
INTRODUCTION
Multiple myeloma (MM) is a hematopoietic neoplasm characterized by an expansion of clonal plasma cells (PC) in the bone marrow (BM) and by an excessive production of monoclonal immunoglobulins (Ig), usually of the IgG- or IgA type [1–3]. Growth, survival, and differentiation of MM cells are triggered by various pro-oncogenic signalling pathways as well as by cytokines and the BM microenvironment [4–9]. Depending on their growth-rate and biologic behaviour, MM cells proliferate and expand in the BM and other organs and subsequently cause organ damage [1–5]. As a result, patients with advanced MM are suffering from pancytopenia, osteolyses, and/or kidney damage. In addition, MM patients often suffer from diffuse osteoporosis and secondary immunoglobulin deficiency [1–4, 10, 11]. During the past 2 decades, several different molecular and cytogenetic risk factors predicting the clinical course and progression in MM have been identified [12–19]. In a considerable number of patients, MM develops from a premalignant condition referred to as monoclonal gammopathy of undetermined significance (MGUS) [20].
Treatment of progressive MM is usually based on cytoreductive agents, glucocorticosteroids, and novel targeted drugs. In the last few years, several attempts have been made to improve treatment of MM by combining novel targeted drugs with each other or with conventional chemotherapy [21–25]. However, despite impressive results, a relatively high percentage of patients develop drug resistances over time. For high risk patients and relapsed MM, more intensive treatments are available, including poly-chemotherapy and autologous or allogeneic stem cell transplantation [21, 26–28]. However, in most cases, MM is still an incurable malignancy. Therefore, there is still a need to identify novel drug-targets and more effective targeted drugs and drug combinations for patients with advanced MM.
Recently, a number of signalling pathways and molecules critically involved in the regulation of growth and survival of MM cells have been identified. These include, among others, the Notch-signalling pathway, the Hedgehog pathway, the phosphoinositide 3-kinase (PI3K), the mammalian target of rapamycin (mTOR) pathway, and the ubiquitin-proteasome pathway [29–36]. Furthermore, the heat shock protein 90 (HSP90) inhibitor 17AAG and the histone deacetylase (HDAC) inhibitor vorinostat were found to exert substantial anti-neoplastic effects on MM cells [37–39]. Other potential drug targets in MM cells are members of the B-cell lymphoma (BCL)-2 family and Polo-like kinase 1 (PLK-1) [40–42].
The concept of neoplastic stem cells has been introduced some time ago, with the intention to explain cellular hierarchies in malignant clones and to define critical target cell populations that exhibit long-term disease-propagating capacity [43–46]. In many neoplasms, these cells are extremely immature and represent a minority in the clone. Based on their selective potential to propagate the malignancy for unlimited time periods, these cells represent a most critical target cell population that needs to be attacked and eliminated in curative treatment approaches [47–49]. In myeloid neoplasms, such as acute myeloid leukemia (AML), the disease-initiating stem- and progenitor cells are considered to reside within a CD34+ compartment of the clone [43–46]. By contrast, in MM, the phenotype of neoplastic stem cells (MM stem cells = MMSC) is a matter of debate [52–60]. In several studies, the mouse-repopulating MMSC were found to reside within a CD138−/CD20+/CD27+ (or a CD19+/CD27+/CD138−) cell population [52–56]. In other studies, MMSC were found to reside within a CD19−/CD45low/CD38+/CD138+ fraction of the clone [56, 57]. In each case, only a small sub-fraction of these cell populations may fulfil true stem cell function. The aims of this study were to examine the effects of various targeted drugs on growth and survival of MM cells, to ask whether novel targeted drugs exert synergistic growth-inhibitory effect, and to explore whether MMSC also respond to these drugs.
RESULTS
Various targeted drugs inhibit proliferation of MM cell lines
As assessed by 3H-thymidine uptake, a number of antineoplastic drugs were found to inhibit growth of MM cells at pharmacologically relevant concentrations (Table 1). Four drugs blocked proliferation in all five MM cell lines tested: the PLK-1 inhibitor BI2536 (IC50 0.01-0.05 μM), the pan-BCL-2 antagonist obatoclax (IC50 0.1-0.5 μM), the HSP90 inhibitor 17AAG (IC50 0.01-1 μM), and the PI3 kinase/mTOR inhibitor BEZ235 (IC50 0.01-1 μM) (Table 1). The Aurora-kinase-targeting drug VX-680 inhibited growth of MM.1S, NCI-H929, and OPM-2 cells (IC50 0.1-1 μM). The HDAC-inhibitor vorinostat suppressed the proliferation of MM.1S, NCI-H929, and U-266 cells (IC50 0.1-1 μM). In the other MM cell lines, vorinostat showed no effects on proliferation below 1 μM. Sunitinib blocked proliferation of MM.1S cells and U-266 cells (IC50 0.1 -1 μM) but did not inhibit growth in the other MM cell lines (Table 1). Similarly, nilotinib suppressed growth of MM.1S and U-266 cells (IC50 0.5-1 μM) but not inhibit growth in the other MM cell lines (IC50 >1 μM). A summary of the drug effects on growth of MM cell lines is shown in Table 1.
Table 1: Effects of various targeted drugs on proliferation of myeloma cell lines
Drug |
IC50 values (μM) produced by various drugs in |
||||
|---|---|---|---|---|---|
MM.1S |
NCI-H929 |
OPM-2 |
RPMI-8226 |
U-266 |
|
BI2536 |
0.01 |
0.01-0.05 |
0.01-0.05 |
0.01-0.05 |
0.01-0.05 |
Obatoclax |
0.1-0.5 |
0.5 |
0.1-0.5 |
0.5 |
0.5 |
BEZ235 |
0.1-0.5 |
<0.01 |
0.1-0.5 |
1 |
0.1-0.5 |
17AAG |
0.01-0.05 |
0.01-0.05 |
0.5-1 |
0.5-1 |
0.1-0.5 |
VX-680 |
0.1-0.5 |
0.5-1 |
0.5-1 |
>10 |
1-5 |
Vorinostat |
0.5 |
0.5-1 |
>1 |
1 |
0.1-0.5 |
Nilotinib |
0.5-1 |
>1 |
>1 |
>1 |
0.5-1 |
Imatinib |
10 |
>10 |
>10 |
>10 |
>10 |
Dasatinib |
5-10 |
>10 |
>10 |
>10 |
5-10 |
Bosutinib |
5-10 |
5 |
1-5 |
5-10 |
1-5 |
Sorafenib |
1-5 |
1-5 |
1-5 |
1-5 |
1-5 |
Sunitinib |
0.1-0.5 |
1-5 |
1-5 |
5-10 |
0.5-1 |
Erlotinib |
1-5 |
>10 |
>10 |
>10 |
5-10 |
Gefitinib |
1-5 |
>10 |
>10 |
>10 |
5-10 |
Lapatinib |
1-5 |
10 |
5-10 |
>10 |
5-10 |
MM cell lines were incubated with increasing concentrations of various drugs (0.001-10 μM) at 37°C for 48 hours. After incubation, 0.5 μCi of 3H-thymidine was added to each well and kept for 16 hours (37°C). Proliferation was measured by analyzing 3H-thymidine uptake. Results are expressed as inhibitory concentration producing 50% inhibition (IC50).
