
INTRODUCTION
Hypoxia is a key micro-environment factor in aggressive tumors that induces angiogenesis to supply cells with oxygen and nutrients. Therapies using antiangiogenic drugs have thus been developed to stop tumors from growing their own blood vessels. Nonetheless, it is reported that antiangiogenic therapy induces acute hypoxic stress, leading to tumor invasion and metastasis because of the acquisition of compensatory mechanisms, which could select specific tumor cell populations able to grow and proliferate in low oxygen environment [1]. In tumor cells, hypoxia was previously shown to induce expression of HLA-G gene [2, 3], an immune checkpoint gene whose function is involved in immune evasion [4]. Otherwise, the use of DNA methylation inhibitors such as 5-aza-2′deoxycytidine (5-aza-dC; decitabine) has been proposed in cancer therapy [5–9] and was demonstrated to induce HLA-G expression in vitro [10, 11]. Therefore, hypoxia environment combined with anticancer therapy might favor HLA-G expression and consequently tumor progression. The identification of molecular mechanisms implicated in the HLA-G induction in these conditions is of crucial importance in the scope of developing future anti-cancer therapies and more particularly immunotherapies.
Human leukocyte antigen G (HLA-G) is a non-classical HLA class I molecule of the major histocompatibility complex [12]. It exhibits low polymorphism [13], and restricted tissue distribution in placenta [14] and immuno-privileged sites such as thymus and cornea [15, 16]. Alternative splicing of the primary transcript leads to 7 mRNAs that can be translated into 7 protein isoforms, membrane bound (G1 to G4) and soluble (G5 to G7) forms [17–19]. HLA-G isoforms regulate the immune responses by modulating the function of natural killer cells, cytotoxic T lymphocytes and antigen presenting cells, through at least two inhibitory receptors IL-T2 [20, 21] and IL-T4 [22]. More particularly, HLA-G induces T regulatory cells (Tregs) and regulatory antigen presenting cells (APCs) that modulate immune response and promote tolerance [23]. Recently, evidence was given that the binding of HLA-G to IL-T2 receptor suppresses B cell responses [24]. Physiologically, HLA-G plays a key role in maintaining immune tolerance at the fetal-maternal interface and has been associated with a lower risk of development of acute and chronic rejection in allograft transplantation [25–27]. This immune checkpoint molecule is also expressed in numerous tumor types, contributing to their immune evasion [28], and its expression has been correlated with poor clinical outcome of patients [11, 28–32]. In agreement, using a xenotumor model or syngeneic tumor cells in mice, it has been reported that HLA-G is involved in tumor immune surveillance in vivo [33, 34]. Therefore, regarding the growing and successful use of antibodies against checkpoints such as anti-CTLA-4 and anti- PD-L1/PD-1 to restore antitumor immunity [35], HLA-G appears as a wider and promising relevant target for cancer immunotherapy [12].
Nonetheless, despite the accumulation of data on the identification of elements controlling HLA-G expression most of them are still to be described. They differ from those of classical HLA class I gene since almost all known regulatory sequences for these genes are disrupted in the HLA-G promoter region [36]. In fact, it is demonstrated that HLA-G gene can be regulated by specific regulatory sequences such as Enhancer L at –12 kb from ATG [37], LINE-1 at –4 kb [38] and LCR at –1.2 kb [39], and specific transcription factors such as CREB1/ATF [40], RREB1[40–42], HSF1[42], IRF1[43] and PR [44]. In addition, we and others have demonstrated the modulation of HLA-G expression by epigenetic mechanisms (DNA methylation and histone modifications) [10, 45], and micro-environmental factors [46–50] . Particularly, HLA-G can be induced by hypoxia [51, 52] or hypoxia-mimicking conditions as shown previously with melanoma cell lines cultured in the presence of desferrioxamine (DFX) [2, 53].
Hypoxia is a physiologically relevant tumor-related stress defined by a decrease of cellular oxygen concentrations which in turn leads to an adaptive response modulating the expression of genes involved in angiogenesis, erythropoiesis and glycolysis [54]. The signal transduction in hypoxic cells is mainly conducted by the basic helix-loop-helix bHLH/PAS hypoxia inducible factor (HIF) [55, 56] comprising a labile HIF-α subunit (HIF-1α and HIF-2α isoforms) whose stability is regulated by an oxygen-dependent hydroxylation, and a stable HIF-β subunit. Under hypoxia or hypoxia-mimicking (DFX) conditions, stabilized HIF-1α translocates into the nucleus where it binds HIF-1β. The α/β heterodimeric transcription factor then recognizes hypoxia responsive elements (HREs) present on target genes causing activation of transcription [57]. Regarding HLA-G, a consensus HRE has been identified by in silico analysis at –242 bp upstream of the ATG target site [53]. Nonetheless, whether or not this site is involved in the DFX-induced HLA-G transcription in melanoma cells [2, 53], is still unsolved.
Moreover, we recently demonstrated that HLA-G expression may be upregulated in grade IV glioblastoma [11], a very aggressive tumor in which hypoxic microenvironment plays a key role in the disease progression [58]. We also reported that 5-aza- dC demethylating treatment induced HLA-G expression in a cellular model of human glioblastoma, the HLA-G negative U251MG glioma cell line [11]. Since melanocytes and glial cells are both derived from similar embryological origin, and their malignant transformed counterparts share many biological properties and express common tumor markers, we thus investigated U251MG cell line for the induction of HLA-G expression and the associated regulatory mechanisms driven by hypoxia-mimicking conditions alone or combined with 5-aza-dC.
RESULTS
HLA-G expression in U251MG glioma cells is induced by hypoxia-mimicking DFX and is upregulated by DFX combined with DNA demethylating treatment
We first investigated the effect of hypoxia-mimicking agent desferrioxamine (DFX) treatment on HLA-G expression in U251MG glioma cells (Figure 1A and Supplementary Figure S1). We observed a significant (P < 0.05) upregulation of HLA-G gene transcription suggesting that HIF could be involved in the activation of HLA-G expression. Giving that demethylating agent 5-aza-dC has been proposed for anti-tumoral therapy, and having previously demonstrated that DNA demethylation is crucial for HLA-G expression, we hypothesized that CpG methylation in HIF target sites (5′-RCGTG-3′) could moderate the observed HLA-G mRNA induction. We thus exposed cells to the demethylating agent 5-aza-dC, using standard conditions, at 100 μM for 72 hours, and then to DFX at 400 μM for an additional 24 hours. Real-time RT-PCR analysis revealed a strong upregulation of HLA-G transcripts following 5-aza-dC treatment alone, and although not significant (P = 0.07) an additional 2× mean-fold enhancement of HLA-G transcript levels was observed following both 5-aza-dC and DFX treatments (Figure 1A). In the latter condition, we also observed by Western blot analysis the upregulation of HLA-G protein expression (Figure 1B). Therefore, hypoxia-mimicking microenvironment may be a pertinent parameter participating in the HLA-G gene activation and in the enhancement of HLA-G gene expression when DNA demethylation is occurring.
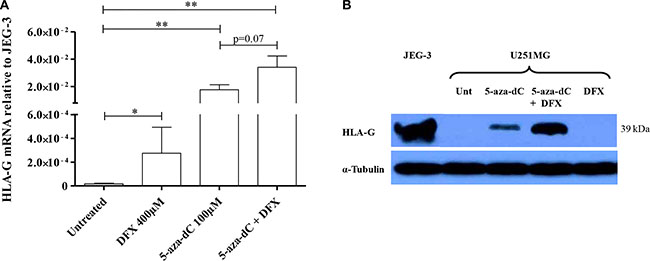
Figure 1: Hypoxia-mimicking conditions induce HLA-G expression in HLA-G negative glioblastoma cell line U251MG. (A) Real-time RT-PCR analysis, targeting all HLA-G mRNA forms, carried out on cells cultured with normal growth conditions or treated with DFX at 400 μM for 24 h, 5-aza-2′-deoxycytidine (5-aza-dC) at 100 μM for 72 h, or 5-aza-dC combined to DFX (3 independent experiments in duplicates). The cells exposed to 5-aza-dC exhibit a ~103 mean-fold increase in HLA-G transcript level, while combined treatments give an additional ~2 mean-fold increase in HLA-G transcript level (3 independent experiments in duplicates). Data is presented as mean ± SEM and statistical analysis was performed using Mann-Whitney U tests (* indicates a p value < 0.05 and ** indicate a p value < 0.01). (B) Representative western blot analysis of HLA-G activation revealed with 4H84 mAb (2 independent experiments). U251MG cells were either treated with conditions described above or not (Unt: untreated). α-Tubulin was used as an internal control. JEG-3: choriocarcinoma cell line expressing HLA-G.
