
Structure and Biochemical properties of SPARC
SPARC is a matricellular glycoprotein that has been associated with extensive tissue remodeling and tumorigenesis. SPARC gene was initially discovered as a bone matrix and an endothelial basement membrane protein (hence the names osteonectin/BM40). SPARC protein is encoded by a single gene in human chromosome 5q31.1 and mouse chromosome 11 [1-3]. Mature SPARC protein has 286 amino acids with three distinct functional domains, including an N-terminus acidic domain (NT), follistatin-like domain (FS) and C-terminus domain (EC). The NT domain, spans the first 52 amino acids, is highly acidic and binds hydroxyapatite with low affinity Ca2+ binding (5-8 Ca2+) [3, 4]. The FS comprises the next 85 amino acids and contains several internal disulfide bonds and N-glycosylation site. The EC domain is 149 amino acids and contains two EF-hand motifs that bind calcium with high affinity and is comprised almost entirely of β-helices (Figure 1).
The biological functions of SPARC were depicted from the phenotypes of Sparc-deficient mice (SP-/-) and were related to defects of fibroblast and myeloid differentiation and plasticity as cataract formation, osteopenia, decreased size and tensile strength of collagen fibers, and increased deposition of adipose tissue [5, 6]. More biological functions evolved when these mice were challenged as accelerated wound and defective organ healing after injury, increased angiogenesis as well as accelerated growth of implanted syngeneic subcutaneous (SC) tumors. All were initially attributed to increased proliferation and angiogenesis with defective matrix assembly and encapsulation [7-19]. Unique contextual biological functions of SPARC were identified from disease/organ-specific models. In cancer, our knowledge of the contextual expression and functions of SPARC was furthered by orthotopic as well as autochthonous (oncogene-driven and carcinogen-induced) models of cancer together with xenografts of human cancer cells in immuno-deficient mice and in vitro 2D and 3D cell culture systems [7, 19-25]. In addition, SPARC is not directly implicated in the cellular transformation and cancer initiation as spontaneous cancers do not develop in mice with germline deletion of SPARC. However, SPARC is significantly implicated in the pathobiology of many cancers where it influences tumor-stromal interactions in both autocrine and paracrine manners modulating tumor progression and response to therapy (summarized in [19]).
The role of SPARC in tumor development and metastasis is contextual. It depends not only on cancer type, but on whether the molecule is produced by cancer cells or surrounding stromal cells, its subcellular localization in a given cell type, the composition of the ECM, as well as on its interactions with the biologically active molecules in a given tumor milieu [19, 23, 26]. Stromal (host) SPARC regulates ECM deposition and modulates tumor growth and progression [7, 8, 19, 22, 23, 27, 28]. SPARC normalizes the tumor microenvironment (TME) through anti-inflammatory properties and regulation of integrin-growth factor receptor interactions [7, 19, 22-24, 26] or through a regulation of MMPs release [27, 29, 30]. SPARC exerts an autocrine and a paracrine inhibition of tumor cell proliferation [7, 19, 22-24, 28, 31-33] evident in cell cycle arrest at G1/S phase [19, 28]. In addition, tumor SPARC is subject to epigenetic silencing through promoter methylation in many cancers [26, 28, 34-41]. Herein, we review the current knowledge of the role and association of SPARC in bladder cancer.
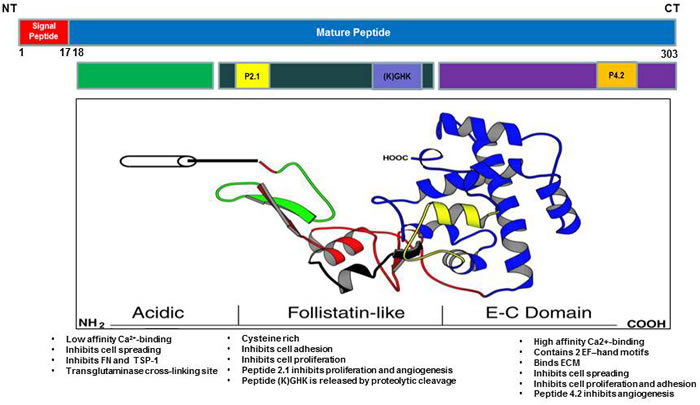
Figure 1: Schematic illustration of SPARC molecule showing the functional domains.