Effects of targeted drugs on in vitro proliferation of primary MM cells
In a next step, we examined the effects of 17AAG, BI2536, BEZ235, and obatoclax on in vitro proliferation of primary neoplastic PC obtained from the BM of patients with MM. The patients’ characteristics are shown in Table 2. We found that all 4 drugs tested exert dose-dependent growth-inhibitory effects in primary MM cells, with pharmacologically relevant IC50 values (Table 3). Figure 1 shows a summary of growth-inhibitory effects obtained with the 4 drugs in the primary cell samples tested. IC50 values obtained with primary BM cells (PC) were found to be within a pharmacological range and to correspond to IC50 values obtained with the MM cell lines tested (Figure 1, Tables 1 and 3).
Table 2: Characteristics of multiple myeloma patients
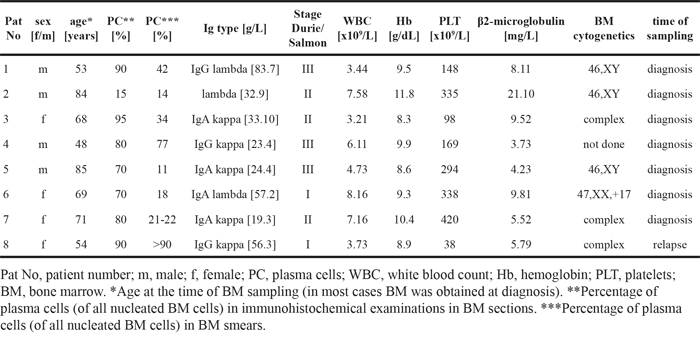
Table 3: Effects of the most effective targeted drugs on proliferation of primary neoplastic BM cells
Pat No |
sex |
age |
PC** [%] |
PC*** [%] |
IC50 (μM) values produced by |
|||
|---|---|---|---|---|---|---|---|---|
17AAG |
BI2536 |
BEZ235 |
obatoclax |
|||||
1 |
m |
53 |
90 |
42 |
0.1-0.5 |
0.05 |
0.01-0.05 |
0.1-0.5 |
2 |
m |
84 |
15 |
14 |
0.5 |
0.005-0.01 |
0.005-0.01 |
0.01-0.05 |
3 |
f |
68 |
95 |
34 |
0.5-1 |
0.1 |
0.01-0.05 |
0.1-0.5 |
6 |
f |
69 |
70 |
18 |
0.1-0.5 |
0.001 |
0.005-0.01 |
0.01-0.05 |
7 |
f |
71 |
80 |
21-22 |
0.1-0.5 |
0.01-0.05 |
0.1-0.5 |
0.1-0.5 |
8 |
f |
54 |
90 |
>90 |
0.5-1 |
0.5 |
0.1-0.5 |
0.5 |
BM, bone marrow; Pat No, patient number; m, male; f, female; PC, plasma cells. **Percent plasma cells (of all nucleated BM cells) are based on immunohistochemical examinations in BM sections. ***Percentage of plasma cells (of all nucleated BM cells) in BM smears. Primary MM cells were incubated with increasing concentrations of 17AAG, BI2536, obatoclax, or BEZ235 (0.001-1 μM) at 37°C for 48 hours. After incubation, 0.5 μCi of 3H-thymidine was added for 16 hours. Proliferation was measured by analyzing 3H-thymidine uptake. Abbreviations: IC50, inhibitory concentration (50%).
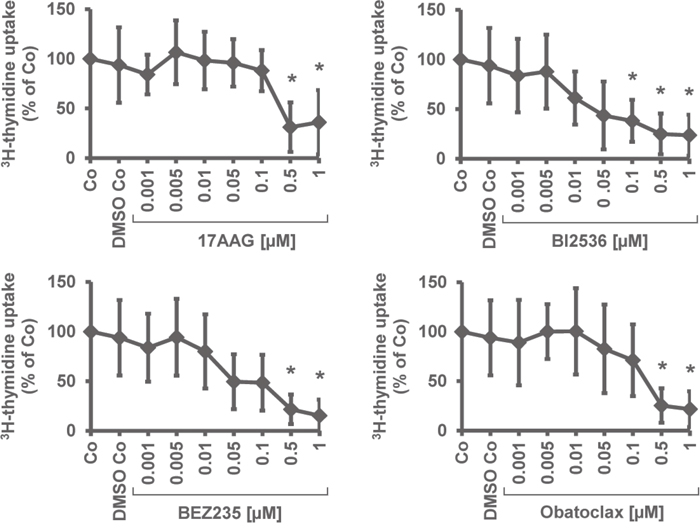
Figure 1: Effects of 17AAG, BI2536, obatoclax, and BEZ235 on proliferation of primary neoplastic MM cells. Primary BM cells obtained from 6 patients with MM were incubated in control medium (Co), DMSO control, or in various concentrations of 17AAG, BI2536, BEZ235, or obatoclax (0.001-1 μM) at 37°C for 48 hours. Thereafter, uptake of 3H-thymidine was measured. Results are expressed as percent of medium control and represent the mean±S.D. from 6 independent experiments. Asterisk (*): p<0.05.
Various targeted drugs induce apoptosis in MM cell lines
To define the mechanism of drug action, we examined drug effects on survival and apoptosis in MM cells. Apoptosis was quantified by analyzing expression of active caspase-3 by flow cytometry (Table 4). BI2536, obatoclax, BEZ235, and 17AAG produced dose-dependent apoptosis in all 5 MM cell lines tested. The most effective drugs were BI2536 (EC50 0.001-0.01 μM) and obatoclax (EC50 0.001-0.5 μM), followed by BEZ235 (EC50 0.01-0.5 μM) and 17AAG (EC50 0.01-1 μM). The HDAC blocker VX-680 induced growth inhibition in OPM-2, RPMI-8226, and U-266 cells (EC50 0.1-1 μM), whereas the other cell lines tested did not respond to VX-680 (Table 4). Vorinostat induced apoptosis in U-266 cells (EC50 0.5-1 μM) but did not produce apoptosis in the other MM cell lines. Sunitinib was found to exert apoptosis-inducing effects in MM.1S, OPM-2, RPMI-8226, and U-266 cells (EC50 0.5-1 μM) (Table 4). The effects of the most potent drugs (17AAG, BI2536, BEZ235) on survival of MM cells was confirmed by Annexin V/PI staining, with similar EC50 values compared to that obtained by staining for active caspase-3 (Figure 2).