HIF-1 is involved in HLA-G gene expression under hypoxia-mimicking conditions
In order to investigate the role of HIF in the upregulation of HLA-G gene expression under hypoxia-mimicking microenvironment we transfected U251MG cells either with a control shRNA (sh-IRR) or with a shRNA directed against the α subunit of the transcription factor (sh-HIF-1α). Western blot analysis performed with the wild type cells or the control shRNA transfectant showed HIF-1α stabilization at 3 h following DFX treatment. On the contrary, HIF-1α was reduced both at the transcriptional and the protein expression levels following DFX treatment in cells transfected with sh- HIF-1α (Figure 2A). Then, to test the effect of HIF-1 on HLA-G gene transcriptional activity, we performed real time RT-PCR analysis on the control shRNA transfectant and the HIF-1α shRNA transfectant, both exposed to DFX and/or the demethylating agent 5-aza-dC using the same conditions as above. Under hypoxia-mimicking conditions, amounts of HLA-G gene transcripts produced by the sh-IRR transfectant were not achieved with the sh-HIF-1α transfectant (Figure 2B), either in the absence or the presence of 5-aza-dC in the culture medium (Figure 2B). These results support that the upregulation of HLA-G transcription in cells cultured in the presence of DFX involves HIF-1.
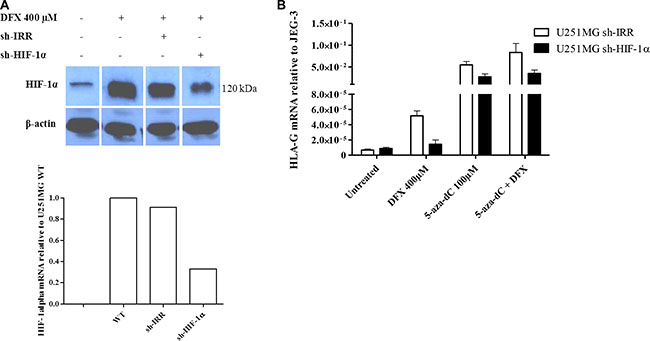
Figure 2: HIF-1 is implicated in the activation of HLA-G expression in hypoxia-mimicking conditions. (A) Representative western blot and real-time RT-PCR analysis of HIF-1α expression in U251MG wild type cells (WT), or transfected with an irrelevant shRNA (sh-IRR) or HIF-1α specific shRNA (sh-HIF-1α). Cells were treated with DFX (400 μM) for 3 h. Western blot was performed with cytoplasmic extracts and β-actin as an internal control. HIF-1α and β-actin bands were collected from the same gel to assemble the picture. qRT-PCR results were compared to those of U251MG WT (assigned a value of 1). (B) Real-time RT-PCR analysis of HLA-G expression in transfected U251MG cells (U251MG sh-IRR and U251MG sh-HIF-1α; 2 independent experiments in duplicates for each transfectant), either treated with 5-aza-dC (100 μM) for 72 h and DFX (400 μM) for 24 h, or not. Results were compared to those of HLA-G positive cell line JEG-3 (assigned a value of 1).
The 1.4 kb-5′ untranslated regulatory region (5′UTR) of HLA-G gene contains ineffective HIF-1 target sites
In silico analysis of the 1.4 kb HLA-G region upstream to the ATG translation initiation site revealed two potential HREs containing the hypoxia binding site (HBS) 5′-RCGTG- 3′ at positions –242 bp (5′-TCCCA GGGCCTCAAGCGTGGCTCTCA-3′) and –966 bp (5′-TA AAAACAGGCAGTGCGTGAGCACTAGTGAGGGG-3′) (Figure 3A). Interestingly, the –966 HBS contains a natural A/G polymorphism (-966(A)/-966(G)) located at the 5′end of the site [59].
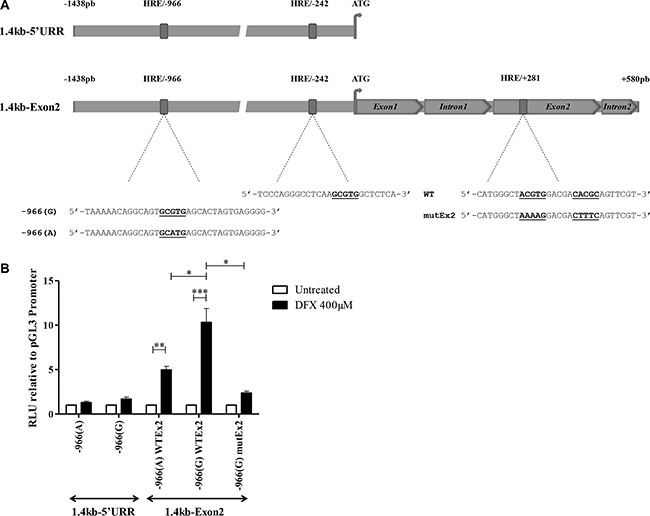
Figure 3: HIF-1α activates HLA-G expression through HREs located on the HLA-G locus. (A) Schematic representation of the location of HREs in the 1.4 kb-5′URR and 1.4 kb-Exon2 fragments of HLA-G cloned in pGL3 basic vector upstream of the firefly luciferase gene. The sequence of each HRE is described. –966 HBS contains a natural G/A polymorphism indicated as –966(G) and –966(A). +281 HRE contains a double HBS that was cloned either in its wild type (WT) or double mutated (mutEx2) forms. (B) Luciferase activity assayed with U251MG transfected with either constructs, treated or not with DFX (400 μM) for 24 h. Results were normalized to luciferase activity in U251MG cells transfected with pGL3 Promoter vector (Promega), and compared to luciferase activity in untreated cells (assigned a value of 1). A co-transfected pRL-TK Renilla Luciferase reporter vector served as internal control for transfection efficiency and extract preparation. Data are presented as mean ± SEM of 3 independent experiments in duplicates, and statistical analysis was performed using Wilcoxon matched-pairs signed rank test (* indicates a p value < 0.05, ** indicate a p value < 0.01 and *** indicate a p value < 0.001).
To assess the functionality of these putative HIF target sites, we first analyzed the activity of the 1.4kb-5′UTR variant having an intact –966 HBS (G form) in U251MG cells. The fragment was subcloned upstream of the firefly luciferase reporter gene into the promoterless pGL3 basic vector (Figure 3A). Extracts from transfected cells, subjected or not to DFX treatment during 24 h, were analyzed for luciferase activity. We observed that the activity of the 1.4 kb-5′UTR was slightly affected by DFX treatment and was not statistically significant, suggesting that HREs in the 1.4 kb HLA-G promoter region are ineffective (Figure 3B).
We also performed EMSA using double stranded 32P labeled oligonucleotides, containing putative HLA-G –242 HBS or –966(G) HBS (Figure 4A). The HIF- 1 stabilization and binding was controlled using a 32P labeled oligonucleotide containing HRE sequence from the transferrin gene promoter (TFHBS), and nuclear protein extracts pre-incubated with an anti-HIF-1α antibody for supershift experiments. In agreement with luciferase experiments, we noted that these two HLA-G HREs were unable to bind HIF-1 while a HIF-1/TFHBS complex was observed (Figure 4A). We also performed competition experiments with a 32P labeled TFHBS oligonucleotide in the presence of an excess of cold HLA-G –242 or –966(G) oligonucleotides. The –242 oligonucleotide did not alter the complex, thus confirming that this HLA-G HRE is nonfunctional. However, we observed that an excess of –966(G) oligonucleotide altered the HIF-1/TFHBS complex, suggesting a slight affinity to HIF- 1 in comparison to –242 HBS, but not strong enough to transactivate HLA-G promoter in luciferase assays (Figure 4B).