Bladder Cancer
Bladder cancer is the most common malignancy affecting the urinary system with estimated 76,960 new cases and projected 16,390 deaths in 2016 in the United States. This represents ~4% increase in new cases and 2.4% increase in mortality from 2015 [42]. Patients with bladder cancer present at or after 5th decade of life with male: female ratio of 3:1 [42, 43]. Environmental risk factors include tobacco smoking, occupational exposure to aromatic amines and polycyclic hydrocarbons, consumption of arsenic-contaminated water, chronic infections, ionizing radiations and therapeutic abuse of phenacetin-containing analgesics. Tobacco smoking is the major environmental risk factor. It is estimated that > 50% of cases of bladder cancer can be attributed to smoking [44]. The most common form of bladder cancer is urothelial carcinoma (UC), formerly known as transitional cell carcinoma that arises from the mucosal lining of the bladder, and frequently occurs as a multifocal disease involving several simultaneous tumors scattered over the urothelium (Figure 2). Urothelial carcinomas often exhibit elements of squamous or glandular differentiation, which is more common in high-grade and high-stage lesions [45]. Other less frequent pathological subtypes include squamous cell carcinoma, adenocarcinoma, and small cell carcinoma [45]. Most patients present with non-muscle invasive disease (NMI) that is treated with bladder preserving endoscopic resection and adjuvant intra-vesical therapies. However, recurrence is high with 50% chances to progress to muscle invasive (MI) disease (reviewed in [46, 47]). Patients who present with MI disease are treated best with aggressive local therapy (e.g. radical cystectomy, radiotherapy) and/or systemic chemotherapy [47]. Half of the patients with MI disease or progressing to it from NMI disease harbor occult metastases with significantly poor prognosis and survival [47]. Because of its protracted natural history, bladder cancer is among the most expensive malignancies to treat from diagnosis to death. Treatments have not advanced in the past 30 years and there is no approved second line of therapy [46, 47], so there is a crucial unmet need for novel therapies, especially for advanced disease.
Numerous genetic and epigenetic alterations were implicated in tumorigenesis and progression of bladder cancer. The Cancer Genome Atlas (TCGA) data [48] revealed statistically significant recurrent mutations in 32 genes, including multiple genes involved in cell-cycle regulation, chromatin regulation, and kinase signaling pathways. However because this disease, like other cancers, consists of biologically heterogeneous cell populations, selection pressures inferred from the microenvironment, favor development of subpopulations of cells with different dormant, invasive and metastatic abilities. In this respect, SPARC represents an interesting molecule as it exerts distinct roles on cancer cells and tissue-specific stromal cells and modulates the interactions of cancer cells with the cellular and acellular components of the surrounding tumor microenvironment (TME).
The expression of SPARC in normal urinary bladder
In the normal bladders, SPARC protein is expressed in basal and luminal surfaces of normal murine and human urothelia and is secreted by primary urothelial cultures [19, 49]. SPARC exerted anti-proliferative and de-adhesive effects on cultured normal human and murine urothelial cells in vitro [50]. SPARC is dispensable for bladder development and function as evidenced by the normal development and function of mice with germline deletion of SPARC. SPARC protein is also expressed in the sub-urothelial stroma [19]. Further studies of the main two stromal components, fibroblasts and macrophages, indicated that both cell types express and secrete SPARC that is required for their differentiation and phenotypic commitment in response to micro-environmental cues [19].
SPARC expression in human bladder cancer is associated with advancer stage and poor outcome
In human bladder cancer tissue microarrays (TMA) that comprised 192 patients’, SPARC protein expression significantly decreased in MI compared to NMI disease [19]. In NMI disease, SPARC is expressed in the cancerous urothelium and adjacent stroma. In contrast, in MI disease, the expression of SPARC exhibited distinctive compartmentalization with decrease in the frequency and intensity of staining in the cancerous tissue while positive staining was mainly observed in the tumor associated stroma. SPARC expression inversely correlated with disease-specific survival (DSS). However, there was no relationship between the intensity and/or frequency of stromal SPARC expression and DSS. Consistently, RNA and copy number analysis data curated from oncomine database (www.oncomine.org) indicated down-regulation/low copy number of SPARC transcript in 15/16 studies (Figure 3).
