
INTRODUCTION
Acute lymphoblastic leukemia (ALL) in infants (<1 year of age) represents an aggressive malignancy, associated with high relapse rates and a poor clinical outcome [1]. The majority (~80%) of these patients carry a leukemia-specific chromosomal translocations involving the Mixed Lineage Leukemia (MLL) gene [1]. MLL-rearranged infant ALL patients fare significantly worse than infant ALL patients who do not carry MLL translocations, with event-free survival rates of 30-40% vs. ~80%, respectively [2]. Recently, we demonstrated that 24% of the infant ALL patients carrying MLL translocation t(4;11), the most frequently observed translocation of MLL among these patients, also carry a RAS mutation. Mutations in NRAS were found in 11% and KRAS mutations in 13% of cases [3]. Moreover, we showed that the presence of a RAS mutation in MLL-rearranged patients represented an independent predictive factor for an even worse clinical outcome in this high-risk group. Nearly all RAS-mutant t(4;11)+ infant ALL patients relapsed within the first year from diagnosis, while still on treatment, and all died within 4 years from diagnosis [3].
Despite this strong association with an exceedingly poor prognosis, a recent study by Emerenciano et al. suggested that RAS mutations in MLL-rearranged infant ALL may not act as driver mutations and are not required for disease progression, but rather act only at disease onset [4]. Yet, our previous data clearly showed that RAS-mutant MLL-rearranged infant ALL patients are at extremely high risk of therapy failure and early death. Moreover, RAS pathway inhibition, including MEK inhibition, was previously shown to effectively inhibit RAS-mutant MLL-rearranged AML in vitro [5, 6]. Therefore, we decided to investigate the potential of RAS pathway inhibition and found that RAS-mutant MLL-rearranged ALL cells are remarkably sensitive to MEK inhibitors.
RESULTS
RAS-mutant MLL-rearranged ALL cells are sensitive to MEK inhibition
Since the previously identified RAS aberrations are all activating mutations (at residues G12, G13 or Q61), we wondered whether small molecule inhibitors targeting RAS pathway components could suppress RAS-mutant leukemic cells [3, 7]. Therefore, 7 RAS pathway inhibitors, already approved for therapeutical use or under clinical investigation for other malignancies with RAS pathway mutations, were selected as therapeutic strategies for the RAS-mutant infant ALL patients. Using 4-day MTS cell viability assays we tested the in vitro anti-leukemic potential of Salirasib (RAS localization inhibitor), Vemurafenib (BRAF inhibitor), Sorafenib (pan-kinase inhibitor), Trametinib, Selumetinib and MEK162 (MEK inhibitors) and Temsirolimus (mTOR inhibitor) against RAS-mutant MLL-rearranged ALL cell line KOPN8, and the RAS-wildtype MLL-rearranged cell lines SEM and RS4;11. Interestingly, the RAS-mutant cell line KOPN8 was more sensitive to the MEK inhibitors MEK162, Selumetinib and Trametinib (Figure 1). Temsirolimus and Sorafenib potently reduced cell viability of both RAS-mutant and RAS-wildtype cell lines. Additionally, Salirasib and Vemurafenib did not substantially reduce cell viability, even at high concentrations (>10μM). To confirm the efficacy of these inhibitors, we performed 4-day MTT cell viability assays on primary diagnostic RAS-mutant (n=6) and RAS-wildtype (n=14) t(4;11)+ infant ALL samples. Interestingly, compared to RAS-wildtype t(4;11)+ ALL cases, the RAS-mutant t(4;11)+ infant ALL cases were significantly more sensitive to all MEK inhibitors (Figure 2A) with median IC50 values of <0.1 μM for MEK162 and Selumetinib and <0.01 μM for Trametinib (Figure 2B). Additionally, all other tested inhibitors (Salirasib, Temsirolimus, Sorafenib and Vemurafenib) reached only IC50 values of >10μM (Supplementary Figure 1).
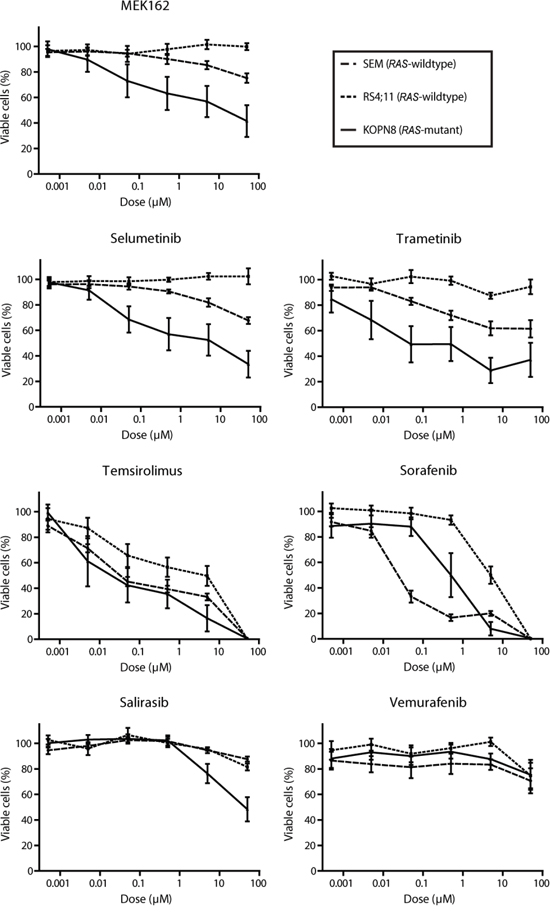
Figure 1: MEK inhibitors specifically impede RAS-mutant MLL-rearranged ALL cell line KOPN8. Cell viability of MLL-rearranged cell lines exposed to MEK162, Selumetinib, Trametinib, Temsirolimus, Sorafenib, Salirasib and Vemurafenib. All cell lines respond to Sorafenib and Temsirolimus, while RAS-mutant KOPN8 (solid line) is more sensitive for MEK162, Selumetinib and Trametinib than RAS-wildtype SEM (large dashed line) or RS4;11 (small dashed line). Data are represented as mean +/- sem. n≥3.