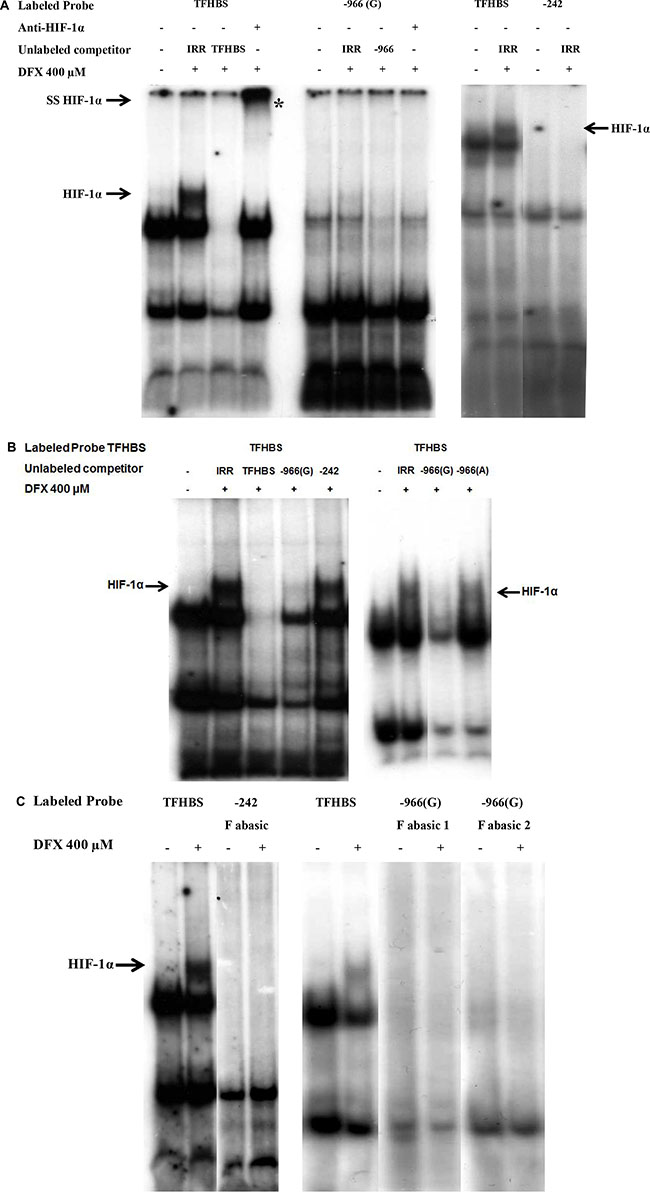
Figure 4: Absence of HIF-1α binding on –966 and –242 HREs of HLA-G promoter in EMSA performed with nuclear extracts of U251MG cells treated (+) or not (–) with DFX (400 μM) for 3 h. Radiolabeled probe TFHBS (transferrin gene HBS) was used as a positive control for HIF-1α binding. (A) EMSA performed with radiolabeled probes –966(G) and –242. Competition experiments were carried out by adding a 100-fold molar excess of unlabeled double stranded irrelevant (IRR) or cold specific probe. HIF-1α is demonstrated by supershift analysis using nuclear extracts incubated overnight with an anti-HIF-1α antibody prior to binding experiments. The supershifted complex is indicated by «SS HIF-1α» and «*». (B) EMSA performed with radiolabeled probe TFHBS and 100-fold molar excess of unlabeled competitors. –966(A) is the mutated form of –966(G) HBS. (C) EMSA performed with radiolabeled probes containing abasic sites: –242 F abasic; –966 (G) F abasic 1; –966 (G) F abasic 2 (see Material and methods). The HIF-1α/Probe complex is indicated by an arrow «HIF-1α» on the gel.
Finally, it is known that hypoxia responsive elements are prone to oxidative damage to DNA, leading to the formation of an abasic site in one strand, with especially high sensitivity to oxidative damage of the terminal guanines in the HIF-1 DNA recognition sequence [60, 61]. Since a greater binding affinity of HIF-1 to these abasic sites was previously observed in vitro, we generated abasic sites for –242 or –966(G) oligonucleotides. EMSA, performed with these oligonucleotides incubated with nuclear extracts of U251MG cells treated or not with DFX during 3 h, did not reveal any HIF-1 binding (Figure 4C). This strongly suggests that efficient HIF target sites at the HLA-G locus, if any, are located outside the 1.4kb promoter sequence.
HIF-1 transactivates HLA-G gene through a HRE located in exon 2
In an attempt to identify functional HIF target sites along the HLA-G locus, we focused on a putative HRE we localized by in silico analysis in the exon 2 (Figure 3A). The sequence contains two HBSs at positions +281 bp (sens) and +291 pb (antisens) from ATG. To investigate this putative HRE, we performed luciferase assays with U251MG cells transfected with the promoterless pGL3 basic vector, in which we inserted a DNA fragment covering both the 1.4kb-5′UTR and a sequence spanning ATG to position +580 pb at the beginning of intron 2 of HLA-G gene (1.4 kb-Exon2 construction) (Figure 3A). The level of luciferase activity observed with extracts from U251MG cells transfected with 1.4 kb-Exon2 and treated with DFX was significantly enhanced in comparison to the level of luciferase activity of untreated transfected cells (Figure 3B). These results strongly argue for a major role of this region in the DFX response. Interestingly, a 5 mean-fold enhancement was observed in the presence of –966(A) SNP and a 10 mean-fold enhancement in the presence of –966(G) SNP. Next, using a mutated 1.4 kb-Exon2 fragment in which the two HBSs at +281 bp and +291 bp have been scrambled, we showed that mutations impaired the upregulation of luciferase activity, thus demonstrating that exon 2 HRE is a major target for factors induced by hypoxia mimicking DFX (Figure 3B).
To further investigate that HLA-G HRE in exon 2 was a direct target for HIF, we first carried out EMSAs with nuclear extracts of U251MG cells exposed or not to DFX during 3 h and 32P-labeled double stranded oligonucleotide containing +281 HBS and +291 HBS (+281 HRE). Incubation of nuclear extracts of cells exposed to DFX with the labeled +281 HRE oligonucleotide revealed a specific complex which was absent when nuclear extracts of untreated cells were used (Figure 5A). This complex disappeared after adding an excess of cold wild type +281 HRE oligonucleotide, and was shifted with an anti-HIF-1α antibody. We also performed experiments with +281 HRE oligonucleotide containing abasic HBSs that generated a DFX-induced specific complex, exhibiting a greater intensity than the wild type sites (Figure 5A). Competition experiments conducted with mutated +281 HRE in which the two HBS have been scrambled did not alter the pattern. In contrast, the competition was partial in the presence of an excess of cold oligonucleotides in which only one of the HBS was mutated (Figure 5B). Finally, supershift experiments performed with nuclear extracts of DFX-treated cells, incubated with 32P-labeled +281 HRE in the presence of anti-HIF-1α and anti-HIF-2α antibodies, identified only the presence of HIF-1α in the complex (Figure 5C).
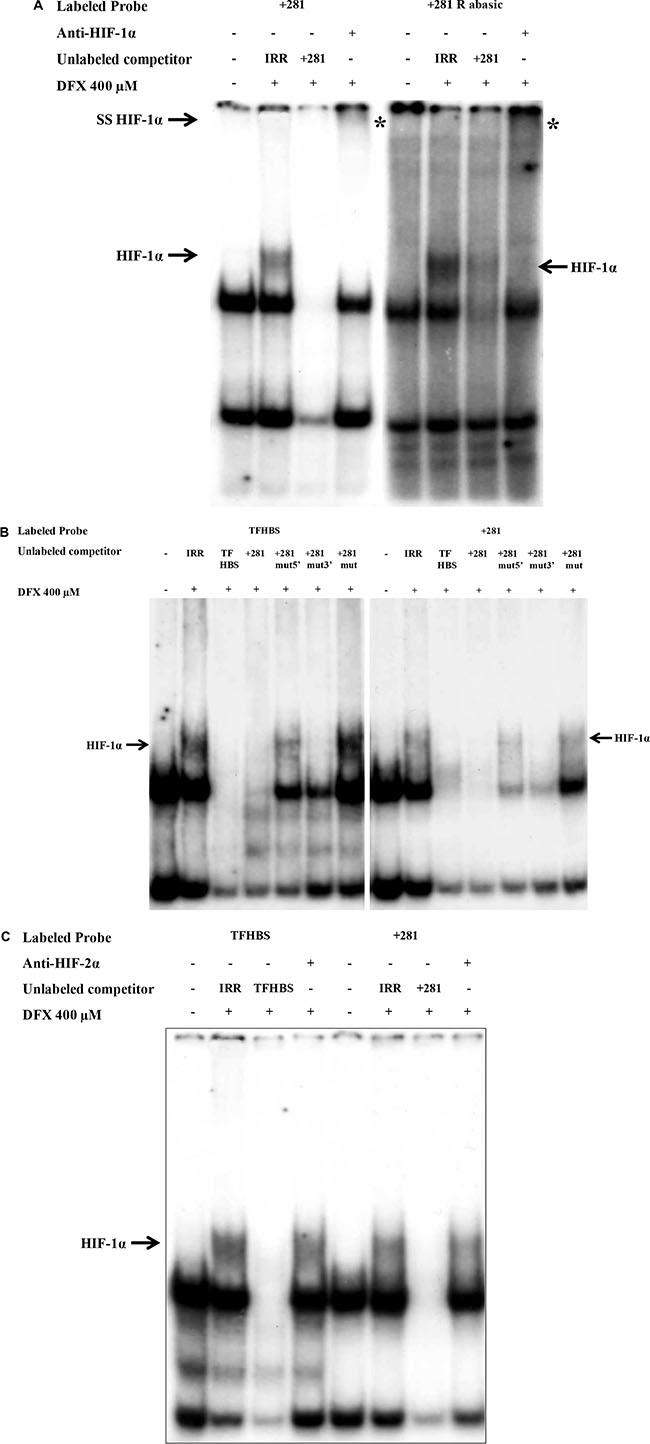
Figure 5: EMSA validation of HIF-1α binding to the HRE located in exon 2 of HLA-G gene (+281) with nuclear extracts of U251MG cells treated (+) or not (–) with DFX (400 μM) for 3 h. (A) EMSA performed with radiolabeled HLA-G probes containing +281 HRE with 2 HBS (5′ and 3′), either wild type (+281) or abasic (+281 R abasic). Competition experiments were realized by adding a 100- fold molar excess of unlabeled double stranded irrelevant or specific cold probe. The HIF-1α/Probe complex is indicated by an arrow «HIF-1α». Nuclear extracts were also incubated overnight with an anti-HIF-1α antibody prior to binding experiments. The supershifted complex is indicated by an arrow «SS HIF-1α» and «*». (B) EMSA performed with radiolabeled control probe TFHBS (Transferrin gene HBS) and +281 HRE in the presence of a 100-fold molar excess of unlabeled competitors: IRR (irrelevant), TFHBS, +281, +281 mut5′ (5′ HBS mutated 5′-CATGGGCTAAAAGGACGACACGCAGTTCGT-3′), 281 mut3′ (3′ HBS mutated 5′-CATGGGCTACGTGGACGACTTTCAGTTCGT-3′) and +281 mut (3′ and 5′ HBS mutated 5′-CATGGGCTAAAAGGACGACTTTCAGTTCGT-3′). (C) EMSA performed with TFHBS and +281 probe with nuclear extracts incubated overnight with anti-HIF-2α antibody prior to binding experiments.