Effect of SPARC on carcinogen-induced urothelial transformation
SPARC transcript and protein expression were reported to be significantly downregulated by carcinogenic heavy metals during malignant transformation of UROtsa cell line [51]. The clinically-related studies appeared descriptive and represent snap shots of archival tumor samples. Therefore, it was imperative to develop multiple preclinical models to comprehensively investigate the roles of SPARC in multi-step carcinogenesis and metastasis cascades [19]. The relevance of SPARC in the hallmarks of bladder cancer was determined using a carcinogen-induced model providing a tobacco metabolite and chemical carcinogen N-nitrosobutyl(4-hydroxybutyl)amine (BBN) to SP-/- and SP+/+ mice for ~ 40 weeks. This model recapitulated human bladder cancer that develops after prolonged exposure to chemical carcinogens and allowed the study the pre-neoplastic lesions that are not clinically encountered [19] (Figure 2). This model in tandem with preclinical models using human cell lines in vitro and in xenografts allowed study of the effect of SPARC on the hallmarks of cancer [52] that encompass fundamental biological capabilities acquired during cell transformation and cancer development including sustained proliferation, evasion of growth suppression, death resistance, replicative immortality, induced angiogenesis, and initiation of invasion and metastasis.
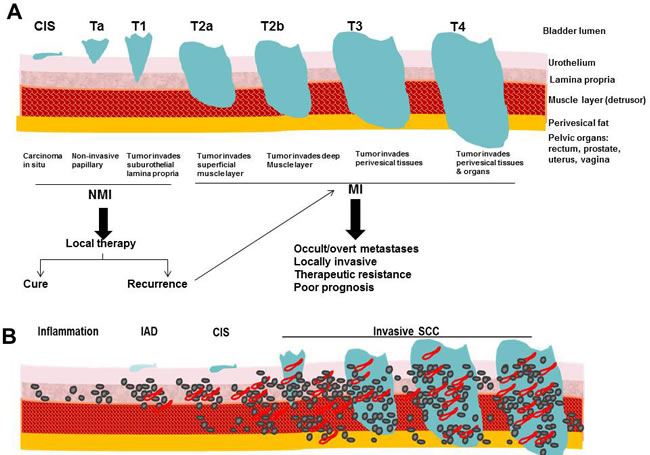
Figure 2: Comparative illustration of: A. Staging of human bladder cancer according to the Tumor-Node-Metastasis (TNM) system, B. pathological stages in carcinogen-induced murine model of bladder cancer.
SPARC restrains cell proliferation and cell cycle progression
Cellular transformation and tumor progression require escape from proliferative suppression and the tightly controlled cell cycle by the G1/S cell cycle checkpoint. SPARC-deficient urothelial lesions exhibited a significant progressive increase in proliferation compared to SPARC-proficient lesions with significantly increasing higher levels of cyclins A1, D1 and E2 that drive cell cycle progression and were positively correlated with the presence, invasion, progression and metastasis of human bladder cancer [53, 54]. Paradoxically, the expression of cell cycle inhibitors p21CIP/WAF1 and p27KIP1 significantly and progressively decreased in SPARC-deficient compared to SPARC-proficient urothelial lesions; a finding that phenocopies their perturbed expression in human urothelial cancer [55-65] and further supports the tumor suppressor effect of SPARC in part through inhibition of cell cycle progression at G1/S.
Interestingly, the expression of SPARC protein and transcript were significantly downregulated in tumorigenic bladder cancer cell line T24T compared to its non-tumorigenic isogenic line T24 [19, 66]. SPARC expression in human bladder cancer cell lines inversely correlated with their proliferation rate, restrained cell cycle progression through slowing G1/S cell cycle proteins, cyclins A1, D1 and E2 with increase of their inhibitors p21CIP/WAF1 and p27KIP1. Of interest is that in human bladder cancer, cyclin D1 (CCND1) is amplified in 20%, whereas inactivating mutations, hemizygous and homozygous deletions of the tumor suppressor CDKN1A gene that encodes p21CIP/WAF1 have been recently reported with higher frequencies of deletion in muscle invasive (MI) disease [44]. The inhibition of activation of cJun/AP1 by SPARC [19] suggests that inhibition of cell cycle deregulation is mediated in part through inhibition of the upstream transcription factor. In addition, TCGA data [48] revealed the enrichment of signaling pathways that were inhibited by SPARC in ovarian cancer [7, 23, 24] as phosphatidylinositol-3-OH kinase/AKT/mTOR pathway and the RTK/MAPK pathway suggesting potential inhibitory effect in bladder cancer milieu.