Table 4: Effects of various targeted drugs on survival (apoptosis) of myeloma cell lines
Drug |
EC50 (μM) values produced by various drugs in |
||||
|---|---|---|---|---|---|
MM.1S |
NCI-H929 |
OPM-2 |
RPMI-8226 |
U-266 |
|
BI2536 |
0.001-0.01 |
0.01 |
0.001-0.01 |
0.001-0.01 |
0.001-0.01 |
Obatoclax |
0.5 |
0.5 |
0.001-0.01 |
0.01-0.1 |
0.5 |
BEZ235 |
0.1-0.5 |
0.5 |
0.01-0.1 |
0.5 |
0.1-0.5 |
17AAG |
0.01-0.1 |
0.1-0.5 |
0.1-0.5 |
0.5 |
1 |
VX-680 |
5 |
1-5 |
0.5-1 |
0.1-0.5 |
0.5 |
Vorinostat |
1-5 |
1-5 |
1 |
1-5 |
0.5-1 |
Nilotinib |
1-5 |
1-5 |
1-5 |
1-5 |
1-5 |
Imatinib |
>10 |
>10 |
>10 |
>10 |
>10 |
Dasatinib |
>10 |
>10 |
5-10 |
>10 |
>10 |
Bosutinib |
1-5 |
5 |
1-5 |
1-5 |
5 |
Sorafenib |
5 |
1-5 |
1-5 |
5 |
1-5 |
Sunitinib |
1 |
1-5 |
0.5-1 |
0.5 |
1 |
Erlotinib |
>10 |
>10 |
>10 |
>10 |
>10 |
Gefitinib |
>10 |
>10 |
>10 |
>10 |
>10 |
Lapatinib |
5-10 |
>10 |
5-10 |
5-10 |
5-10 |
Myeloma cell lines were incubated with increasing concentrations of various drugs (0.001-10 μM) at 37°C for 48 hours. Apoptosis was determined by active caspase-3 staining and flow cytometry as described in the text. Abbreviations: EC50, effective concentration (50%).
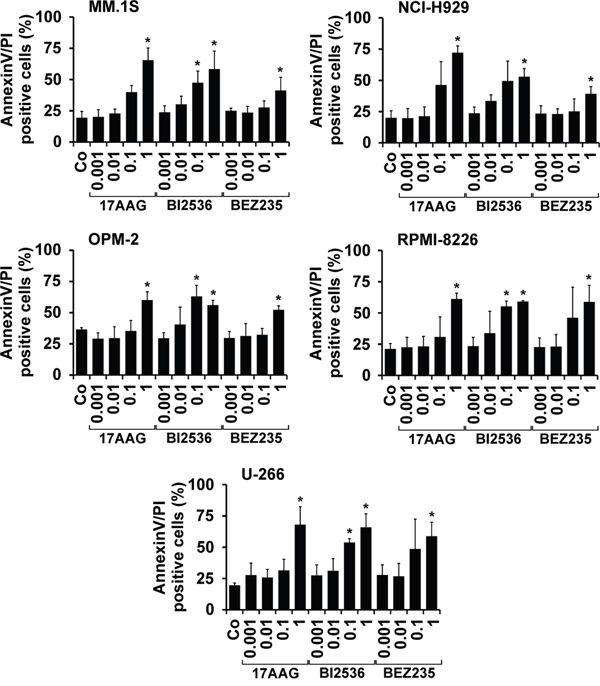
Figure 2: Effects of 17AAG, BI2536, and BEZ235 on growth of MM cell lines. MM cell lines (MM.1S, NCI-H929, OPM-2, RPMI-8226, U-266) were incubated in control medium (Co) or in various concentrations of 17AAG, BI2536, or BEZ235 (0.001-1 μM) at 37°C for 48 hours. Then, the percentage of apoptotic cells was determined by AnnexinV/PI staining and flow cytometry. Results show the percentage of AnnexinV/PI+ cells and represent the mean±S.D. from 3 independent experiments. Asterisk (*): p<0.05.
Drug effects on survival (apoptosis) of primary MM cells, putative MM stem cells (MMSC), CD34+/CD38− cells, and CD34+/CD38+ cells
In a next step, the most potent drugs (17AAG, BI2536, BEZ235) were examined for their effects on survival of primary BM-derived plasma cells, putative neoplastic MM stem cells, CD34+/CD38− hematopoietic stem cells (HSC) and CD34+/CD38+ hematopoietic progenitor cells by staining for active caspase-3 and Annexin V/DAPI. As shown in Figure 3, all three drugs tested (17AAG, BI2536, BEZ235) induced apoptosis in CD138+ MM cells as well as in CD138−/CD20+/CD27+ MMSC-containing cell fractions in all donors tested (Figure 3). However, we also found that all three drugs produce apoptosis in presumably normal CD34+/CD38− HSC and CD34+/CD38+ progenitor cells (Figure 3).
Figure 3: Effects of 17AAG, BI2536, and BEZ235 on survival of primary MM cells. Primary BM cells derived from 6 patients with MM were incubated in control medium (Co) or in medium containing 17AAG, BI2536, or BEZ235 (each 1 μM) at 37°C for 48 hours. Thereafter, cells were stained with antibodies against AnnexinV A. or active caspase-3 B. by multicolor flow cytometry as described in the text. The following subsets of cells were examined: CD138+ MM cells (black bars), CD138−/CD27+/CD20+ putative MMSC (grey bars), CD34+/CD38− hematopoietic stem cells (open bars), and CD34+/CD38+ cells (hatched bars). Results are expressed as percent AnnexinV+ cells (A) or percent active caspase-3+ cells (B) and represent the mean±S.D. from 6 independent experiments. Asterisk (*): p<0.05.
17-AAG, BI2536, and BEZ235 inhibit cell cycle progression in MM cells
We next examined the effects of 17AAG, BI2536, and BEZ235 on cell cycle progression in the 5 MM cell lines tested. As expected, the PI3 kinase/mTOR blocker BEZ235 produced a G1 cell cycle arrest in all MM cell lines tested, whereas 17AAG and BI2536 induced a G2 cell cycle arrest in these cells (Figure 4). The effects of 17AAG and BI2536 on cell cycle progression were weaker compared to the effects produced by BEZ235 (Figure 4).
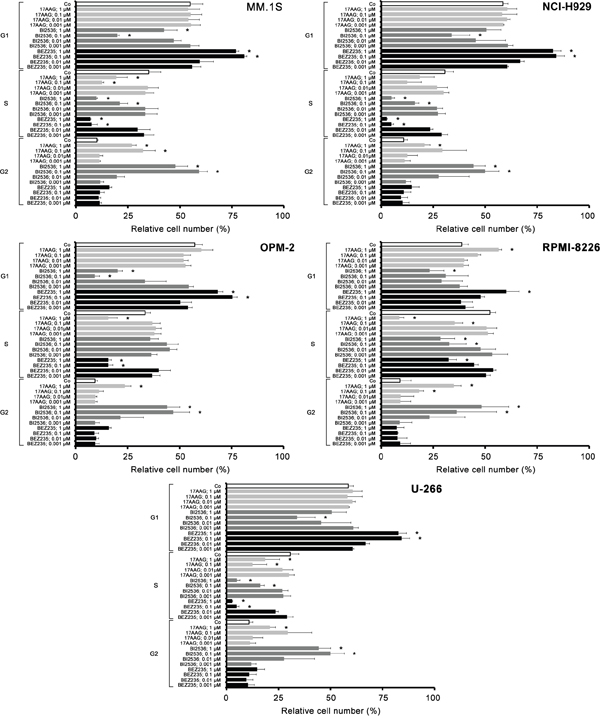
Figure 4: Effects of 17AAG, BI2536, and BEZ235 on cell cycle progression in MM cell lines. MM.1S cells (upper left panel), NCI-H929 cells (upper right panel), OPM-2 cells (middle left panel), RPMI-8226 cells (middle right panel) and U-266 cells (lower panel) were incubated in control medium (Co) or various concentrations of 17AAG, BI2536, or BEZ235 (0.001-1 μM each) at 37°C for 48 hours. Then, cell cycle distribution was analyzed by flow cytometry as described in the text. Asterisk (*): p<0.05.