Additionally, we analyzed the HIF-1 binding to the HLA-G locus by ChIP experiments on U251MG cells. PCR targeting HLA-G exon 2 revealed that stabilized HIF-1 was positioned at this chromatin region of the HLA-G locus when cells were cultured with conditions allowing both HIF-1α stabilization (DFX during 3 h) and high level of HLA-G gene induction (5-aza-dC treatment) (Figure 6).
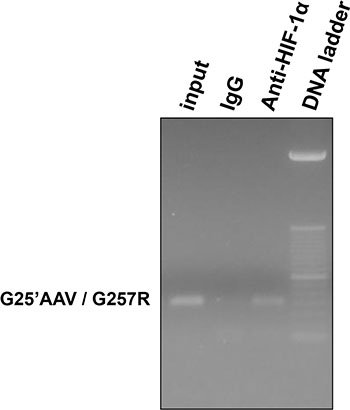
Figure 6 : HIF-1 binding to the HLA-G gene in U251MG cells treated with 100 μM 5-aza-dC (72 hours) and 400 μM DFX (additional 3 hours). ChIP experiment performed with anti-HIF-1α antibody or control Rabbit IgG. G25′AAV / G257R indicates primer set used for PCR targeting exon 2 of HLA-G gene; Input indicates input chromatin used as PCR control.
Altogether, these results demonstrate that HRE in exon 2 of HLA-G gene is a functional target site for HIF-1 in response to hypoxia-mimicking conditions, and strongly suggest that –966(G) SNP may influence the level of the response.
HLA-G HBS in U251MG cells are differentially methylated
Having demonstrated the impact of demethylating agent in the enhancement of HLA-G transcriptional activity in DFX-treated U251MG cells, we finally analyzed the methylation status of HLA-G HREs using primer sets AMP1 and AMP2 targeting 5′ UTR, and primer set AMP3 targeting exon 2. Sodium bisulfite conversion and pyrosequencing of DNA from U251MG cells (2 independent experiments) revealed 100 % conversion of cytosines located outside CpG dinucleotide context (data not shown). Notably we observed that the percentage of CpG methylation was 91% at +281 HBS, 66.5% at +291 HBS (+281 HRE) and 91.4% at –966 HRE (Table 1). Lower CpG methylation at + 291 HBS is thus in agreement with HIF-1-mediated gene activation in cells treated with DFX alone. In addition these results support that demethylation treatment is likely to favor HIF-1 binding following DFX treatment.
Table 1: HLA-G HBS methylation in U251MG cells analyzed by pyrosequencing of bisulfite treated DNA (mean of 2 independent experiments)
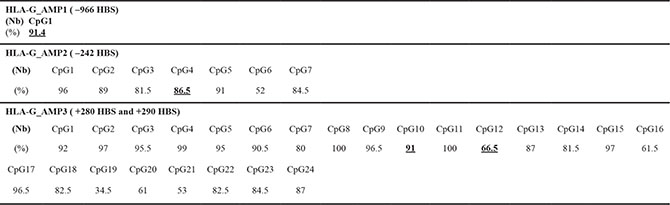
Percentage of methylation (%) and number of CpG (Nb) analyzed for each amplicon from 5′ to 3′. Bold and underlined values correspond to CpG located within the HLA-G HBS.
DISCUSSION
HLA-G immune checkpoint plays a role in maintaining immune tolerance at the fetal-maternal interface, and was shown to participate in tumor immune escape [4, 33]. Hypoxia is a common micro-environmental factor to these major contexts of HLA-G expression [62, 63]. We here demonstrate the induction of HLA-G in glioma cells under hypoxia-mimicking conditions and, for the first time, a functional HIF-1 target site. It is noteworthy that binding of HIF-1 to a transcriptionally active HRE was previously demonstrated for the gene encoding another checkpoint molecule, PD-L1 [64], suggesting that a close interaction between hypoxia and immune regulation exits. Although the amount of HLA-G transcription and protein expression was predominantly controlled by DNA demethylation processes, we also observed that the magnitude of HLA-G expression was still boosted by superimposing hypoxia-mimicking conditions. Therefore, the present work and previous ones, demonstrating that HLA-G gene expression in melanoma cells is induced by hypoxic conditions [2, 53], clearly support the contribution of this micro-environment factor in the expression and the level of HLA-G expression. Hypoxia-mimicking conditions were also shown to activate factors associated with extracellular matrix degradation and invasion of glioma cells in vitro [65, 66]. Moreover, hypoxia was recently demonstrated to induce in vivo macrophage polarization and re-education toward an M2 phenotype in U251MG glioblastoma model [67]. Therefore hypoxia appears as a key factor in the processes that could contribute to glioma evasion.
Unexpectedly, luciferase assays and EMSA experiments revealed that despite the identification of 2 putative HREs in the 5′ UTR, HLA-G gene induction occurred more particularly through the HRE located in exon 2. It is a fact that regulatory elements located within the transcribed portions of genes are common, but such elements in coding exons are much less frequent. Some of the documented examples include c-myc (exon 1) [68], rabbit α-globin (fragment extending from exon 1 to intron 2) [69] and keratin-18 (exon 6) [70], and concern both positive and negative regulatory elements. Regarding classical HLA class I genes, a negative regulatory element has been described in exon 1 [71]. Moreover, a large number of potential high affinity HIF-binding sites have been identified across the genome in promoter regions, exons and introns, and many remote from known promoters [72–74]. Demonstration has also been made that HIF DNA binding can have an effect on target gene regulation over great distances, and that the hypoxia-inducible gene expression was qualitatively and quantitatively similar, independently of the position of HIF binding on the gene locus [72].
Many studies have characterized the robustness of HIF binding sites. In the binding regions, the A predominates over G at the R position of the core 5′-RCGTG-3′ motif, and there are HIF sequence preferences beyond the HBS. Indeed, HIF binding regions are GC rich, and there is a HIF preference for T at –1 bp of the R position and for a C or G at –2 bp of the R position [72, 75]. It is remarkable that none of these preferences are observed for –242 putative HBS, which could explain the lack of HIF binding to this region in EMSA. On the other hand, all the requirements are met in the HRE we identified in the exon 2 and also at –966 bp. In addition, unlike the –966(G), the +281 HRE region contains a double HBS in close vicinity to each other, which may explain the great affinity of this region to HIF binding. Such an interesting feature of the HIF DNA binding sequence was previously described for the transferrin gene enhancer [76]. In fact, in this same study, the authors showed that a single HBS derived from the Epo gene was not sufficient to induce expression of a reporter gene, and that concatamerized HBSs were necessary to achieve it. They argued that a single HBS in isolation is not sufficient to convey full hypoxic activation of oxygen-regulated genes and that it requires additional cis-acting elements. Such elements differ between genes and could be a CACA box described for the Epo [55] and VEGF genes [77], an activator protein (AP-1) site [78], a cAMP-responsive element [79], but also the HIF site itself.
On the other hand, we have shown that the labeled –966(G) oligonucleotide was not able to bind alone HIF- 1 in EMSA experiments. However an excess of cold oligonucleotide –966(G) competed both with the TFHBS and +281 HRE. Moreover, in luciferase assays, the presence of –966(A) (–966(A) WTEx2 construct) instead of –966 (G) (–966(G) WTEx2 construct) within the 1.4kb-Exon2 construct, provided a significantly lower luciferase activity upon DFX treatment. These results indicate that, although alone it was not able to bind HIF, the –966(G) HBS plays a role in HLA-G gene regulation in hypoxia-mimicking environment.
Collectively, these findings strongly suggest that the primary HIF-1 binding site is the +281 HRE, and that it might connect with the transcriptional complex at the promoter of HLA-G by DNA looping, thus rendering the –966 site accessible for HIF-1 binding. DNA looping was previously documented over large distances, making transcriptional activity independent of physical distance along chromosomes [80–83]. Thus –966 G/A polymorphism could contribute to the magnitude of HLA-G expression [84] during pregnancy and cancer, more particularly glioblastoma.