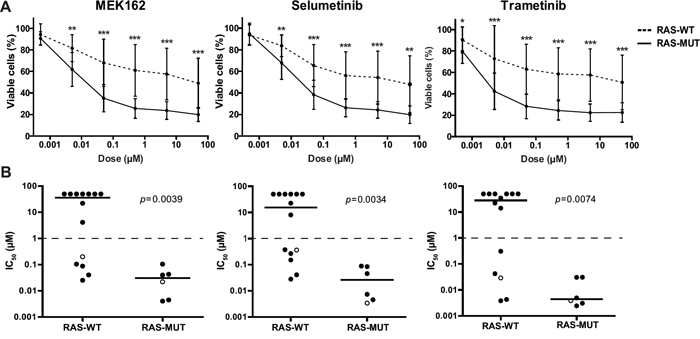
Figure 2: Primary RAS-mutant MLL-rearranged ALL cells are sensitive to MEK inhibitors. A. Patient derived t(4;11)+ infant ALL cells exposed to MEK inhibitors indicate RAS-mutant samples (solid line, n=6) are more sensitive compared to RAS-wildtype samples (dashed line, n=14). Data are represented as median +/- sd. *0.01<p<0.05; **0.001<p<0.01; ***p<0.001. B. The IC50 (concentration needed to inhibit 50% of the leukemic cell viability) of the individual t(4;11)+ infant ALL patient samples shown in A. Median IC50 values, represented by horizontal bars, confirm strong sensitivity of RAS-mutant patient samples compared to the majority of RAS-wildtype samples. Open circles indicate matched diagnosis (wildtype) and relapse (mutant) samples. The tick lines indicate separation between MEK inhibitor sensitive and resistant patient samples (IC50<1μM and IC50>1μM, respectively).
Also, we included one matched pair of diagnostic/relapse t(4;11)+ samples. For this particular patient, no RAS mutation was present at diagnosis, but a RAS mutation could be identified at relapse. Indeed, the RAS-mutant relapse sample of this patient was more sensitive to all three MEK inhibitors tested than the RAS-wildtype diagnostic sample (Figure 2B).
Enhanced RAS activation in t(4;11)+ infant ALL cells carrying RAS mutations
The MEK inhibitors MEK162, Selumetinib and Trametinib significantly reduce viability of RAS-mutant MLL-rearranged ALL cells. Notably, a subset of the RAS-wildtype primary t(4;11)+ infant ALL samples also responded favorably to the MEK inhibitors (Figure 2B). We wondered whether other biomarkers, besides RAS mutation status, could predict MEK inhibitor sensitivity in MLL-rearranged ALL. Wildtype RAS proteins are under regulation of upstream signaling events, often involving tyrosine kinase receptors, while mutant RAS proteins are less dependent on upstream activation due to reduced GTPase activity, rendering a surplus of activated GTP-bound RAS. Therefore, we determined the RAS protein levels and RAS activity in our primary t(4;11)+ infant ALL cells. No significant difference in RAS protein levels was observed between the RAS-mutant and RAS-wildtype t(4;11)+ infant ALL samples using Western blot analysis (Figure 3A). Next, we investigated the level of active (GTP-bound) RAS in these samples by precipitation with RAF-1 RAS interaction peptide, followed by immunoblotting. As expected, the RAS-mutant t(4;11)+ infant ALL samples showed significant (p=0.013) higher levels of RAS activation as compared to RAS-wildtype samples (Figure 3B). No differences in RAS activation were observed between RAS-wildtype samples that were sensitive or resistant to MEK inhibition.
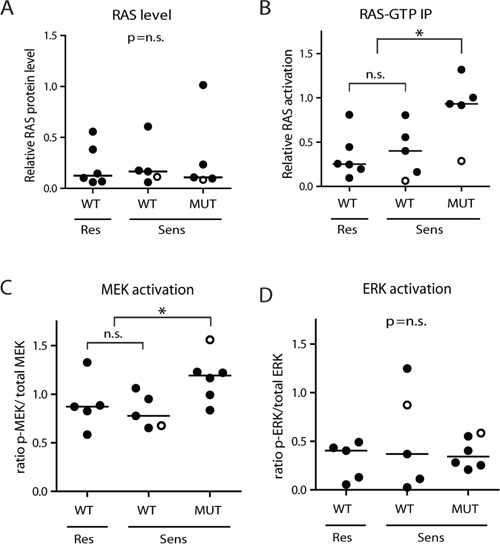
Figure 3: RAS-mutant t(4;11)-positive ALL cells have enhanced downstream activation. A. RAS protein level, relative to β-actin, determined by western blotting in t(4;11)+ infant ALL samples, subdivided according to RAS mutation status (WT or MUT) and MEK inhibitor sensitivity (Res or Sens). No differences in median protein level (horizontal bars) are observed between the different subgroups. B. Relative RAS activation is enhanced in RAS-mutant t(4;11)+ patient samples, though no difference is observed between the MEK inhibitor resistant and sensitive RAS-wildtype subgroups. C. Ratio p-MEK/total MEK in RAS-mutant (MUT) and RAS-wildtype (WT) t(4;11)-rearranged infant ALL samples shows increased MEK activation in RAS-mutant samples, while the MEK inhibitor resistant and sensitive RAS-wildtype samples have comparable MEK activation. D. Ratio p-ERK/total ERK in RAS-mutant and RAS-wildtype t(4;11)-rearranged infant ALL samples shows no significant differences in ERK activation between subgroups. Open circles indicate matched diagnosis (wildtype) and relapse (mutant) samples. Horizontal bars present group medians. Open circles indicate matched diagnosis (wildtype) and relapse (mutant) samples. *p<0.05.