Identification of drug combinations producing synergistic growth-inhibitory effects
Based on the encouraging results obtained with several of the targeted drugs applied and because of their potential toxicity, we screened for cooperative anti-neoplastic drug effects in the 5 MM cell lines employed. Drug combinations were classified as additive, antagonistic, or synergistic by CalcuSyn software. A summary of drug-combination effects is shown in Table 5. Clear synergistic growth-inhibitory effects were observed when applying the drug combinations 17AAG+BI2536 and 17AAG+BEZ235 in MM.1S, OPM-2, RPMI-8226, and U-266 cells, 17AAG+obatoclax and BI2536+BEZ235 in MM.1S, NCI-H929, OPM-2, and RPMI-8226 cells, BI2536+obatoclax in MM.1S, OPM-2, and RPMI-8226 cells, and BEZ235+obatoclax in MM.1S and RPMI-8226 cells (Table 5). Figure 5 shows examples of synergistic drug interactions obtained in drug combination experiments in MM cell lines.
Table 5: Effects of various drug combinations on proliferation of myeloma cell lines
Drug combinations |
Drug interactions produced in |
||||
|---|---|---|---|---|---|
MM.1S |
NCI-H929 |
OPM-2 |
RPMI-8226 |
U-266 |
|
17AAG and BI2536 |
s |
an |
s |
s |
s |
17AAG and BEZ235 |
s |
an |
s |
s |
s |
17AAG and obatoclax |
s |
s |
s |
s |
an |
BI2536 and BEZ235 |
s |
s |
s |
s |
an |
BI2536 and obatoclax |
s |
an |
s |
s |
an |
BEZ235 and obatoclax |
s |
ad |
an |
s |
an |
To determine potential additive or synergistic drug effects, MM cell lines were exposed to 17AAG, BI2536, BEZ235, and obatoclax, alone or in combination at a fixed ratio of drug concentrations at 37°C for 48 hours. Proliferation was measured by analyzing 3H-thymidine uptake. Drug interactions (synergistic = s, additive = ad, and antagonistic = an) were calculated by CalcuSyn software. A combination index (CI) of <1 indicates synergism, CI values of 1 indicate additive effects, and CI values >1 indicate antagonistic drug effects.
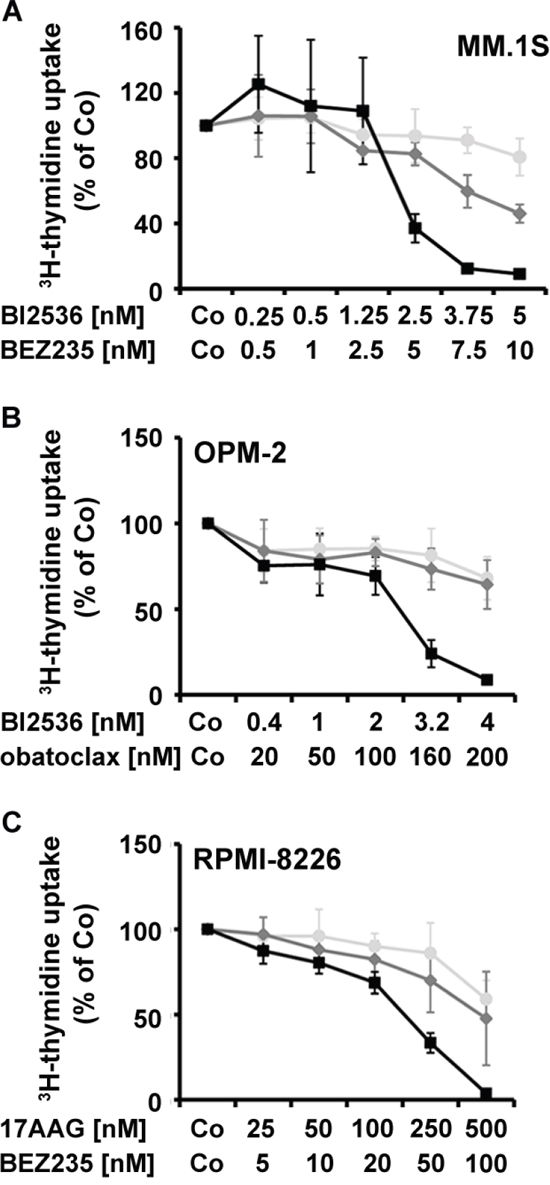
Figure 5: Effects of drug combinations on proliferation in MM cells. MM.1S cells A., OPM-2 cells B., and RPMI-8226 cells C. were incubated in control medium (Co), in medium containing individual drugs alone, or in medium containing drug combinations (at fixed ratio) at 37°C for 48 hours. Then, uptake of 3H-thymidine was measured. MM.1S cells (A) were incubated in various concentrations of BI2536 (●-●), BEZ235 (♦-♦), or combinations of both drugs (■-■). OPM-2 cells (B) were incubated with various concentrations of BI2536 (●-●) or obatoclax (♦-♦) or combinations of both drugs (■-■). RPMI-8226 cells (C) were incubated with various concentrations of 17AAG (●-●) or BEZ235 (♦-♦) or combinations of both drugs (■-■). Results show the percentage of 3H-thymidine uptake compared to medium control and represent the mean±S.D. of one typical experiment.
Identification of molecular targets expressed in MM cell lines
As assessed by qPCR, all MM cell lines expressed transcripts specific for PLK-1, PI3K, mTOR, BCL-2, and the myeloid cell leukemia 1 (MCL-1) antigen (Table 6). BCL-xL mRNA was detected in MM.1S, RPMI-8226, and U-266 cells, but not in NCI-H929 and OPM-2 cells. No definitive correlation between expression of molecular targets (or target-patterns) and responses to individual drugs or drug combinations was found.
Table 6: Molecular targets expressed in myeloma cell lines
Drug targets |
% of human ABL mRNA |
||||
|---|---|---|---|---|---|
MM.1S |
NCI-H929 |
OPM-2 |
RPMI-8226 |
U-266 |
|
PLK-1 |
++ |
++ |
++ |
++ |
++ |
PI3K |
++ |
++ |
+ |
++ |
++ |
mTOR |
++ |
++ |
++ |
++ |
++ |
BCL-2 |
++ |
++ |
++ |
++ |
+ |
BCL-xL |
+/- |
- |
- |
+ |
++ |
MCL-1 |
++ |
++ |
++ |
++ |
++ |
To quantify mRNA expression levels in myeloma cell lines, qPCR was performed using primers specific for PLK-1, PI3K, mTOR, BCL-2, BCL-xL, MCL-1, and ABL (Table 7). mRNA levels were expressed as percentage of ABL. The following score of mRNA expression was applied (% of ABL): <10%: -; 10%-29%: +/-; 30%-100%:+; >100%: ++.
PLK-1: polo-like kinase 1; PI3K: phosphoinositide 3-kinase; mTOR: mammalian target of rapamycin; BCL-2: B-cell lymphoma 2; BCL-xL: B-cell lymphoma-extra large; MCL-1: myeloid cell leukemia 1.