We observed that the amount of HLA-G transcription and protein expression was predominantly controlled by DNA demethylation processes. This result is compatible with previous findings on the role of DNA methylation in HLA-G expression in tumor cells [45, 85–87]. However, it is striking that the effect of DFX was strong enough to still be observed when cells were cultured in medium containing both hypoxia-mimicking and demethylating agents. In agreement with the influence of epigenetics in HIF-mediated response, it is demonstrated the importance of the basal transcriptional permissive state of the genes under normal growth conditions, notably RNA Pol II occupancy on the promoter, and/or histone modifications [72, 88]. Consistently, it is documented that DNA demethylation, as hypoxia, is a key process during pregnancy [89–91] and cancer [92, 93]. Global DNA hypomethylation occurs in the early embryo that expresses HLA-G [94]. On the contrary, an overall progressive increase in average methylation is observed from first to third trimester placenta [95, 96] with a decrease in HLA-G expression, while global hypomethylation has been associated with tumor progression [97, 98]. In addition to the effect of changes in chromatin structure, HREs contain CpG dinucleotides that are potentially methylated, and HREs could become functional following 5-aza-dC treatment. Recent reports showed that HIF binds HREs that are hypomethylated in genes where DNA methylation has been documented as an important gene modulation mechanism [99]. Interestingly, following 5-aza-dC treatment of ovarian cancer cells (BG-1) HLA-G –242 site was shown to remain methylated, while hypomethylation of sequences was observed within 5′ HLA-G regulatory region [85].
In conclusion, our results support that HLA-G could be involved in glioblastoma evasion to antiangiogenic therapy [100, 101], since de novo acute hypoxic stress will lead to HIF-1 stabilization and then binding to the exon 2 HRE. In such a situation –966 G/A polymorphism could modulate the magnitude of HLA-G expression and thus should be taken into account. Besides, 5-aza-dC treatment has been proposed to develop tumor antigen specific active immunotherapy or to reduce telomerase expression in glioblastoma [5]. In such a situation, resident hypoxia and DNA demethylation acting both at the HLA-G promoter region and HRE sites, might provide favorable conditions for ectopic HLA-G expression and then tumor invasion.
MATERIALS AND METHODS
Cell line and culture
The U251MG glioblastoma cells (kindly provided by Dr H. Wiendl, Department of Neurology, University of Wuerzburg, Wuerzburg, Germany) were maintained in DMEM (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% FBS (Sigma-Aldrich, Saint Louis, MO), Gentamycin at 0.02 mg/ml and Fungizone Amphotericin B at 0.25 μg/ml.
Cells were grown in the appropriate medium with hypoxia-mimicking conditions provided by the presence of desferrioxamine (DFX) (Sigma-Aldrich) at 200 μM and 400 μM [2, 53]. (Supplementary Figure S1). Experiments combining both hypoxia-mimicking conditions and DNA demethylation were conducted by treating cells with 5-aza-dC (Sigma-Aldrich) at 100 μM during 72 h and then DFX at 400 μM (concentration providing significant HLA-G upregulation) during 24 h.
Plasmids and transfections
Constructions with the promoterless pGL3 basic vector were performed as previously described [45] using 1438 bp of HLA-G promoter region 5′ to the ATG initiation codon (1.4 kb-5′UTR construction). The DNA fragments covering both the 1.4 kb HLA-G region and a region spanning ATG to 87 bp of intron 2 (1.4 kb-Exon2 construction) were obtained by PCR using the following specific reverse oligonucleotide 5′-TTTTAGATCTCAGA CCCGGAGACTCGGGAG-3′ that contains a BglII restriction site. Mutated HBSs were introduced within the 1.4 kb-Exon2 construction using complementary mutated oligonucleotides carrying the same mutations as in EMSA. Site-directed mutagenesis of the two HBSs in exon 2 was performed by MilleGen (Labège, France). In all luciferase assays, the cells were co-transfected with the pRL-TK Renilla Luciferase reporter vector (Promega, Charbonnières-les-Bains, France) used as a control of the variations of the transfection efficiency and protein extracts preparation.
The HIF-1α shRNA plasmid and the control shRNA Plasmid A (Sc-44225 and Sc-108060, Santa Cruz Biotechnology, Inc, Santa Cruz, CA) were used to silence HIF-1α expression in U251MG cells according to manufacturer’s instructions. Briefly, transfections with shRNA plasmids were facilitated by the Lipofectamin 2000 reagent (Invitrogen Life Technologies). The Lipofectamin-DNA mixture was added to the cells incubated in serum and antibiotic free medium that was replaced by supplemented medium after 4 h. In transient expression, the assays were executed 48 h after transfection. In stable expression, cells were cultured in the medium supplemented with Puromycin (Invivogen, Toulouse, France) at 3.5 μg/ml.
QRT-PCR analysis
Total RNA was extracted using TRIzol® reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions. Residual DNA was eliminated by DNAse I treatment (New England Biolabs, Evry, France) for 10 min at 37°C. 5 μg of total RNA from each sample was retrotranscribed to cDNA with Moloney murine leukemia virus (M-MLV)-Reverse Transcriptase (Invitrogen Life Technologies) at 42°C during 1 h. 0.5 μg of cDNA samples was used as template for amplification reactions carried out with the TaqMan real-time PCR assay in an ABI 7000 sequence detection system (Applied Biosystems Life Technologies, Villebon sur Yvette, France). For each experiment, mixed PCR reaction has been performed in duplicates, in the presence of GAPDH specific oligonucleotides as an internal standard (Applied Biosystems Life Technologies), HLA-G specific primers 5′-CTGGTTGTCCTTGCAGCTGTAG-3′ (forward) and 5′-CCTTTTCAATCTGAGCTCTTCTTTCT-3′ (reverse) (Eurogentec, Seraing, Belgium) and the TaqMan TAMRA probe 5′-CACTGGAGCTGCGGTCGCTGCT-3′ (Applied Biosystems Life Technologies). Quantification relative to HLA-G+ JEG-3 (assigned a value of 1) was determined by the ΔΔCt method [10].
HIF-1α expression analysis was assessed using specific primers 5′-CCTTCGATCAGTTGTCACCA TTAGA-3′ (reverse) 5′-ACTGTAACTGTGCTTTGAGGA CTTG-3′ (forward) and the TaqMan FAM probe 5′-CAGTTCCGCAAGCCCT-3′ (Applied Biosystems Life Technologies). GAPDH was used as a reporter gene (Applied Biosystems Life Technologies). Each experiment was repeated at least three independent times.
Protein extraction and western blot
Protein extracts from cells, either cultured or not with DFX and/or 5-aza-dC, were isolated using the ProteoJET Cytoplasmic and Nuclear Protein Extraction kit (Fermentas Thermo Fisher Scientific, Villebon sur Yvette, France), in the presence of protease and phosphatase inhibitors according to manufacturer’s instructions. The extracts were quantified using the BCA Protein Assay kit (Pierce Thermo Fisher Scientific, Illkirch, France). Proteins were then separated on 4 %–8 % or 4 %–12 % gradient polyacrylamid gels in TG-SDS 1× Buffer. Proteins were transferred on a Hybond-C Extra membrane (Amersham Biosciences, Velizy-Villacoublay, France) in TG 1x-Ethanol 20% Buffer. The membranes were blocked for 30 min in 5% non-fat milk in phosphate buffered saline (PBS) containing 0.2 % Tween 20, and incubated with the primary antibody overnight at 4°C: HIF-1α (BD Transduction Laboratories), HLA-G 4H84 clone at 1 μg/ml (Exbio, Praha, Czech Republic), and β-actin at 0.5 μg/ml (Sigma-Aldrich). After washing in PBS-Tween, membranes were incubated for 1 h at room temperature with peroxidase-labeled secondary antibodies. The immunoreactive bands were visualized by chemiluminescence (Amersham Biosciences).
Electrophoretic mobility shift assay (EMSA)
γ-32P (Amersham) double stranded oligonucleotides (Eurogentec) encompassing the HREs of HLA-G and transferrin (control) were as follow (only the top strands are shown; HBSs are underlined): HLA-G promoter region at –966 bp 5′-TAAAAACAGGCAGTGCG TGAGCACTAGTGAGGGG-3′ and –242 bp 5′-TCCCAG GGCCTCAAGCGTGGCTCTCA-3′ [53]; HLA-G exon 2 at +281 bp and +291 bp 5′-CATGGGCTACGTGGACGAC ACGCAGTTCGT-3′, and a consensus HBS from transferrin gene promoter TFHBS 5′-TTCCTGCACGTAC ACACAAAGCGCACGTATTTC-3′ [76]. Oligonucleotides which allowed us to create a uniquely located abasic site in the duplex at the HRE binding contained a dU replacing a G in the HBS: –966 bp F (Forward) abasic 1 5′-TAAAAACAGGCAGTdUCGTGAGCACTAGTGA GGGG-3′, –966 bp F abasic 2 5′-TAAAAACAGGCAGTG CGTdUAGCACTAGTGAGGGG-3′, and –242 bp F abasic 5′-TCCCAGGGCCTCAAGCGTdUGCTCTCA-3′, +281 bp F abasic 5′-CATGGGCTACGTdUGACGACACGCAGT TCGT-3′ and +281 bp R (Reverse) abasic 5′-ACG AACTGCGTdUTCGTCCACGTAGCCCATG-3′). The dU-containing oligonucleotide annealed to its wild type complement and abasic sites were generated by treating duplexes with Uracil N-glycosylase (New England Biolabs) during 30 min.