Subsequently, we determined phosphorylation levels of MEK (p-MEK) and ERK (p-ERK) by immunoblotting (Supplementary Figure 2A and 2B, respectively). Quantification of the blots indicated a significantly higher level of p-MEK in our RAS-mutant samples, compared to RAS-wildtype samples (p=0.0312, Figure 3C), although there was no difference in p-MEK levels between the MEK inhibitor resistant and sensitive RAS-wildtype subgroups. Still, we did find a higher p-MEK level in the mutated relapse sample compared to its matched wildtype diagnosis sample. Additionally, no differences in p-ERK levels were found between RAS-wildtype and RAS-mutant samples (Figure 3D), nor between RAS-wildtype cells that were sensitive or resistant to MEK inhibition.
In MLL-rearranged AML, MEK inhibitor resistance can occur through activation of tyrosine kinase receptor (TKR) signaling (i.e. involving VEGFR-2) [5]. Furthermore, we previously found MLL-rearranged ALL is characterized by high expression of Fms-like tyrosine kinase 3 (FLT3) [8]. Therefore, we interrogated available gene expression profiles of primary samples for possible differences in TKR expression levels between the MEK inhibitor sensitive and resistant subgroups (Supplementary Figure 3). Interestingly, apart from FLT3, expression of TKRs is relatively low in the different patient samples. Surprisingly, FGFR-1 expression is significantly lower in MEK inhibitor resistant RAS-wildtype samples (p=0.02), while there are no significant differences in expression of FLT3, VEGFR (1-3), FGFR (2-4), EGFR and ERBB (2-4), PDGFR (A-B) or Lck and Src.
MEK inhibition results in reduced ERK phosphorylation
Next, we exposed MLL-rearranged ALL cell lines SEM and KOPN8 to the MEK inhibitors (Selumetinib, MEK162 and Trametinib) and determined p-ERK and p-MEK levels by immunoblotting (Figure 4). Interestingly, p-ERK levels were drastically reduced in both SEM and KOPN8, already after 6 hours of exposure, and this effect was sustained for at least 48 hours, regardless of the inhibitor used (Figure 4A). Furthermore, prolonged exposure (24 and 48 hours) to the MEK inhibitors Selumetinib and MEK162 resulted in an increase of p-MEK in SEM and KOPN8 (Figure 4B). Additionally, we determined phosphorylation of ERKs downstream effector ELK-1, but ELK-1 activation was not influenced by MEK inhibition (Supplementary Figure 4A).
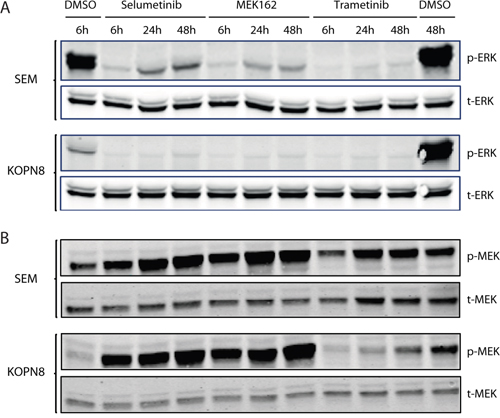
Figure 4: MEK inhibition results in reduced ERK phosphorylation. A. Western blot analysis of SEM and KOPN8 (upper and lower panels, respectively) exposed to 500 nM of MEK inhibitor or vehicle control (DMSO) for 6, 24 and 48 hours. Both cell lines almost completely lose ERK phosphorylation (p-ERK), while total ERK (t-ERK) levels remain unaffected. B. Analysis of MEK phosphorylation (p-MEK) suggests exposure to MEK162 and Selumetinib results in enhanced MEK phosphorylation in both cell lines, whereas total MEK (t-MEK) levels remain constant.
Since SEM cells responded modestly to MEK inhibition but did show a significant loss of p-ERK levels, we investigated whether these cells could circumvent loss of ERK activation by upregulating RAS-mediated PI3K-Akt-mTOR signaling. Therefore, the downstream phosphorylation of Akt (Ser437) and p70S6K (Thr389) was assessed by immunoblotting. However, no differences in Akt and p70S6K phosphorylation were observed in response to MEK inhibitor exposure (Supplementary Figure 4B and 4C).
MEK inhibitors induce apoptosis
Subsequently, we investigated the phenotypic effects of the MEK inhibitors on SEM and KOPN8 through analysis of early and late apoptosis markers (Annexin-V and 7-AAD, respectively), using flow-cytometry. Interestingly, both RAS-wildtype SEM and RAS-mutant KOPN8 undergo early apoptosis, after treatment with MEK inhibitor (Figure 5A and 5B, respectively). However, while late apoptosis is barely observed for SEM (Figure 5C), late apoptosis in MEK inhibitor exposed KOPN8 cells is enhanced substantially, especially after prolonged exposure (Figure 5D), suggesting the response to MEK inhibition is characterized by increased apoptosis. Furthermore, MEK inhibitor exposure induced protein levels of pro-apoptotic BIM, most evidently for KOPN8, while p53 levels remained unaffected (Supplementary Figure 5). Additionally, we investigated cell cycle progression under influence of MEK inhibition. Interestingly, no considerable differences in SEM or KOPN8 cell cycle progression were observed after 96 hours exposure to MEK162, Selumetinib or Trametinib (Figure 5E and 5F, respectively), nor after exposure for 24, 48 and 72 hours (Supplementary Figure 6A-6B, 6C-6D and 6E-6F, respectively).
