee of Science and Technology of China (15JC1401500, 14140904200, 15ZR1411100).
REFERENCES
1. Halwani R, Al-Muhsen S and Hamid Q. Airway remodeling in asthma. CurrOpinPharmacol. 2010; 10:236-245.
2. Hamid Q and Tulic M. Immunobiology of asthma. AnnuRevPhysiol. 2009; 71:489-507.
3. Holgate ST. Innate and adaptive immune responses in asthma. NatMed. 2012; 18:673-683.
4. Carlsen KH. Childhood asthma and the environment. Pediatric allergy and immunology. 1994; 5:48-51.
5. Kanchongkittiphon W, Gaffin JM and Phipatanakul W. The indoor environment and inner-city childhood asthma. Asian Pacific Journal of Allergy and Immunology. 2014; 32:103-110.
6. Leavy O. Asthma and allergy: Diet and airway inflammation. Nature reviews Immunology. 2014; 14:64-65.
7. Fahy JV. Type 2 inflammation in asthma—present in most, absent in many. Nature reviews Immunology. 2015; 15:57-65.
8. Amin K. The role of mast cells in allergic inflammation. RespirMed. 2012; 106:9-14.
9. Tantisira KG and Drazen JM. Genetics and pharmacogenetics of the leukotriene pathway. The Journal of allergy and clinical immunology. 2009; 124:422-427.
10. Duroudier NP, Tulah AS and Sayers I. Leukotriene pathway genetics and pharmacogenetics in allergy. Allergy. 2009; 64:823-839.
11. Vitiello L, Gorini S, Rosano G and la Sala A. Immunoregulation through extracellular nucleotides. Blood. 2012; 120:511-518.
12. Eltzschig HK, Sitkovsky MV and Robson SC. Purinergic signaling during inflammation. The New England journal of medicine. 2012; 367:2322-2333.
13. Muller T, Vieira RP, Grimm M, Durk T, Cicko S, Zeiser R, Jakob T, Martin SF, Blumenthal B, Sorichter S, Ferrari D, Di Virgillio F and Idzko M. A potential role for P2X7R in allergic airway inflammation in mice and humans. American journal of respiratory cell and molecular biology. 2011; 44:456-464.
14. Muller T, Robaye B, Vieira RP, Ferrari D, Grimm M, Jakob T, Martin SF, Di Virgilio F, Boeynaems JM, Virchow JC and Idzko M. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy. 2010; 65:1545-1553.
15. Stachon P, Peikert A, Michel NA, Hergeth S, Marchini T, Wolf D, Dufner B, Hoppe N, Ayata CK, Grimm M, Cicko S, Schulte L, Reinohl J, von zur Muhlen C, Bode C, Idzko M, et al. P2Y6 deficiency limits vascular inflammation and atherosclerosis in mice. Arteriosclerosis, thrombosis, and vascular biology. 2014; 34:2237-2245.
16. Vieira RP, Muller T, Grimm M, von Gernler V, Vetter B, Durk T, Cicko S, Ayata CK, Sorichter S, Robaye B, Zeiser R, Ferrari D, Kirschbaum A, Zissel G, Virchow JC, Boeynaems JM, et al. Purinergic receptor type 6 contributes to airway inflammation and remodeling in experimental allergic airway inflammation. American journal of respiratory and critical care medicine. 2011; 184:215-223.
17. Liu GD, Ding JQ, Xiao Q and Chen SD. P2Y6 receptor and immunoinflammation. Neuroscience bulletin. 2009; 25:161-164.
18. Ben Yebdri F, Kukulski F, Tremblay A and Sevigny J. Concomitant activation of P2Y(2) and P2Y(6) receptors on monocytes is required for TLR1/2-induced neutrophil migration by regulating IL-8 secretion. European journal of immunology. 2009; 39:2885-2894.
19. Zhang Z, Wang Z, Ren H, Yue M, Huang K, Gu H, Liu M, Du B and Qian M. P2Y(6) agonist uridine 5’-diphosphate promotes host defense against bacterial infection via monocyte chemoattractant protein-1-mediated monocytes/macrophages recruitment. Journal of immunology. 2011; 186:5376-5387.
20. Alberto AV, Faria RX, de Menezes JR, Surrage A, da Rocha NC, Ferreira LG, Frutuoso Vda S, Martins MA and Alves LA. Role of P2 Receptors as Modulators of Rat Eosinophil Recruitment in Allergic Inflammation. PloS one. 2016; 11:e0145392.
21. Hansen A, Alston L, Tulk SE, Schenck LP, Grassie ME, Alhassan BF, Veermalla AT, Al-Bashir S, Gendron FP, Altier C, MacDonald JA, Beck PL and Hirota SA. The P2Y6 receptor mediates Clostridium difficile toxin-induced CXCL8/IL-8 production and intestinal epithelial barrier dysfunction. PloS one. 2013; 8:e81491.
22. Pascual RM and Peters SP. Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. The Journal of allergy and clinical immunology. 2005; 116:477-486; quiz 487.
23. Shifren A, Witt C, Christie C and Castro M. Mechanisms of remodeling in asthmatic airways. Journal of allergy. 2012; 2012:316049.
24. Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunological reviews. 2011; 242:205-219.