Nuclear extracts were incubated alone for 15 min at room temperature in Darnell binding buffer (KCl 400 mM, Hepes pH 7.5 200 mM, MgCl2 10 mM, EGTA 1 mM, DTT 5 mM and Ficoll 4%). Binding reactions were carried out by incubating nuclear extracts (2–4 μl) with radiolabeled oligonucleotide probes (1–2 ng; 1.5 × 105 cpm) for further 20 min at room temperature in the presence of 2 μg/μl sonicated salmon sperm DNA (Invitrogen Life Technologies) to reduce nonspecific binding. Competition experiments were assessed by adding a 100-fold molar excess of unlabeled double stranded oligonucleotides corresponding to irrelevant (5′-GCTCCTTCTGAGTATCTTTACA-3′), specific competitors and oligonucleotides with mutation introduced at the HBS: –966(A) 5′-TAAAAACAGG CAGTGCATGAGCACTAGTGAGGGG-3′, +281 mut 5′ 5′-CATGGGCTAAAAGGACGACACGCAGTTCGT-3′, +281 mut 3′ 5′-CATGGGCTACGTGGACGACTTTCA GTTCGT-3′ and +281 mut 5′-CATGGGCTAAAAGGAC GACTTTCAGTTCGT-3′). In order to identify HIF-1α and HIF-2α in the DNA-Protein complexes, supershift experiments were conducted with 2 μg of rabbit anti-HIF- 1α (sc-10790, Santa Cruz Biotechnology) and anti-HIF-2α (ab199, Abcam, Paris, France) antibodies incubated with the nuclear extracts before the addition of radiolabeled probe.
The DNA-Protein complexes were then subjected to electrophoresis on a 3 % non-denaturing polyacrylamide gel at room temperature in 0.5X TBE at 200 V for 2 h, and processed for autoradiography.
Luciferase reporter assays
Reporter assays were conducted using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Aliquots of cell lysates were transferred into a 96 well microplate and incubated successively with the Luciferase and Renilla substrates. Luminescence was recorded using the FluoStar Optima plate reader (BMG Labtech, Champigny sur Marne, France) for each reaction on the same aliquot. Luciferase activities were then corrected by normalization to the corresponding Renilla activities, to control the variations of the transfection efficiency and protein extracts preparation. All assays were performed in duplicates from three independent experiments.
DNA methylation analysis by pyrosequencing
Quantitative DNA methylation analysis was performed by pyrosequencing of bisulfite treated DNA [102]. One μg of DNA was bisulfite converted using the EpiTect 96 Bisulfite kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Regions of interest for validation were amplified using 30 ng of bisulfite treated human genomic DNA and 5 to 7.5 pmol of forward and reverse primer, one of them being biotinylated. Sequences for oligonucleotides for PCR amplification and pyrosequencing are indicated in Table 2. Reaction conditions were 1x HotStar Taq buffer supplemented with 1.6 mM MgCl2, 100 μM dNTPs and 2.0 U HotStar Taq polymerase (Qiagen) in a 25 μl volume. The PCR program consisted of a denaturing step of 15 min at 95°C followed by 50 cycles of 30 s at 95°C, 30 s at the respective annealing temperature and 20 s at 72°C, with a final extension of 5 min at 72°C. 10 μl of PCR product were rendered single-stranded as previously described [103] and 4 pmol of the respective sequencing primer were used for analysis. Quantitative DNA methylation analysis was carried out on a PSQ 96 MD system with the PyroGold SQA Reagent Kit (Qiagen) and results were analyzed using the PyroMark CpG software (V.1.0.11.14, Qiagen).
Table 2: Primers used in quantitative DNA methylation analysis performed by pyrosequencing of bisulfite treated U251MG DNA
Gene |
Size |
PCR primer forward |
PCR primer reverse |
Pyrosequencing primer(s) |
CpGs |
|---|---|---|---|---|---|
HLA-G_AMP1 chr6:29794412-29794951 |
114 |
Biotin-TGGATATTTTTT AAAAATAGGTAGTG |
TATTACAACCAAAAA CCAACACAAA |
TCAATACAATCA CAATACCC |
1 |
HLA-G_AMP2 chr6:29795132-29795671 |
222 |
GATTTAGGGAGA TATTGAGATAGAA |
Biotin-CACCTAATAAAAA TAAAAACTAAAACC |
GTTTGGTATAAGA GTAG |
1–7 |
HLA-G_AMP3 chr6:29795612-29796151 |
234 |
GTTTTTATTTTATG AGGTATTTTAG |
Biotin-ACAAATTCATTCT ATCAATCTATAC |
TTTATTTTATGAGG TATTTT |
1–21 |
GGAGTAGGAGGGGT |
22–24 |
Chromatin immunoprecipitation assays (ChIP)
To analyze in situ the binding of HIF-1α to the HLA-G locus we used CHIP-IT Express HT Assays (Active Motif, La Hulpe, Belgium) according to the manufacturer’s recommendation. Briefly, formaldehyde cross-linked chromatin from DFX- and 5-aza-dC-treated U251MG cells was sonicated and immunoprecipitated with either anti-HIF-1α antibody (NB100-105, Novus Biologicals, Littleton, CO, USA) or control Rabbit IgG, and protein G-coated magnetic beads. Immunoprecipitated DNA was amplified using specific exon 2 HLA-G primers (G25′AAV Forward: 5′-TCCATGAGGTATTTCAGCGC and G.257 Reverse: 5′-TGTTCCGTGTCTCCTCTTCC) and visualized with Ethidium Bromide coloration after agarose gel migration.
Statistical analysis
GraphPad Prism was used for statistical analysis. Data are presented as mean ± SEM and statistical analysis was performed for at least three independent experiments using Mann-Whitney U test or Wilcoxon matched-pairs signed rank test. A p value < 0.05 was considered significant.
ACKNOWLEGMENTS AND FUNDING
This work was supported by the Commissariat à l′Energie Atomique et aux Energies Alternatives (CEA), The Coordenação de Aperfeiçoamento do Pessoal de Nível Superior (CAPES; PDEE 3260/06-2), the binational research cooperation programs CAPES/Brazil-COFECUB/France (project Me 653/09), Programa Ciência sem Fronteiras « Pesquisador Visitante Especial » (CNPq grant 406594/2013-9) and Université Sorbonne Paris Cité « Mobilité Internationale Brésil 2015 ». L.Y was supported by IRTELIS fellowship from CEA, Fondation ARC pour la Recherche sur le Cancer and CAPES-COFECUB.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011; 8:210–221.
2. Mouillot G, Marcou C, Zidi I, Guillard C, Sangrouber D, Carosella ED, Moreau P. Hypoxia modulates HLA-G gene expression in tumor cells. Hum Immunol. 2007; 68:277–285.
3. Chang CC, Murphy SP, Ferrone S. Differential in vivo and in vitro HLA-G expression in melanoma cells: potential mechanisms. Hum Immunol. 2003; 64:1057–1063.
4. Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA, Avril MF, Dausset J, Guillet JG, Carosella ED. HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci USA. 1998; 95:4510–4515.
5. Patel R, Shervington L, Lea R, Shervington A. Epigenetic silencing of telomerase and a non-alkylating agent as a novel therapeutic approach for glioma. Brain Res. 2008; 1188:173–181.
6. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004; 64:4980–4986.
7. Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002; 21:5496–5503.
8. Ecke I, Petry F, Rosenberger A, Tauber S, Monkemeyer S, Hess I, Dullin C, Kimmina S, Pirngruber J, Johnsen SA, Uhmann A, Nitzki F, Wojnowski L, et al. Antitumor effects of a combined 5-aza-2′deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res. 2009; 69:887–895.
9. Everson RG, Antonios JP, Lisiero DN, Soto H, Scharnweber R, Garrett MC, Yong WH, Li N, Li G, Kruse CA, Liau LM, Prins RM. Efficacy of systemic adoptive transfer immunotherapy targeting NY-ESO-1 for glioblastoma. Neuro Oncol. 2016; 18:368–378.