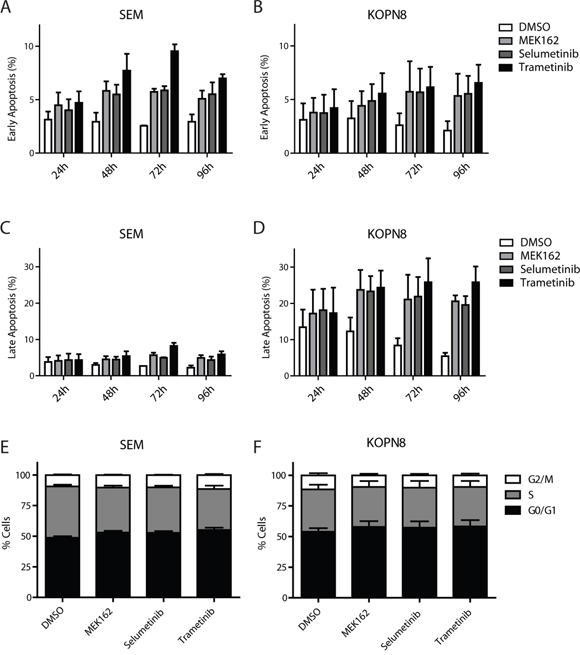
Figure 5: MEK inhibitors induce apoptosis. A. and B. Early apoptosis (percentage AnnexinV single positive of total) of SEM and KOPN8 cells (respectively) after exposure to DMSO vehicle (white bars) or 500 nM MEK162, Selumetinib or Trametinib (light grey, dark grey and black bars, respectively), indicates MEK inhibition slightly induces early apoptosis. Data are represented as mean +/- sem. n=3. C. and D. Late apoptosis (percentage AnnexinV and 7-AAD double positive cells of total) of SEM and KOPN8 (respectively) show that while SEM cells have no induction of late apoptosis in response to MEK inhibition, compared to the DMSO controls, KOPN8 clearly undergoes apoptosis, especially after prolonged exposure (>48 hours). Data are represented as mean +/- sem. n=3. E. and F. Cell cycle analysis of SEM and KOPN8 (respectively) after 96 hours exposure to vehicle (DMSO) or 500 nM MEK162, Selumetinib or Trametinib indicates MEK inhibition does not impinge on the cell cycle progression. Stacked bar graph indicates percentage of cells in G0/G1 (black), S (grey) and G2/M (white) cell cycle stages. Data are represented as mean +/- sem. n=3.
MEK inhibition enhances prednisolone sensitivity
In our previous study, we found that MLL-rearranged infant ALL patient samples harboring RAS mutations are more resistant to prednisolone [3]. Therefore, we examined whether inhibition of MEK could enhance prednisolone sensitivity of RAS-mutant cells. As shown in Figure 6A, prednisolone alone decreased cell viability of SEM cells to only ~50%. Interestingly, while Trametinib by itself induced only minor cell viability decrease in SEM cells (Figure 6B), the combination of Trametinib and prednisolone greatly enhanced the efficacy of prednisolone, especially at higher concentrations (Figure 6A). The combination of prednisolone and Trametinib also strongly decreased cell viability in KOPN8 more potently than either drug alone; low concentrations of Trametinib nearly eradicated all KOPN8 cells that did not respond to prednisolone treatment (Figure 6C). A similar sensitizing effect was observed when exposing SEM and KOPN8 to MEK162 or Selumetinib in combination with prednisolone (Supplementary Figure 7A-7D and 7F-7I, respectively). Since Trametinib alone already effectively decreases viability of KOPN8 cells (Figure 6D), we quantified the combinatorial effect of MEK inhibitors and prednisolone using the synergy factor (FSyn) calculation, as previously described [9, 10]. The plot in Figure 6E shows the fractional effect (i.e. the relative decrease of cell viability) induced by the combination of Trametinib with prednisolone, and the corresponding Synergy Factor. Interestingly, in both SEM and KOPN8 cells we observed FSyn values < 0.5, indicating strong synergy between Trametinib and prednisolone. Also combining MEK162 or Selumetinib with prednisolone resulted in moderate to strong synergistic effects (Supplementary Figure 7E and 7J, respectively). Additionally, we investigated whether this enhanced effect was related to differential expression of the glucocorticoid receptor (GR), the target of prednisolone. However, MEK inhibitor exposure did not alter GR protein levels in either SEM or KOPN8 cells (Supplementary Figure 7K).
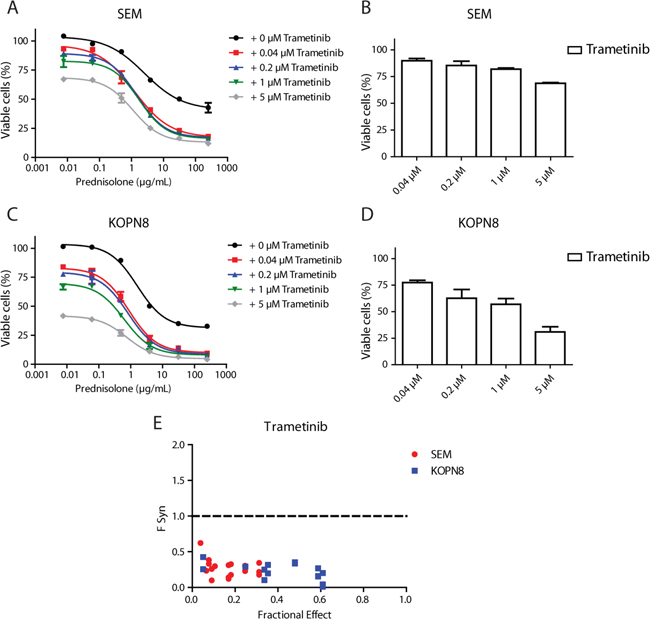
Figure 6: MEK inhibition enhances prednisolone sensitivity. A. Dose-response curves of the SEM cell line exposed to prednisolone alone (black curve) or in combination with 0.04 μM, 0.2 μM, 1 μM or 5 μM Trametinib (red, blue, green and grey curves, respectively). Low concentrations of Trametinib particularly sensitize cells that escape high concentrations of prednisolone. Data are represented as mean +/- sem. n=3. B. Response of SEM to the single Trametinib concentrations used in A. n=3. C. Dose-response curves of KOPN8 treated with prednisolone (black curve), or in combination with the aforementioned Trametinib concentrations (shown in red, blue, green and grey, respectively). KOPN8 cells are also sensitized towards prednisolone by co-exposure with low concentrations of Trametinib. Data are represented as mean +/- sem. n=3 D. KOPN8 exposed to single Trametinib concentrations. Data are represented as mean +/- sem. n=3. E. Combined exposure to prednisolone and Trametinib (merged data from 3 separate experiments) was quantified using FSyn calculations (FSyn<1 indicates synergy) and plotted against fractional effect (i.e. inhibition of cell viability). In SEM (red) moderate to strong synergy was observed, while all combinations of Trametinib and prednisolone result in strong to very strong synergy in KOPN8 (blue).