DISCUSSION
During the past few years, therapy of MM improved considerably and resulted in better progression-free and overall survival. However, still, not all patients with MM enter long-term progression-free survival after therapy. Overall, there is a need to develop new more effective targeted drugs and drug combinations for these patients. In the past few years, several novel targeted drugs have shown promising results in preclinical studies [21–25, 29, 31–33, 36–42]. In the current study, we have extended these analyses by examining the effects of various targeted drugs on growth and survival of primary MM cells and putative MMSC, and by combining most effective targeted drugs with each other. Of the 15 drugs tested in this study, 17AAG, BI2536, BEZ235, and obatoclax were identified as potent inhibitors of growth and survival of MM cells. In addition, we found that all 4 drugs block growth and survival of primary MM cells, and that drug combinations elicit additive or even synergistic growth-inhibitory effects. These data may have clinical implications and may pave the way for the development of novel more effective anti-MM therapies.
A number of different signaling pathways and survival molecules contribute to growth and survival of MM cells [29–36]. During the past 15 years, targeted drugs have been developed that interfere with these pathways and block specific signaling molecules or survival-related molecules in neoplastic cells [32, 33, 37–42]. In our study, 15 targeted drugs were applied, and 4 of these drugs were found to act as potent inhibitors of growth and survival of MM cells. These drugs are the Hsp90 targeting drug 17AAG, the PLK-1 inhibitor BI2536, the dual PI3 kinase/mTOR blocker BEZ235, and the pan BCL-2 inhibitor obatoclax. The IC50 values produced by these drugs were found to be in a pharmacologically relevant range. In addition, these drugs suppressed cell cycle progression and induced apoptosis in MM cells. We were also able to show that the molecular targets through which these drugs exert their growth-inhibitory effects, namely PLK-1, PI3K, mTOR, BCL-2, BCL-xL, and MCL-1, are expressed in MM cells. Whereas most targets were expressed abundantly in MM cell lines, BCL-xL mRNA was only expressed at low levels or was not detected in the MM cell lines tested. We also tried to correlate (synergistic) drug effects with target expression patterns in MM cells. However, no clear correlation between expression of molecular targets and responses to individual drugs or drug combinations was found. This is best explained by the fact that several different (known and unknown) targets are recognized by these drugs and contribute to the observed drug effects.
Cell line models are a useful tool for screening potential drug effects in various cancer entities. However, primary cells may behave differentially and often show different responses to targeted drugs. Therefore, we examined drug effects on primary MM cells. We found that 17AAG, BI2536, BEZ235, and obatoclax induce growth inhibition in primary BM-derived MM cells in all patients tested. The IC50 values were comparable among patients, independent of the percentage of neoplastic cells in BM samples, type of MM, or disease status (diagnostic sample versus relapse). Although all 4 drugs exerted potent effects, slightly lower IC50 values were obtained with BI2536 and BEZ235 compared to 17AAG and obatoclax. It is also noteworthy that the IC50 values obtained with primary MM cells corresponded to IC50 values obtained in MM cell lines.
In the past 15 years, neoplastic stem cells have been identified as a novel relevant target of therapy in malignant blood cell disorders [47–49]. Any type of therapy can only be curative when eliminating these disease-propagating cells. In MM, the phenotype of neoplastic stem cells (MMSC) is still a matter of debate [45, 52–60]. In the past few years evidence has accumulated to suggest that these cells may reside as rare subset in a CD138−/CD20+/CD27+ population of the clone [52–56]. In the present study, we examined the effects of the 4 most potent drugs on survival of these putative MMSC. We found that these drugs induce apoptosis not only in the bulk of MM cells but also in putative MMSC, which may have clinical implications. However, these studies were performed in vitro, and additional studies using these drugs in suitable xenotransplantation assays performed with primary MM cells would be required to confirm their MMSC-eliminating activity. Unfortunately, however, no robust xenotransplantation model is available in MM. Once such model is available, we will examine drug effects on MMSC engraftment in future studies.
Most of the targeted drugs examined may also exert growth-inhibitory effects on normal blood and BM cells. In the current study, we examined the effects of the 4 most potent targeted drugs on survival of CD34+/CD38− HSC and CD34+/CD38+ progenitor cells. In these experiments all 4 drugs induced apoptosis in normal BM stem and progenitor cells at 1 μM. These data suggest that most of these drugs can produce cytopenia in patients which corresponds with clinical observations. An alternative possibility may be that some of the CD34+ cells were immature clonal cells. However, although this hypothesis has been propagated by some investigators [61, 62] others have concluded that clonal MM progenitors do not reside within CD34+ cells [63].
Based on the obvious risk of side effects and the observation that synergistic drug effects can often be obtained, anti-MM therapy usually consists of various drug combinations. In the present study, we were interested to learn whether the most effective targeted drugs identified would also show cooperative or even synergistic anti-proliferative effects on MM cells when applied in combination. Indeed, we found that various combinations of the targeted drugs applied produce clear synergistic effects on growth of MM cell lines. The most potent synergistic effects were seen when combining BI2536 and BEZ235 in MM.1S or BI2536 and obatoclax in OPM-2 and 17AAG and BEZ235 in RPMI-8226 cells. Remarkably, in MM.1S cells and RPMI-8226 cells, all drug combinations applied showed synergistic effects. These data suggest that it may be reasonable to apply such drug combinations also in vivo once the individual drugs have shown to act anti-neoplastic in patients. By employing such combination strategies, drug-induced toxicity may also be reduced.
In conclusion, our data show that various targeted drugs exert major growth-inhibitory and apoptosis-inducing effects on primary MM cells, their putative stem cells, and MM cell lines, and that these effects can be further augmented by applying drug combinations. Clinical trials are now warranted in order to confirm these effects in vivo in patients with MM. The most obvious clinical need may be patients with relapsed or refractory MM [64, 65].
MATERIALS AND METHODS
Reagents
A number of anti-neoplastic drugs were tested for their ability to inhibit growth of MM cells: the tyrosine kinase inhibitors (TKI) bosutinib, dasatinib, imatinib, sorafenib, sunitinib, and nilotinib, the ErbB-receptor inhibitors lapatinib, erlotinib, and gefitinib, the Aurora-kinase inhibitor VX-680, the HSP90 inhibitor 17AAG, the PLK-1 inhibitor BI2536, the pan-BCL-2 antagonist obatoclax, and the HDAC-inhibitor vorinostat were purchased from Chemietek (Indianapolis, IN, USA). The PI3 kinase/mTOR inhibitor BEZ235 was obtained from Selleck Chemicals (Houston, TX, USA). Stock solutions of drugs were prepared by dissolving in dimethylsulfoxide, DMSO (Merck, Darmstadt, Germany). RPMI 1640 medium and fetal calf serum (FCS) were purchased from PAA Laboratories (Pasching, Austria), and 3H-thymidine from PerkinElmer (Waltham, MA, USA). FITC-labeled CD34 monoclonal antibody (mAb) 581, PE-labeled CD34 mAb 581, FITC-labeled CD138 mAb MI15, PE-labeled CD138 mAb DL-101, PerCP-labeled CD45 mAb 2D1, APC-labeled CD38 mAb HIT2, PE-labeled and Alexa Fluor® 647-labeled active caspase-3 mAb C92-605 were purchased from BD Biosciences (San Jose, CA, USA). The PerCP-labeled CD20 mAb 2H7 and the APC-labeled CD27 mAb O323 were obtained from Biolegend (San Diego, CA, USA), and an Annexin V/FITC kit from eBioscience (San Diego, CA, USA).