25. Kobayashi T, Soma T, Noguchi T, Nakagome K, Nakamoto H, Kita H and Nagata M. ATP drives eosinophil effector responses through P2 purinergic receptors. Allergology international. 2015; 64 Suppl:S30-36.
26. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nature reviews Immunology. 2011; 11:201-212.
27. Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MAM, Kool M, Muskens F and Lambrecht BN. Inflammatory dendritic cells-not basophils-are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. Journal of Experimental Medicine. 2010; 207:2097-2111.
28. Bradding P. Asthma: eosinophil disease, mast cell disease, or both? Allergy Asthma ClinImmunol. 2008; 4:84-90.
29. Ro J, Lee YS, Kim SY and Hong G. Transglutaminase 2 expressed in mast cell activation induces IgE production in B cells, airway inflammation and remodeling via up-regulating CD40L and cytokine expression in mouse allergic asthma. Journal of immunology. 2012; 188.
30. Hong GU, Park BS, Park JW, Kim SY and Ro JY. IgE production in CD40/CD40L cross-talk of B and mast cells and mediator release via TGase 2 in mouse allergic asthma. Cell Signal. 2013; 25:1514-1525.
31. Hong GU, Kim NG, Kim TJ and Ro JY. CD1d expressed in mast cell surface enhances IgE production in B cells by up-regulating CD40L expression and mediator release in allergic asthma in mice. Cellular Signalling. 2014; 26:1105-1117.
32. Okayama Y, Ra C and Saito H. Role of mast cells in airway remodeling. Current opinion in immunology. 2007; 19:687-693.
33. Bugajev V, Halova I, Draberova L, Bambouskova M, Potuckova L, Draberova H, Paulenda T, Junyent S and Draber P. Negative regulatory roles of ORMDL3 in the FcepsilonRI-triggered expression of proinflammatory mediators and chemotactic response in murine mast cells. Cellular and molecular life sciences. 2016; 73:1265-1285.
34. Wang Q, Zhao DY, Xu H, Zhou H, Yang QY, Liu F and Zhou GP. Down-regulation of microRNA-223 promotes degranulation via the PI3K/Akt pathway by targeting IGF-1R in mast cells. PloS one. 2015; 10:e0123575.
35. Takayama G, Ohtani M, Minowa A, Matsuda S and Koyasu S. Class I PI3K-mediated Akt and ERK signals play a critical role in FcepsilonRI-induced degranulation in mast cells. International immunology. 2013; 25:215-220.
36. Cohn L, Tepper JS and Bottomly K. IL-4-independent induction of airway hyperresponsiveness by Th2, but not Th1, cells. JImmunol. 1998; 161:3813-3816.
37. Park S, Baek H, Jung KH, Lee G, Lee H, Kang GH, Lee G and Bae H. Bee venom phospholipase A2 suppresses allergic airway inflammation in an ovalbumin-induced asthma model through the induction of regulatory T cells. Immunity, inflammation and disease. 2015; 3:386-397.
38. Shi JP, Li XN, Zhang XY, Du B, Jiang WZ, Liu MY, Wang JJ, Wang ZG, Ren H and Qian M. Gpr97 Is Dispensable for Inflammation in OVA-Induced Asthmatic Mice. PloS one. 2015; 10:e0131461.
39. Ano S, Morishima Y, Ishii Y, Yoh K, Yageta Y, Ohtsuka S, Matsuyama M, Kawaguchi M, Takahashi S and Hizawa N. Transcription factors GATA-3 and RORgammat are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. Journal of immunology. 2013; 190:1056-1065.
40. Ikeda RK, Miller M, Nayar J, Walker L, Cho JY, McElwain K, McElwain S, Raz E and Broide DH. Accumulation of peribronchial mast cells in a mouse model of ovalbumin allergen induced chronic airway inflammation: modulation by immunostimulatory DNA sequences. Journal of immunology. 2003; 171:4860-4867.
41. Dahlin JS, Malinovschi A, Ohrvik H, Sandelin M, Janson C, Alving K and Hallgren J. Lin- CD34hi CD117int/hi FcepsilonRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood. 2016; 127:383-391.
42. Catalli A, Karpov V, Erdos LE, Tancowny BP, Schleimer RP and Kulka M. Stimulus-selective regulation of human mast cell gene expression, degranulation and leukotriene production by fluticasone and salmeterol. PloS one. 2014; 9:e96891.
43. Carlens J, Wahl B, Ballmaier M, Bulfone-Paus S, Forster R and Pabst O. Common gamma-chain-dependent signals confer selective survival of eosinophils in the murine small intestine. Journal of immunology. 2009; 183:5600-5607.
44. Zhang M, Wang H, Teng H, Shi J and Zhang Y. Expression of SHH signaling pathway components in the developing human lung. Histochemistry and cell biology. 2010; 134:327-335.
45. Demo SD, Masuda E, Rossi AB, Throndset BT, Gerard AL, Chan EH, Armstrong RJ, Fox BP, Lorens JB, Payan DG, Scheller RH and Fisher JM. Quantitative measurement of mast cell degranulation using a novel flow cytometric annexin-V binding assay. Cytometry. 1999; 36:340-348.