DISCUSSION
MLL-rearranged ALL in infants is a high-risk hematologic malignancy, characterized by a high incidence of relapse and high mortality rate [11]. Recently, we showed that 14-24% of these patients carry a RAS mutation, as an independent predictor of extremely poor outcome [3]. In the present study, we demonstrate that the MEK inhibitors Trametinib, Selumetinib and MEK162 display strong anti-leukemic effects against RAS-mutant MLL-rearranged ALL cells. Considering the dismal prognosis for infants suffering from MLL-rearranged ALL with additional RAS mutations, our data supports application of these inhibitors in the treatment of this patient group. Recently, Irving et al. already showed that Selumetinib effectively inhibits leukemia progression in an in vivo model of RAS-mutant BCP-ALL, and Burgess et al. found Trametinib to prolong the survival of mice transplanted with NRASG12D AML cells [12, 13]. Moreover, Trametinib has recently been approved for the treatment of adult BRAF-mutated melanoma, while different clinical trials with Selumetinib and MEK162 show promising results in adult patients with RAS/RAF mutation positive melanoma and non-small-cell lung cancer [14–18]. Even though most clinical trials focused on solid tumors in adult patients, pediatric clinical trials are underway for neurofibromas and gliomas, and could expedite clinical application of these MEK inhibitors in MLL-rearranged infant ALL.
Interestingly, while all RAS-mutant MLL-rearranged ALL patient samples are susceptible to MEK inhibition, patients without RAS mutations also might benefit from MEK inhibitor treatment, since a subgroup of RAS-wildtype patient samples appears sensitive to MEK inhibition. While in our previous study, we identified RAS mutations and found no BRAF aberrations, mutations of other upstream regulators, i.e. tyrosine kinase receptors, can occur in other malignancies [3]. Andersson et al. recently showed that additional somatic mutations in MLL-rearranged infant ALL, like (sub-)clonal RAS/PI3K pathway aberrations, occur in up to 50% of the cases, supporting our previous observation that RAS mutations in MLL-rearranged infant ALL frequently occur at a sub-clonal level [3, 19]. These findings do not support the hypothesis that other (upstream) mutations are driving RAS-MEK-ERK signaling, but also do not explain observed extensive MEK inhibitor sensitivity of all (subclonal) RAS-mutant and specified RAS-wildtype patient samples. While we found enhanced RAS and MEK activation in RAS-mutant samples, these biomarkers could not differentiate MEK inhibitor sensitive and resistant RAS-wildtype samples. Interestingly, Kampen et al. recently proposed a MEK inhibitor escape mechanism in MLL-rearranged AML, which was mediated by VEGFR-2 and PI3K-signaling, and we wondered whether this could play a role in the MEK inhibitor resistance of our wildtype patient cells [5]. However, we observed no difference in downstream PI3K-signalling (i.e. Akt or p70S6K phosphorylation) in response to MEK inhibitor exposure. Additionally, we discovered no significant tyrosine kinase receptor expression differences in MLL-rearranged infant ALL patient samples that could explain the MEK inhibitor response of RAS-wildtype samples. Surprisingly, FGFR-1 expression was lower in MEK inhibitor resistant samples, but it is unclear how this would explain MEK inhibitor resistance. Alternatively, Minjgee et al. report that RAS-mutant transfected cells can induce downstream RAS signaling in a paracrine manner, through excretion of cytokines [20]. Interestingly, Nakanishi et al. previously demonstrated that MLL-fusion proteins can induce ERK phosphorylation through regulating EphA7 receptor tyrosine kinase expression, but this was not accompanied by increased RAF or MEK phosphorylation [21]. Still, their data shows that leukemic cells carrying the t(4;11) translocation are sensitive to small molecule inhibitors of ERK phosphorylation. These findings indicate alternative regulatory mechanisms for ERK signaling in MLL-rearranged leukemia could explain the MEK inhibitor sensitivity we observe in RAS-wildtype cells.
Loss of ERK phosphorylation in response to MEK162, Selumetinib or Trametinib exposure confirmed the effect of MEK inhibition. Interestingly, prolonged exposure of cells to MEK162 or Selumetinib resulted in increased MEK phosphorylation. Previously, Hatzivassiliou et al. showed that the aromatic fluorine of allosteric MEK inhibitor GDC-0973 interacts with MEK residue S212 [22]. Their data indicate this interaction results in exposure of the phosphorylation sites S218/S222, which are then susceptible to RAF mediated phosphorylation. Since MEK162 and Selumetinib both have this aromatic fluorine, the mechanism of interaction with MEK is probably similar to GDC-0973. Hence, although MEK activation in presence of GDC-0973, MEK162 or Selumetinib can still occur, the transduction of the signal by MEK-mediated phosphorylation of ERK is no longer possible, as we show in Figure 4.