Culture of MM cells
The MM cell lines NCI-H929, OPM-2, RPMI-8226 and U-266 were obtained from the German Collection of Microorganisms and Cell Cultures (DMSZ; Braunschweig, Germany) and MM.1S from American Type Culture Collection (ATCC; Manassas, VA, USA). Cell lines were cultured in RPMI1640 with 10% FCS and antibiotics at 5% CO2 and 37°C. Cells were passaged every 2-3 days and re-thawed from an original stock every 6-8 weeks. The biologic stability of these cell lines was checked by cell surface phenotyping (flow cytometry). Primary BM cells were obtained (routine investigations) from 8 patients with MM after written informed consent was given. Samples were collected at diagnosis, or relapse (Table 2). The study was approved by the ethics committee of the Medical University of Vienna. Primary BM cells were either analyzed by multicolor flow cytometry or were fractionated using Ficoll, in order to obtain mononuclear cells (MNC).
Flow cytometry and characterization of MMSC
Heparinized BM cells (106/tube) were incubated with combinations of mAb for 15 minutes. PC were defined as CD138+ cells and the MMSC-containing cell fractions were defined as CD138−/CD20+/CD27+ cells using FITC-labeled CD138 mAb, APC-labeled CD27 mAb, and PerCP-labeled CD20 mAb. Hematopoietic stem cells (HSC) were defined as CD34+/CD38- cells and hematopoietic progenitors as CD34+/CD38+ cells, using FITC-labeled CD34 mAb and APC-labeled CD38 mAb. After erythrocyte-lysis using FACS-Lysing-Solution (BD Biosciences) PC, MMSC, HSC, and hematopoietic progenitor cells were analyzed by multicolor flow cytometry on a FACSCalibur (BD Biosciences) using FlowJo software (TreeStar, Ashland, OR, USA). To study drug effects on survival (apoptosis) of primary PC, MMSC, HSC, and progenitors cells, primary BM MNC of 6 MM patients were incubated in control medium, 17AAG, BI2536, or BEZ235 (1 μM each) at 37°C for 48 hours. Thereafter, cells were washed and examined for apoptosis by combined staining for surface antigens (to define cell populations) and either AnnexinV-FITC plus 4′, 6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA) or with an antibody against active caspase-3 as described [66]. Apoptosis was quantified by measuring the percentage of AnnexinV+ cells and active caspase-3+ cells in various gated cell fractions, namely CD138+ PC, CD138-/CD20+/CD27+ MMSC, CD34+/CD38- HSC, and CD34+/CD38+ hematopoietic progenitor cells.
Evaluation of drug effects on proliferation of MM cell lines and primary MM cells
To study the effects of various drugs on proliferation of MM cell lines, 3H-thymidine incorporation experiments were performed. For this purpose, MM.1S, NCI-H929, OPM-2, RPMI-8226 and U-266 cells (104/well) were seeded in 96-well plates (TPP, Trasadingen, Switzerland) and incubated with increasing drug concentrations (0.001-10 μM) at 37°C for 48 hours. In a different set of experiment, primary BM derived cells obtained from 6 patients with MM were incubated in control medium or various concentrations of 17AAG, BI2536, obatoclax, and BEZ235 (0.001-1 μM) at 37°C for 48 hours. After incubation, 0.5 μCi of 3H-thymidine was added to each well and kept for 16 hours (37°C). Cells were then harvested on filter membranes (Perkin Elmer, Waltham, MA, USA) in a FilterMate Harvester (Perkin Elmer). Filters were air-dried and the bound radioactivity counted in a ß-counter (MicroBeta2 2450 Microplate Counter, Perkin Elmer). To determine potential additive or synergistic drug effects, MM cells were exposed to various combinations of 17AAG, BI2536, BEZ235, and obatoclax at a fixed ratio of drug concentrations [67]. All experiments were performed in triplicates.
Evaluation of drug-induced apoptosis in MM cell lines
To assess the effects of targeted drugs on expression of activated caspase-3, flow cytometry experiments were performed using MM cell lines and an antibody against active caspase-3 (C92-605; BD Biosciences). In these experiments, cells were cultured in the presence of control medium or targeted drugs at various concentrations (0.001-10 μM) at 37°C for 48 hours. Prior to staining, cells were fixed in formaldehyde (2%), and permeabilized using methanol (100%) at -20°C for 30 minutes. Expression of active caspase-3 was analyzed on a FACSCalibur (BD Biosciences) as decribed [66]. To confirm apoptosis in cell lines after drug exposure, Annexin V/PI staining was performed with the three most effective drugs 17AAG, BI2536, BEZ235 in all five MM cell lines as reported [67]. For cell cycle studies with the three potent drugs 17AAG, BI2536 and BEZ235, drug-exposed cells were resuspended in 500 μL permeabilization buffer (0.1% Na-acetate and 0.1% Triton X-100). Then 40 μl PI were added, and cell cycle distribution analyzed on a FACSCalibur. For obatoclax, the AnnexinV/PI staining and cell cycle analyis was not possible due to the autofluoresence of the drug.
Quantitative PCR (qPCR)
Total RNA was isolated from the five MM cell lines (MM1.S, NCI-H929, OPM-2, RPMI-8226, U-266) using RNeasy MinElute™ Cleanup Kit (Qiagen, Hilden, Germany). cDNA was synthesized using Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA, USA), random primers, First-Strand-Buffer, dNTPs, and RNasin (all from Invitrogen) according to the manufacturer’s instructions. qPCR was performed as described [66] using iTaq SYBR-Green-Supermix with ROX (Bio-Rad, Hercules, CA) and primers specific for BCL-2, BCL-xL, MCL-1, PI3K, mTOR and PLK-1 (Table 7). BCL-2, BCL-xL, MCL-1, PI3K, mTOR, and PLK-1 mRNA levels were expressed as percentage of ABL transcript levels [66].
Table 7: Oligonucleotide sequences of primers used for quantitative PCR (qPCR)
Gene |
Sequence |
|---|---|
hu PLK-1-fwd |
5′-CCC ATC TTC TGG GTC AGC AAG-3′ |
hu PLK-1-rev |
5′-AAG AGC ACC CCC ACG CTG TT-3′ |
hu PI3K-fwd |
5′-TAG CTA TTC CCA CGC AGG AC-3′ |
hu PI3K-rev |
5′-TTG CTT TGA GCT GTT CTT TGT C-3′ |
hu mTOR-fwd |
5′-CCC ACG TTC CTT AAC GTC AT-3′ |
hu mTOR-rev |
5′-GGC TCT TCA CAA AGG ACA CC-3′ |
hu BCL-2-fwd |
5′-TTG ACA GAG GAT CAT GCT GTA CTT-3′ |
hu BCL-2-rev |
5′-TCA GTC TAC TTC CTC TGT GAT GTT GT-3′ |
hu BCL-xL-fwd |
5′-CTC CTC TCC CGA CCT GTG AT-3′ |
hu BCL-xL-rev |
5′-AAG ATT CTG AAG GGA GAG AAA GAG A-3′ |
hu MCL-1-fwd |
5′-GTG CAG CGC AAC CAC GAG-3′ |
hu MCL-1-rev |
5′-CGA TTT CAC ATC GTC TTC GTT T-3′ |
hu ABL-fwd |
5′-TGT ATG ATT TTG TGG CCA GTG GAG-3′ |
hu ABL-rev |
5′-GCC TAA GAC CCG GAG CTT TTC A-3′ |
hu: human; fwd: forward; rev, reverse; PLK-1: polo-like kinase 1; PI3K: phosphoinositide 3-kinase; mTOR: mammalian target of rapamycin; BCL-2: B-cell lymphoma 2; BCL-xL: B-cell lymphoma-extra large; MCL-1: myeloid cell leukemia 1; ABL: Abelson murine leukemia viral oncogene homolog.