10. Moreau P, Mouillot G, Rousseau P, Marcou C, Dausset J, Carosella ED. HLA-G gene repression is reversed by demethylation. Proc Natl Acad Sci USA. 2003; 100:1191–1196.
11. Wastowski IJ, Simoes RT, Yaghi L, Donadi EA, Pancoto JT, Poras I, Lechapt-Zalcman E, Bernaudin M, Valable S, Carlotti CG Jr, Flajollet S, Jensen SS, Ferrone S, et al. Human leukocyte antigen-G is frequently expressed in glioblastoma and may be induced in vitro by combined 5-aza-2′-deoxycytidine and interferon-gamma treatments: results from a multicentric study. Am J Pathol. 2013; 182:540–552.
12. Carosella ED, Rouas-Freiss N, Roux DT, Moreau P, LeMaoult J. HLA-G: An Immune Checkpoint Molecule. Adv Immunol. 2015; 127:33–144.
13. Castelli EC, Mendes-Junior CT, Veiga-Castelli LC, Roger M, Moreau P, Donadi EA. A comprehensive study of polymorphic sites along the HLA-G gene: implication for gene regulation and evolution. Mol Biol Evol. 2011; 28:3069–3086.
14. Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, HLA-G, expressed in human trophoblasts. Science. 1990; 248:220–223.
15. Crisa L, Mc Master MT, Ishii JK, Fisher SJ, Salomon DR. Identification of a thymic epithelial cell subset sharing expression of the class Ib HLA-G molecule with fetal trophoblasts. J Exp Med. 1997; 186 :289–298.
16. Le Discorde M, Moreau P, Sabatier P, Legeais JM, Carosella ED. Expression of HLA-G in human cornea, an immune-privileged tissue. Hum Immunol. 2003; 64:1039–1044.
17. Ishitani AG, D. E. Alternative splicing of HLA-G transcripts yields proteins with primary structures resembling both class I and class II antigens. Proc Natl Acad Sci USA. 1992; 89:3947–3951.
18. Kirszenbaum M, Moreau P, Gluckman E, Dausset J, Carosella E. An alternatively spliced form of HLA-G mRNA in human trophoblasts and evidence for the presence of HLA-G transcript in adult lymphocytes. Proc Natl Acad Sci USA. 1994; 91:4209–4213.
19. Paul P, Cabestre FA, Ibrahim EC, Lefebvre S, Khalil-Daher I, Vazeux G, Quiles RM, Bermond F, Dausset J, Carosella ED. Identification of HLA-G7 as a new splice variant of the HLA-G mRNA and expression of soluble HLA-G5, -G6, and -G7 transcripts in human transfected cells. Hum Immunol. 2000; 61:1138–1149.
20. Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, Angman L, Cella M, Lopez-Botet M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med. 1997; 186:1809–1818.
21. Wu D, Kuiaste I, Moreau P, Carosella E, Yotnda P. Rescuing lymphocytes from HLA-G immunosuppressive effects mediated by the tumor microenvironment. Oncotarget. 2015; 6:37385–37397. doi: 10.18632/oncotarget.6044.
22. Colonna M, Samaridis J, Cella M, Angman L, Allen RL, O’Callaghan CA, Dunbar R, Ogg GS, Cerundolo V, Rolink A. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol. 1998; 160:3096–3100.
23. Carosella ED, Gregori S, LeMaoult J. The tolerogenic interplay(s) among HLA-G, myeloid APCs, and regulatory cells. Blood. 2011; 118:6499–6505.
24. Naji A, Menier C, Morandi F, Agaugue S, Maki G, Ferretti E, Bruel S, Pistoia V, Carosella ED, Rouas-Freiss N. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell responses. J Immunol. 2014; 192:1536–1546.
25. Carosella ED, Favier B, Rouas-Freiss N, Moreau P, Lemaoult J. Beyond the increasing complexity of the immunomodulatory HLA-G molecule. Blood. 2008; 111:4862–4870.
26. Deschaseaux F, Delgado D, Pistoia V, Giuliani M, Morandi F, Durrbach A. HLA-G in organ transplantation: towards clinical applications. Cell Mol Life Sci. 2011; 68:397–404.
27. Brugiere O, Thabut G, Krawice-Radanne I, Rizzo R, Dauriat G, Danel C, Suberbielle C, Mal H, Stern M, Schilte C, Pretolani M, Carosella ED, Rouas-Freiss N. Role of HLA-G as a predictive marker of low risk of chronic rejection in lung transplant recipients: a clinical prospective study. Am J Transplant. 2015; 15:461–471.
28. Amiot L, Ferrone S, Grosse-Wilde H, Seliger B. Biology of HLA-G in cancer: a candidate molecule for therapeutic intervention? Cell Mol Life Sci. 2011; 68:417–431.
29. Nuckel H, Rebmann V, Durig J, Duhrsen U, Grosse-Wilde H. HLA-G expression is associated with an unfavorable outcome and immunodeficiency in chronic lymphocytic leukemia. Blood. 2005; 105:1694–1698.
30. Yan WH, Lin A, Chen BG, Luo WD, Dai MZ, Chen XJ, Xu HH, Li BL. Unfavourable clinical implications for HLA-G expression in acute myeloid leukaemia. J Cell Mol Med. 2008; 12:889–898.
31. de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol. 2010; 185:7452–7459.
32. Guo ZY, Lv YG, Wang L, Shi SJ, Yang F, Zheng GX, Wen WH, Yang AG. Predictive value of HLA-G and HLA-E in the prognosis of colorectal cancer patients. Cell Immunol. 2015; 293:10–16.
33. Agaugue S, Carosella ED, Rouas-Freiss N. Role of HLA-G in tumor escape through expansion of myeloid-derived suppressor cells and cytokinic balance in favor of Th2 versus Th1/Th17. Blood. 2011; 117:7021–7031.
34. Loumagne L, Baudhuin J, Favier B, Montespan F, Carosella ED, Rouas-Freiss N. In vivo evidence that secretion of HLA-G by immunogenic tumor cells allows their evasion from immunosurveillance. Int J Cancer. 2014; 135:2107–2117.
35. Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A Systematic Review of Immunotherapy in Urologic Cancer: Evolving Roles for Targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol. 2015; 68:267–279.
36. Solier C, Mallet V, Lenfant F, Bertrand A, Huchenq A, Le Bouteiller P. HLA-G unique promoter region: functional implications. Immunogenetics. 2001; 53:617–625.
37. Ferreira LM, Meissner TB, Mikkelsen TS, Mallard W, O’Donnell CW, Tilburgs T, Gomes HA, Camahort R, Sherwood RI, Gifford DK, Rinn JL, Cowan CA, Strominger JL. A distant trophoblast-specific enhancer controls HLA-G expression at the maternal-fetal interface. Proc Natl Acad Sci USA. 2016; 113:5364–5369.
38. Ikeno M, Suzuki N, Kamiya M, Takahashi Y, Kudoh J, Okazaki T. LINE1 family member is negative regulator of HLA-G expression. Nucleic Acids Res. 2012; 40:10742–10752.
39. Schmidt CM, Ehlenfeldt RG, Athanasiou MC, Duvick LA, Heinrichs H, David CS, Orr HT. Extraembryonic expression of the human MHC class I gene HLA-G in transgenic mice. Evidence for a positive regulatory region located 1 kilobase 5′ to the start site of transcription. J Immunol. 1993; 151:2633–2645.
40. Gobin SJ, Biesta P, de Steenwinkel JE, Datema G, van den Elsen PJ. HLA-G transactivation by cAMP-response element-binding protein (CREB). An alternative transactivation pathway to the conserved major histocompatibility complex (MHC) class I regulatory routes. J Biol Chem. 2002; 277:39525–39531.
41. Flajollet S, Poras I, Carosella ED, Moreau P. RREB-1 Is a Transcriptional Repressor of HLA-G. J Immunol. 2009; 183:6948–6959.
42. Ibrahim EC, Morange M, Dausset J, Carosella ED, Paul P. Heat shock and arsenite induce expression of the nonclassical class I histocompatibility HLA-G gene in tumor cell lines. Cell Stress Chaperon. 2000; 5:207–218.
43. Lefebvre S, Berrih-Aknin S, Adrian F, Moreau P, Poea S, Gourand L, Dausset J, Carosella ED, Paul P. A specific interferon (IFN)-stimulated response element of the distal HLA-G promoter binds IFN-regulatory factor 1 and mediates enhancement of this nonclassical class I gene by IFN-beta. J Biol Chem. 2001; 276:6133–6139.
44. Yie SM, Li LH, Li GM, Xiao R, Librach CL. Progesterone enhances HLA-G gene expression in JEG-3 choriocarcinoma cells and human cytotrophoblasts in vitro. Hum Reprod. 2006; 21:46–51.
45. Mouillot G, Marcou C, Rousseau P, Rouas-Freiss N, Carosella ED, Moreau P. HLA-G gene activation in tumor cells involves cis-acting epigenetic changes. Int J Cancer. 2005; 113:928–936.