Recently, we found the presence of RAS mutations in MLL-rearranged infant ALL cells correlated with prednisolone resistance, an obstacle in the treatment of infant ALL [2, 3]. Remarkably, our present data shows that MEK inhibition strongly enhances the sensitivity of both RAS-wildtype and RAS-mutant MLL-rearranged ALL cells to prednisolone, also further exemplifying the possible value of MEK inhibitors for RAS-mutant, as well as RAS-wildtype, MLL-rearranged infant ALL patients. The prednisolone-sensitizing effect of MEK inhibitors proposes a possible role for RAS-MEK-ERK signaling in the response to glucocorticoids. Recent work by Jones et al. shows that MEK plays a key role in drug resistance in relapsed pediatric ALL, and that MEK inhibition can sensitize ALL relapse samples to chemotherapeutics, including methylprednisolone [23]. Moreover, Ariës et al. found Trametinib could restore prednisolone sensitivity in RAS-mutant BCP-ALL patient samples, whereas Rambal et al. showed that MEK activation reduces dexamethasone sensitivity, and the MEK inhibitor PD183452 enhanced dexamethasone responses in ALL cells in a BIM-dependent manner [24, 25]. Activated ERK can phosphorylate BIM, targeting it for proteasomal degradation, and thereby diminishing apoptosis induced by dexamethasone [26]. Moreover, we established that, while glucocorticoid receptor expression remains constant, MEK inhibition upregulates pro-apoptotic BIM, which implies that inhibiting MEK, resulting in abrogation of ERK phosphorylation, may result in prolonged maintenance of pro-apoptotic BIM activity upon prednisolone exposure, leading to enhanced prednisolone sensitivity. This is further supported by our previous study showing that in MLL-rearranged ALL, prednisolone sensitization mediated by pan-BCL-2 family inhibitors was largely driven by the up-regulation of pro-apoptotic BID and BIM [27].
In summary, our data shows that RAS-mutant MLL-rearranged infant ALL patients may benefit from therapeutic strategies administering small-molecule MEK inhibitors. Furthermore, since MEK inhibition sensitizes MLL-rearranged ALL cells to prednisolone regardless of the RAS mutations status, RAS-wildtype MLL-rearranged infant ALL patients may also benefit from MEK inhibitor treatment through enhanced sensitivity to prednisolone.
MATERIALS AND METHODS
Patient samples and cell lines
Bone marrow and peripheral blood samples from untreated infant ALL patients were collected at the Sophia Children’s Hospital (Rotterdam, The Netherlands) as part of the international collaborative INTERFANT treatment protocol [2]. Approval for these studies was obtained from the Erasmus MC Institutional Review Board. Informed consent was obtained according to the Declaration of Helsinki. All samples were processed within 24 hours after sampling as described before, with optional removal of contaminating non-leukemic cells by immunomagnetic beads, to ensure leukemic blast content for all samples was >90% [8]. The t(4;11)-rearranged ALL cell line SEM and t(11;19)-rearranged ALL cell line KOPN8 were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany), while the t(4;11)-rearranged ALL cell line RS4;11 was purchased from The Global Biosource Center (ATCC, Middlesex, UK). All cell lines were cultured in suspension in RPMI-1640 with GlutaMAX (Invitrogen Life Technologies, Waltham, MA, USA) supplemented with 10% Fetal Calf Serum, 100 IU/mL penicillin, 100 IU/mL streptomycin and 0.125 μg/mL amphotericin B (Invitrogen Life Technologies) at 37°C under 5% CO2 atmosphere.
In vitro cytotoxicity assay and small molecule inhibitors
The in vitro cytotoxicity of MEK162, Selumetinib and Trametinib (MedChem Express, Stockholm, Sweden) was tested by MTS and MTT assays. All inhibitors were weighed, dissolved in dimethyl sulfoxide (DMSO) and stored at -20°C until use. Cytotoxicity assay dilutions were prepared in cell culture medium, keeping final DMSO concentration <0.5%. Final concentrations of the small molecule inhibitors ranged from 50 μM to 0.15 nM, indicated in the respective figures. The in vitro sensitivity of cell lines was assessed by using 4-day MTS conversion assays, as described previously [28]. In vitro cytotoxicity of patient cells was assessed by using a 4-day MTT conversion assay, as described before [8]. Data were normalized to vehicle (DMSO) controls.
Western blot analysis
Protein extracts (25 μg) were electrophoretically resolved on pre-cast SDS-polyacrylamide gels (anyKD, TGX, Bio-Rad, Veenendaal, The Netherlands) and transferred to nitrocellulose membranes. Membranes were blocked with 5% bovine serum albumin and subsequently probed with antibodies directed against total or phosphorylated ERK, MEK, ELK-1, Akt, or p70S6K (Cell Signaling, Danvers, MA, USA). Membranes were counterstained with IRDye® 680/800 conjugated secondary antibodies (Li/COR, Leusden, The Netherlands) and were scanned by an Odyssey imaging system (Li/COR). Membranes were re-probed with mouse monoclonal anti-β-actin antibodies (Sigma-Aldrich, St. Louis, MO, USA) as loading control. Fluorescence was quantified using the Odyssey 3.0 application software.
RAS activation
RAS activation was analyzed using the RAS Activation Assay Kit (17-218, Merck-Millipore, Amsterdam, The Netherlands). Briefly, 1x107 cells were isolated and lysed with Mg2+ Lysis Buffer (MLB), and stored at -80°C until use. GST-fused RAF-1 RBD bead slurry was added to the lysate and incubated for 1 hour at 4°C while agitating. Beads were isolated by centrifugation and washed with MLB, and precipitated protein was denatured with Laemmli buffer at 95°C before immunoblotting. As a positive control, total cell lysate was included in the immunoblotting procedure. The provided RAS antibody (05-516, Merck-Millipore) was used, and GST (Cell Signaling) and β-actin (Sigma-Aldrich) antibodies were used as loading controls for the beads and total protein, respectively. Fluorescence was quantified using the Odyssey 3.0 application software.