Statistical evaluation of data
To determine the significance in differences in growth and apoptosis, the Student’s t test for dependent samples was applied. Results were considered significant when p<0.05. Drug interactions (additive, synergistic, and antagonistic) were determined by calculating combination index (CI) values using CalcuSyn software (Biosoft, Cambridge, U.K.) [68]. A CI value of 1 indicates an additive effect, whereas CI values below 1 indicate synergistic drug effects, and CI values >1 antagonistic effects.
CONFLICTS OF INTEREST
The authors have no conflicts of interests to declare.
FUNDING
This study was supported by a Cancer Stem Cell Grant of the Medical University of Vienna.
REFERENCES
1. Kastrinakis NG, Gorgoulis VG, Foukas PG, Dimopoulos MA, Kittas C. Molecular aspects of multiple myeloma. Ann Oncol. 2000; 11:1217-1228.
2. Seidl S., Kaufmann H., Drach J. New insights into the pathophysiology of multiple myeloma. Lancet Oncol. 2003; 4:557-564.
3. Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004; 351:1860-1873.
4. Barlogie B, Epstein J, Selvanayagam P, Alexanian R. Plasma cell myeloma--new biological insights and advances in therapy. Blood. 1989; 73:865-879.
5. Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006; 42:1564-1573.
6. Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006; 20:193-199.
7. Katz BZ. Adhesion molecules--The lifelines of multiple myeloma cells. Semin Cancer Biol. 2010; 20:186-195.
8. Lemaire M, Deleu S, De Bruyne E, Van Valckenborgh E, Menu E, Vanderkerken K. The microenvironment and molecular biology of the multiple myeloma tumor. Adv Cancer Res. 2011; 110:19-42.
9. Wang J, De Veirman K, De Beule N, Maes K, De Bruyne E, Van Valckenborgh E, Vanderkerken K, Menu E. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget. 2015; 6:43992-44004. doi: 10.18632/oncotarget.6083.
10. Barillé-Nion S, Bataille R. New insights in myeloma-induced osteolysis. Leuk Lymphoma. 2003; 44:1463-1467.
11. Yaccoby S. Advances in the understanding of myeloma bone disease and tumour growth. Br J Haematol. 2010; 149:311-321.
12. Königsberg R, Zojer N, Ackermann J, Krömer E, Kittler H, Fritz E, Kaufmann H, Nösslinger T, Riedl L, Gisslinger H, Jäger U, Simonitsch I, Heinz R, et al. Predictive role of interphase cytogenetics for survival of patients with multiple myeloma. J Clin Oncol. 2000; 18:804-812.
13. Hideshima T, Bersagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004; 104:607-618.
14. San Miguel JF, García-Sanz R. Prognostic features of multiple myeloma. Best Pract Res Clin Haematol. 2005; 18:569-583.
15. Shaughnessy JD Jr, Haessler J, van Rhee F, Anaissie E, Pineda-Roman M, Cottler-Fox M, Hollmig K, Zangari M, Mohiuddin A, Alsayed Y, Grazziutti M, Epstein J, Crowley J, et al. Testing standard and genetic parameters in 220 patients with multiple myeloma with complete data sets: superiority of molecular genetics. Br J Haematol. 2007; 137:530-536.
16. Kyle RA, Rajkumar SV. Multiple Myeloma. Blood. 2008; 111:2962-2972.
17. Landgren O, Kyle RA, Rajkumar SV. From myeloma precursor disease to multiple myeloma: new diagnostic concepts and opportunities for early intervention. Clin Cancer Res. 2011; 17:1243-1252.
18. Munshi NC, Anderson KC, Bergsagel PL, Shaughnessy J, Palumbo A, Durie B, Fonseca R, Stewart AK, Harousseau JL, Dimopoulos M, Jagannath S, Hajek R, Sezer O, et al. Consensus recommendations for risk stratification in multiple myeloma: report of the International Myeloma Workshop Consensus Panel 2. Blood. 2011; 117:4696-4700.
19. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, Proszek PZ, Johnson DC, Kaiser MF, Melchor L, Aronson LI, Scales M, Pawlyn C, et al. Mutational sectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015; 33:3911-3920.
20. Kyle RA, Rajkumar SV. Monoclonal gammopathies of undetermined significance: a review. Immunol Rev. 2003; 194:112-139.
21. Kyle RA, Rajkumar SV. Treatment of multiple myeloma: an emphasis on new developments. Ann Med. 2006; 38:111-115.
22. Ocio EM, Mateos MV, Maiso P, Pandiella A, San-Miguel JF. New drugs in multiple myeloma: mechanisms of action and phase I/II clinical findings. Lancet Oncol. 2008; 9:1157-1165.
23. Morabito F, Recchia AG, Mazzone C, Gentile M. Targeted therapy of multiple myeloma: the changing paradigm at the beginning of the new millennium. Curr Cancer Drug Targets. 2012; 12:743-756.
24. Ocio EM, Richardson PG, Rajkumar SV, Palumbo A, Mateos MV, Orlowski R, Kumar S, Usmani S, Roodman D, Niesvizky R, Einsele H, Anderson KC, Dimopoulos MA, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. 2014; 28:525-542.
25. Le Ray E, Jagannath S, Palumbo A. Advances in targeted therapy for the treatment of patients with relapsed/refractory multiple myeloma. Expert Rev Hematol. 2016; 9:91-105.
26. Nishihori T, Alsina M. Advances in the autologous and allogeneic transplantation strategies for multiple myeloma. Cancer Control. 2011; 18:258-267.
27. Bensinger W. Allogeneic stem cell transplantation for multiple myeloma. Hematol Oncol Clin North Am. 2014; 28:891-902.
28. Shah N, Callander N, Ganguly S, Gul Z, Hamadani M, Costa L, Sengsayadeth S, Abidi M, Hari P, Mohty M, Chen YB, Koreth J, Landau H, et al. Hematopoietic Stem Cell Transplantation for Multiple Myeloma: Guidelines from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2015; 21:1155-1166.
29. Hideshima T, Chauhan D, Richardson P, Anderson KC. Identification and validation of novel therapeutic targets of multiple myeloma. J Clin Oncol. 2005; 23:6345-6350.
30. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, Watkins DN, Matsui W. Hedgehog signalling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007; 104:4048-4053.
31. Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of Notch signalling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008; 111:2220-2229.
32. Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res. 2009; 315:485-497.
33. McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, Jakubikova J, Maira SM, Garcia-Echeverria C, Schlossman R, Munshi NC, Richardson PG, Anderson KC, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res. 2009; 69:5835-5842.
34. Anderson KC. New insights into therapeutic targets in myeloma. Hematology Am Soc Hematol Educ Program. 2011; 2011:184-190.