46. Urosevic M, Kurrer MO, Kamarashev J, Mueller B, Weder W, Burg G, Stahel RA, Dummer R, Trojan A. Human leukocyte antigen G up-regulation in lung cancer associates with high-grade histology, human leukocyte antigen class I loss and interleukin-10 production. Am J Pathol. 2001; 159:817–824.
47. Lefebvre S, Moreau P, Guiard V, Ibrahim EC, Adrian-Cabestre F, Menier C, Dausset J, Carosella ED, Paul P. Molecular mechanisms controlling constitutive and IFN-gamma-inducible HLA-G expression in various cell types. J Reprod Immunol. 1999; 43:213–224.
48. Wagner SN, Rebmann V, Willers CP, Grosse-Wilde H, Goos M. Expression analysis of classic and non-classic HLA molecules before interferon alfa-2b treatment of melanoma. Lancet. 2000; 356:220–221.
49. Zidi I, Guillard C, Marcou C, Krawice-Radanne I, Sangrouber D, Rouas-Freiss N, Carosella ED, Moreau P. Increase in HLA-G1 proteolytic shedding by tumor cells: a regulatory pathway controlled by NF-kappaB inducers. Cell Mol Life Sci. 2006; 63:2669–2681.
50. Moreau P, Flajollet S, Carosella ED. Non-classical transcriptional regulation of HLA-G: an update. J Cell Mol Med. 2009; 13:2973–2989.
51. Bourguignon M, Yaghi L, Flajollet S, Radanne-Krawice I, Rouas-Freiss N, Lugrin D, Richalet JP, Carosella ED, Moreau P. Increased soluble human leukocyte antigen-G levels in peripheral blood from climbers on Mount Everest. Hum Immunol. 2010; 71:1105–1108.
52. Reppel L, Margossian T, Yaghi L, Moreau P, Mercier N, Leger L, Hupont S, Stoltz JF, Bensoussan D, Huselstein C. Hypoxic culture conditions for Mesenchymal Stromal/Stem Cells from Wharton’s jelly: a critical parameter to consider in a therapeutic context. Curr Stem Cell Res Ther. 2014; 9:306–318.
53. Chang CC, Ferrone S. HLA-G in melanoma: can the current controversies be solved? Semin Cancer Biol. 2003; 13:361–369.
54. Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002; 16:1151–1162.
55. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992; 12:5447–5454.
56. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995; 92:5510–5514.
57. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010; 20:51–56.
58. Karsy M, Guan J, Jensen R, Huang LE, Colman H. The impact of hypoxia and mesenchymal transition on glioblastoma pathogenesis and cancer stem cells regulation. World Neurosurg. 2016; 88:222–236.
59. Tan Z, Shon AM, Ober C. Evidence of balancing selection at the HLA-G promoter region. Hum Mol Genet. 2005; 14:3619–3628.
60. Gibbs PE, Miralem T, Maines MD. Characterization of the human biliverdin reductase gene structure and regulatory elements: promoter activity is enhanced by hypoxia and suppressed by TNF-alpha-activated NF-kappaB. FASEB J. 2010; 24:3239–3254.
61. Ziel KA, Grishko V, Campbell CC, Breit JF, Wilson GL, Gillespie MN. Oxidants in signal transduction: impact on DNA integrity and gene expression. FASEB J. 2005; 19:387–394.
62. Thrall DE, Rosner GL, Azuma C, McEntee MC, Raleigh JA. Hypoxia marker labeling in tumor biopsies: quantification of labeling variation and criteria for biopsy sectioning. Radiother Oncol. 1997; 44:171–176.
63. Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, Fisher SJ. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004; 114:744–754.
64. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014; 211:781–790.
65. Zagzag D, Nomura M, Friedlander DR, Blanco CY, Gagner JP, Nomura N, Newcomb EW. Geldanamycin inhibits migration of glioma cells in vitro: a potential role for hypoxia-inducible factor (HIF-1alpha) in glioma cell invasion. J Cell Physiol. 2003; 196:394–402.
66. Frieboes HB, Zheng X, Sun CH, Tromberg B, Gatenby R, Cristini V. An integrated computational/experimental model of tumor invasion. Cancer Res. 2006; 66:1597–1604.
67. Leblond MM, Gerault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, Valable S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology. 2016; 5:e1056442.
68. Yang JQ, Remmers EF, Marcu KB. The first exon of the c-myc proto-oncogene contains a novel positive control element. EMBO J. 1986; 5:3553–3562.
69. James-Pederson M, Yost S, Shewchuk B, Zeigler T, Miller R, Hardison R. Flanking and intragenic sequences regulating the expression of the rabbit alpha-globin gene. J Biol Chem. 1995; 270:3965–3973.
70. Neznanov N, Umezawa A, Oshima RG. A regulatory element within a coding exon modulates keratin 18 gene expression in transgenic mice. J Biol Chem. 1997; 272:27549–27557.
71. Sim BC, Hui KM. A HLA class I cis-regulatory element whose activity can be modulated by hormones. Int J Cancer. 1994; 59:646–656.
72. Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011; 117:e207–217.
73. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci USA. 2009; 106:4260–4265.
74. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009; 284:16767–16775.
75. Ortiz-Barahona A, Villar D, Pescador N, Amigo J and del Peso L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010; 38:2332–2345.
76. Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997; 272:20055–20062.
77. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996; 16:4604–4613.
78. Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995; 270:13333–13340.
79. Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J Biol Chem. 1995; 270:21021–21027.
80. Schmucker AC, Wright JB, Cole MD, Brinckerhoff CE. Distal interleukin-1beta (IL-1beta) response element of human matrix metalloproteinase-13 (MMP-13) binds activator protein 1 (AP-1) transcription factors and regulates gene expression. J Biol Chem. 2012; 287:1189–1197.
81. Chambeyron S, Bickmore WA. Does looping and clustering in the nucleus regulate gene expression? Curr Opin Cell Biol. 2004; 16:256–262.
82. Marenduzzo D, Faro-Trindade I, Cook PR. What are the molecular ties that maintain genomic loops? Trends Genet. 2007; 23:126–133.
83. Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007; 128:787–800.
84. Donadi EA, Castelli EC, Arnaiz-Villena A, Roger M, Rey D, Moreau P. Implications of the polymorphism of HLA-G on its function, regulation, evolution and disease association. Cell Mol Life Sci. 2011; 68:369–395.
85. Menendez L, Walker LD, Matyunina LV, Totten KA, Benigno BB, McDonald JF. Epigenetic changes within the promoter region of the HLA-G gene in ovarian tumors. Mol Cancer. 2008; 7:43.
86. Polakova K, Bandzuchova E, Kuba D, Russ G. Demethylating agent 5-aza-2′-deoxycytidine activates HLA-G expression in human leukemia cell lines. Leuk Res. 2009; 33:518–524.
87. Seliger B, Schlaf G. Structure, expression and function of HLA-G in renal cell carcinoma. Semin Cancer Biol. 2007; 17:444–450.
88. Xia X, Kung AL. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009; 10:R113.
89. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007; 447:425–432.
90. Sitras V, Fenton C, Paulssen R, Vartun A, Acharya G. Differences in gene expression between first and third trimester human placenta: a microarray study. PloS one. 2012; 7:e33294.
91. Brunner AL, Johnson DS, Kim SW, Valouev A, Reddy TE, Neff NF, Anton E, Medina C, Nguyen L, Chiao E, Oyolu CB, Schroth GP, Absher DM, et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009; 19:1044–1056.
92. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002; 3:415–428.
93. Shahrzad S, Bertrand K, Minhas K, Coomber BL. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics. 2007; 2:119–125.
94. Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012; 484:339–344.
95. Novakovic B, Saffery R. DNA methylation profiling highlights the unique nature of the human placental epigenome. Epigenomics. 2010; 2:627–638.
96. Fulka H, Mrazek M, Tepla O, Fulka J, Jr. DNA methylation pattern in human zygotes and developing embryos. Reproduction. 2004; 128:703–708.
97. Fraga MF, Herranz M, Espada J, Ballestar E, Paz MF, Ropero S, Erkek E, Bozdogan O, Peinado H, Niveleau A, Mao JH, Balmain A, Cano A, et al. A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res. 2004; 64:5527–5534.
98. Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006; 66:8469–8476.
99. Horiuchi A, Hayashi T, Kikuchi N, Hayashi A, Fuseya C, Shiozawa T, Konishi I. Hypoxia upregulates ovarian cancer invasiveness via the binding of HIF-1alpha to a hypoxia-induced, methylation-free hypoxia response element of S100A4 gene. Int J Cancer. 2012; 131:1755–1767.
100. Rose SD, Aghi MK. Mechanisms of evasion to antiangiogenic therapy in glioblastoma. Clin Neurosurg. 2010; 57:123–128.
101. Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009; 15:4589–4599.
102. Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc. 2007; 2:2265–2275.
103. Tost J, Gut IG. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol Biol. 2007; 373:89–102.