Annexin-V/7-AAD apoptosis and cell cycle assays
For assessment of early and late apoptosis, the PE Annexin-V Apoptosis Detection Kit (BD Pharmingen, Breda, The Netherlands) was used according to the manufacturer’s protocol. Briefly, drug-exposed cells were isolated, washed with PBS and re-suspended in binding buffer. Cells were stained with PE Annexin V and/or 7-AAD for 15 minutes, and sorted using fluorescence activated cell sorting (FACS). Cell cycle progression was assessed by permeabilization of isolated cells through hypotonic lysis. Subsequently, RNAse treatment was performed, and DNA was stained using Propidium Iodide, after which FACS determined DNA content. Data was analyzed using FlowJo software (FlowJo, Ashland, OR, USA).
Gene expression data
Recently published gene expression data (Affymetrix HU133plus2.0) for part of the t(4;11)+ patient samples was available (i.e. for 6 of 9 MEK inhibitor resistant RAS-wildtype samples, 4 of 5 sensitive RAS-wildtype samples and 3 of 6 RAS-mutant samples) [29]. This data is available in GEO database19 (accession number GSE19475) and was acquired as previously described [29]. Tyrosine kinase receptor expression was derived from this dataset, using the following probe sets: 206674_at (FLT3), 204406_at (VEGFR-1), 203934_at (VEGFR-2), 234379_at (VEGFR-3), 210973_s_at (FGFR-1), 208225_at (FGFR-2), 204380_s_at (FGFR-3), 204579_at (FGFR-4), 211551_at (EGFR), 210930_s_at (ERBB2), 226213_at (ERBB3), 214053_at (ERBB4), 205463_s_at (PDGFR-A), 217112_at (PDGFR-B), 204891_s_at (Lck) and 213324_at (Src).
Statistical analysis
Statistical analyses were performed with SPSS Statistics version 17.0 (SPSS Inc. Chicago, IL, USA). All tests were two-tailed and p-values < 0.05 were considered significant. The effect of combining drugs (i.e. synergy, additivity or antagonism) was assessed using Berenbaums criteria, as previously described [9, 10]. Briefly, we calculated the Synergy Factor (FSyn) with the formula FSyn = ([Drug Xin combination with Y]/[Drug X]) + ([Drug Yin combination with X]/[Drug Y]) for a particular fractional effect. If the drug combination results in FSyn < 1, this is considered synergy.
ACKNOWLEDGMENTS
The authors would like to thank the members and participating institutes of the INTERFANT-99 study for generously providing leukemic samples.
Members of the INTERFANT-99 study are as follows: M. Campbell (Programa Infantil Nacional de Drogas Atineoplasicas (PINDA)), M. Felice (Argentina), A. Ferster (Children’s Leukemia Group (CLCG)), I. Hann and A. Vora (UK Children’s Cancer Study Group (UKCCSG)), L. Hovi (Nordic Society of Paediatric Haematology and Oncology (NOPHO)), G. Janka-Schaub (Cooperative Study Group for Treatment of ALL (COALL)), C.K. Li (Hong Kong), G. Mann (Berlin-Frankfurt-Munster Group-Austria (BFM-A)), T. LeBlanc (French ALL Group (FRALLE)), R. Pieters (Dutch Childhood Oncology Group (DCOG)), G. de Rossi and A. Biondi (Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP)), J. Rubnitz (St Jude Children’s Research Hospital (SJCRH)), M. Schrappe (Berlin-Frankfurt-Munster Group-Germany (BFM-G)), L. Silverman (Dana-Farber Cancer Institute (DFCI)), J. Stary (Czech Paediatric Haematology (CPH)), R. Suppiah (Australian and New Zealand Children’s Haematology/Oncology Group (ANZCHOG)), T. Szczepanski (Polish Paediatric Leukemia and Lymphoma Study Group (PPLLSG)), and M. Valsecchi and P. de Lorenzo (Trial Operating Center (CORS)).
We thank P. Garrido Castro and D. Geerts for scientific input and reading the manuscript.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
GRANT SUPPORT
This work was financially supported by research funding from KIKA (Kinderen Kankervrij). Furthermore, R.W. Stam was financially supported by the Dutch Cancer Society (KWF Kankerbestrijding).
REFERENCES
1. Greaves MF. Infant leukaemia biology, aetiology and treatment. Leukemia. 1996; 10:372–77. doi:
2. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, Hovi L, LeBlanc T, Szczepanski T, Ferster A, Janka G, Rubnitz J, Silverman L, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007; 370:240–50. doi:10.1016/S0140-6736(07)61126-X.
3. Driessen EM, van Roon EH, Spijkers-Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, Pieters R, Stam RW. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013; 98:937–44. doi:10.3324/haematol.2012.067983.
4. Emerenciano M, Barbosa TC, de Almeida Lopes B, Meyer C, Marschalek R, Pombo-de-Oliveira MS. Subclonality and prenatal origin of RAS mutations in KMT2A (MLL)-rearranged infant acute lymphoblastic leukaemia. Br J Haematol. 2015; 170:268–71. doi:10.1111/bjh.13279.
5. Kampen KR, Ter Elst A, Mahmud H, Scherpen FJ, Diks SH, Peppelenbosch MP, de Haas V, Guryev V, de Bont ES. Insights in dynamic kinome reprogramming as a consequence of MEK inhibition in MLL-rearranged AML. Leukemia. 2014; 28:589–99. doi:10.1038/leu.2013.342.