35. Colombo M, Galletti S, Garavelli S, Platonova N, Paoli A, Basile A, Taiana E, Neri A, Chiaramonte R. Notch signaling deregulation in multiple myeloma: A rational molecular target. Oncotarget. 2015; 6:26826-26840. doi: 10.18632/oncotarget.5025.
36. Lub S, Maes K, Menu E, Bruyne E, Vanderkerken K, Valckenborgh EV. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget, 2016; 7:6521-6537. doi: 10.18632/oncotarget.6658.
37. Duus J, Bahar HI, Venkataraman G, Ozpuyan F, Izban KF, Al-Masri H, Maududi T, Toor A, Alkan S. Analysis of expression of heat shock protein-90 (HSP90) and the effects of HSP90 inhibitor (17-AAG) in multiple myeloma. Leuk Lymphoma. 2006; 47:1369-1378.
38. Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, Garcia-Vargas J, Sanz-Rodriguez C, Rizvi S. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009; 2:31.
39. Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA, Anderson KC. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br J Haematol. 2011 ; 152:367-379.
40. Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood. 2007; 109:5430-5438.
41. Evans RP, Dueck G, Sidhu R, Ghosh S, Toman I, Loree J, Bahlis N, Klimowicz AC, Fung J, Jung M, Lai R, Pilarski LM, Belch AR, et al. Expression, adverse prognostic significance and therapeutic small molecule inhibition of Polo-like kinase 1 in multiple myeloma. Leuk Res. 2011; 35:1637-1643.
42. Stewart HJ, Kishikova L, Powell FL, Wheatley SP, Chevassut TJ. The polo-like kinase inhibitor BI 2536 exhibits potent activity against malignant plasma cells and represents a novel therapy in multiple myeloma. Exp Hematol. 2011; 39:330-338.
43. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367:645-648.
44. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3:730-737.
45. Johnsen HE, Kjeldsen MK, Urup T, Fogd K, Pilgaard L, Boegsted M, Nyegaard M, Christiansen I, Bukh A, Dybkaer K. Cancer stem cells and the cellular hierarchy in haematological malignancies. Eur J Cancer. 2009; 45:194-201.
46. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, Chomienne C, Ishikawa F, Schuringa JJ, Stassi G, Huntly B, Herrmann H, Soulier J, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012; 12:767-775.
47. ten Cate B, de Bruyn M, Wei Y, Bremer E, Helfrich W. Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr Drug Targets. 2010; 11:95-110.
48. Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets. 2011; 11:56-71.
49. Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011; 30:1009-1019.
50. Sinclair A, Latif AL, Holyoake TL. Targeting survival pathways in chronic myeloid leukaemia stem cells. Br J Pharmacol. 2013; 169:1693-1707.
51. Zhou J, Chng WJ. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J Stem Cells. 2014; 6:473-484.
52. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, Watkins DN, Huff CA, Jones RJ. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008; 68:190-197.
53. Basak GW, Carrier E. The search for multiple myeloma stem cells: the long and winding road. Biol Blood Marrow Transplant. 2010; 16:587-594.
54. Abe M, Harada T, Matsumoto T. Concise review: Defining and targeting myeloma stem cell-like cells. Stem Cells. 2014; 32:1067-1073.
55. Franqui-Machin R, Wendlandt EB, Janz S, Zhan F, Tricot G. Cancer stem cells are the cause of drug resistance in multiple myeloma: fact or fiction? Oncotarget 2015; 6:40496-40506. doi: 10.18632/oncotarget.5800.
56. Johnsen HE, Bøgsted M, Schmitz A, Støve Bødker J, El-Galaly TC, Johansen P, Valent P, Zojer N, De Bruyne E, Vanderkerken K, van Duin M, Sonneveld P, Perez-Andres M, et al. The myeloma stem cell concept, revisited: from phenomenology to operational terms. Manuscript submitted.
57. Hosen N, Matsuoka Y, Kishida S, Nakata J, Mizutani Y, Hasegawa K, Mugitani A, Ichihara H, Aoyama Y, Nishida S, Tsuboi A, Fujiki F, Tatsumi N, et al. CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia. 2012; 26:2135-2141.
58. Paíno T, Ocio EM, Paiva B, San-Segundo L, Garayoa M, Gutiérrez NC, Sarasquete ME, Pandiella A, Orfao A, San Miguel JF. CD20 positive cells are undetectable in the majority of multiple myeloma cell lines and are not associated with a cancer stem cell phenotype. Haematologica. 2012; 97:1110-1114.
59. Paiva B, Paino T, Sayagues JM, Garayoa M, San-Segundo L, Martín M, Mota I, Sanchez ML, Bárcena P, Aires-Mejia I, Corchete L, Jimenez C, Garcia-Sanz R, et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood. 2013; 122:3591-3598.
60. Thiago LS, Perez-Andres M, Balanzategui A, Sarasquete ME, Paiva B, Jara-Acevedo M, Barcena P, Sanchez ML, Almeida J, González M, San Miguel JF, Garcia-Sanz R, Orfao A. Circulating clonotypic B cells in multiple myeloma and monoclonal gammopathy of undetermined significance. Haematologica. 2014; 99:155-162.
61. Takishita M, Kosaka M, Goto T, Saito S. Cellular origin and extent of clonal involvement in multiple myeloma: genetic and phenotypic studies. Br J Haematol. 1994; 87:735-742.
62. Pilarski LM, Masellis-Smith A, Szczepek A, Mant MJ, Belch AR. Circulating clonotypic B cells in the biology of multiple myeloma: speculations on the origin of myeloma. Leuk Lymphoma. 1996; 22:375-383.
63. Rasmussen T, Jensen L, Honoré L, Andersen H, Johnsen HE. Circulating clonal cells in multiple myeloma do not express CD34 mRNA, as measured by single-cell and real-time RT-PCR assays. Br J Haematol. 1999; 107:818-824.
64. Boegsted M, Holst JM, Fogd K, Falgreen S, Sørensen S, Schmitz A, Bukh A, Johnsen HE, Nyegaard M, Dybkaer K. Generation of a predictive melphalan resistance index by drug screen of B-cell cancer cell lines. PLoS One. 2011; 6:e19322.
65. Bøgsted M, Bilgrau AE, Wardell CP, Bertsch U, Schmitz A, Bødker JS, Kjeldsen MK, Goldschmidt H, Morgan GJ, Dybkaer K, Johnsen HE. Proof of the concept to use a malignant B cell line drug screen strategy for identification and weight of melphalan resistance genes in multiple myeloma. PLoS One. 2013; 8:e83252.
66. Herrmann H, Kneidinger M, Cerny-Reiterer S, Rülicke T, Willmann M, Gleixner KV, Blatt K, Hörmann G, Peter B, Samorapoompichit P, Pickl W, Bharate GY, Mayerhofer M, et al. The Hsp32 inhibitors SMA-ZnPP and PEG-ZnPP exert major growth-inhibitory effects on CD34(+)/CD38(+) and CD34(+)/CD38(-) AML progenitor cells. Curr Cancer Drug Targets. 2012; 12:51-63.
67. Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Böhm A, Gruze A, Samorapoompichit P, Manley PW, Fabbro D, Pickl WF, Sillaber C, Valent P. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006; 107:752-759.
68. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22:27-55.