6. Lavallée VP, Baccelli I, Krosl J, Wilhelm B, Barabé F, Gendron P, Boucher G, Lemieux S, Marinier A, Meloche S, Hébert J, Sauvageau G. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat Genet. 2015; 47:1030–37. doi:10.1038/ng.3371.
7. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72:2457–67. doi:10.1158/0008-5472.CAN-11-2612.
8. Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, van der Voort E, Valsecchi MG, de Lorenzo P, Sallan SE, Armstrong SA, Pieters R. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005; 106:2484–90. doi:10.1182/blood-2004-09-3667.
9. Spijkers-Hagelstein JA, Pinhanços SS, Schneider P, Pieters R, Stam RW. Chemical genomic screening identifies LY294002 as a modulator of glucocorticoid resistance in MLL-rearranged infant ALL. Leukemia. 2014; 28:761–69. doi:10.1038/leu.2013.245.
10. Berenbaum MC. Synergy, additivism and antagonism in immunosuppression. A critical review. Clin Exp Immunol. 1977; 28:1–18. doi:
11. Biondi A, Cimino G, Pieters R, Pui CH. Biological and therapeutic aspects of infant leukemia. Blood. 2000; 96:24–33. doi:
12. Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, Swidenbank I, Ponthan F, Kirschner-Schwabe R, Groeneveld-Krentz S, Hof J, Allan J, Harrison C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014; 124:3420–30. doi:10.1182/blood-2014-04-531871.
13. Burgess MR, Hwang E, Firestone AJ, Huang T, Xu J, Zuber J, Bohin N, Wen T, Kogan SC, Haigis KM, Sampath D, Lowe S, Shannon K, Li Q. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014; 124:3947–55. doi:10.1182/blood-2014-05-574582.
14. Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V, Kemsley K, Dummer R. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012; 18:555–67. doi:10.1158/1078-0432.CCR-11-1491.
15. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, et al, and METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012; 367:107–14. doi:10.1056/NEJMoa1203421.
16. Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013; 14:249–56. doi:10.1016/S1470-2045(13)70024-X.
17. Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, Lacouture ME, Panageas KS, Wolchok JD, Carvajal RD, Schwartz GK, Rosen N, Chapman PB. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013; 19:2257–64. doi:10.1158/1078-0432.CCR-12-3476.
18. Jänne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, Smith I, Crinò L. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013; 14:38–47. doi:10.1016/S1470-2045(12)70489-8.
19. Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, Nakitandwe J, Holmfeldt L, Parker M, Easton J, Huether R, Kriwacki R, Rusch M, et al, and St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015; 47:330–37. doi:10.1038/ng.3230.
20. Minjgee M, Toulany M, Kehlbach R, Giehl K, Rodemann HP. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys. 2011; 81:1506–14. doi:10.1016/j.ijrobp.2011.05.057.
21. Nakanishi H, Nakamura T, Canaani E, Croce CM. ALL1 fusion proteins induce deregulation of EphA7 and ERK phosphorylation in human acute leukemias. Proc Natl Acad Sci USA. 2007; 104:14442–47. doi:10.1073/pnas.0703211104.
22. Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013; 501:232–36. doi:10.1038/nature12441.
23. Jones CL, Gearheart CM, Fosmire S, Delgado-Martin C, Evensen NA, Bride K, Waanders AJ, Pais F, Wang J, Bhatla T, Bitterman DS, de Rijk SR, Bourgeois W, et al. MAPK signaling cascades mediate distinct glucocorticoid resistance mechanisms in pediatric leukemia. Blood. 2015; 126:2202–12. doi:10.1182/blood-2015-04-639138.
24. Ariës IM, van den Dungen RE, Koudijs MJ, Cuppen E, Voest E, Molenaar JJ, Caron HN, Pieters R, den Boer ML. Towards personalized therapy in pediatric acute lymphoblastic leukemia: RAS mutations and prednisolone resistance. Haematologica. 2015; 100:e132–36. doi:10.3324/haematol.2014.112995.
25. Rambal AA, Panaguiton ZL, Kramer L, Grant S, Harada H. MEK inhibitors potentiate dexamethasone lethality in acute lymphoblastic leukemia cells through the pro-apoptotic molecule BIM. Leukemia. 2009; 23:1744–54. doi:10.1038/leu.2009.80.
26. Iglesias-Serret D, de Frias M, Santidrián AF, Coll-Mulet L, Cosialls AM, Barragán M, Domingo A, Gil J, Pons G. Regulation of the proapoptotic BH3-only protein BIM by glucocorticoids, survival signals and proteasome in chronic lymphocytic leukemia cells. Leukemia. 2007; 21:281–87. doi:10.1038/sj.leu.2404483.
27. Spijkers-Hagelstein JA, Schneider P, Pinhanços SM, Garrido Castro P, Pieters R, Stam RW. Glucocorticoid sensitisation in Mixed Lineage Leukaemia-rearranged acute lymphoblastic leukaemia by the pan-BCL-2 family inhibitors gossypol and AT-101. Eur J Cancer. 2014; 50:1665–74. doi:10.1016/j.ejca.2014.03.011.
28. Hartsink-Segers SA, Zwaan CM, Exalto C, Luijendijk MW, Calvert VS, Petricoin EF, Evans WE, Reinhardt D, de Haas V, Hedtjärn M, Hansen BR, Koch T, Caron HN, et al. Aurora kinases in childhood acute leukemia: the promise of aurora B as therapeutic target. Leukemia. 2013; 27:560–68. doi:10.1038/leu.2012.256.
29. Stam RW, Schneider P, Hagelstein JA, van der Linden MH, Stumpel DJ, de Menezes RX, de Lorenzo P, Valsecchi MG, Pieters R. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood. 2010; 115:2835–44. doi:10.1182/blood-2009-07-233049.