Figure 3: Comparison of SPARC expression/copy number across 16 Analyses of gene expression/copy number analysis in human bladder cancer.
Anti-oxidant and anti-inflammatory
In carcinogen-induced urothelial lesions, SPARC inhibited the progressive generation of ROS and markers of DNA, protein and lipid oxidative damage; a scenario that was significantly augmented in SP-/- urothelial lesions. SPARC-proficient (SP+/+) lesions exhibited significantly decreased activation of the downstream signaling cascades that converged in the activation of AP-1 and NFκB, the major orchestrators of inflammation, carcinogenesis, invasiveness and metastasis [19, 67-69]. High levels of ROS (H2O2) are generated by urothelial cancer cells compared to normal urothelial cells implying that ROS are generated upon carcinogen exposure and from enhanced metabolism of the rapidly proliferating transformed cells [19]. This explains, in part, the significantly higher levels of ROS in the rapidly proliferating SP-/- urothelial cells. High ROS concentrations are generated by cancer cells and by the surrounding juxta-tumoral stromal cells mainly TAMs and CAFs [19, 70] (Figure 4).
The increased ROS in the evolving urothelial cancer milieu was manifested by progressive increased levels of 8-hydroxy-2’ -deoxyguanosine (8-OHdG) that contributes to genome stability and promotes carcinogenesis [71]. Chronic oxidative stress through DNA damage has been shown to induce to double strand breaks (DSBs) with complex DNA ends. Repairing such complex DSBs with non-homologous end joining (NHEJ) may be critical for the production of spontaneous mutations and inflammation-related cancers [72]. Mitochondrial DNA and nuclear DNA undergo several alterations that may result in mutation accumulation and genomic instability [72, 73]. In lieu of these reports we can speculate that SPARC, through its anti-oxidant and anti-inflammatory effects may exert a protective role against carcinogen- and ROS-induced genome instability and mutations.
Oncogenic transformation induces metabolic reprogramming of cancer cells with increased glucose and glutamine metabolism and, consequently, increased proliferation and ROS production [73-75]. Oncogenic transcription factors as NFκB, and AP-1 are activated by ROS and in synergy with the specific signaling from their downstream target genes may further perturbation of cellular energy and metabolism [73-75]. The rapidly proliferating cells in the absence of SPARC along with activation of pro-oncogenes and loss of tumor suppressors generate increased levels of ROS, a manifestation of altered cellular metabolism, may induce aerobic glycolysis or Warburg effect [49].
The kinetics of SPARC protein expression during differentiation of primary fibroblasts and macrophages revealed that it increases during early differentiation, then decreases to basal levels in macrophages but remained steady in differentiated fibroblasts. Mechanistic studies using heterotypic co-cultures of normal and cancerous urothelial cells with normal and tumor associated stromal cells indicated that SPARC inhibited the acquisition inflammatory secretory phenotype of tumor associated macrophages (TAMs) and cancer associated fibroblasts (CAFs) through inhibition of the activation of NFκB and AP-1 with subsequent decrease in their secreted cytokines and cancer cell invasiveness. These findings suggested that SPARC markedly inhibited the inflammatory feed-forward loop that is reciprocated and maintained among cancer cells, TAMs, and CAFs through secreted inflammasomes (ROS, bioactive lipids, cytokines, chemokines and growth factors) that act as a double-edged sword in the tumor microenvironment. On the one hand, they sustain cancer cell proliferation, invasiveness, angiogenesis, and metastasis. On the other hand, they play a critical role in recruitment and differentiation of stromal cells.
In addition, the inflammatory TME in low/absence of SPARC can contribute to immune suppression enabling tumor cells to evade the immune system and disseminate [76]. Therefore, the kinetics of SPARC expression in cancerous and stromal cells in human and murine urothelial cancer implicate SPARC in the intricate tightly-regulated programs of cellular recruitment, proliferation, differentiation/de-differentiation of stromal cells [5, 6, 10, 19, 77, 78]. The differential compartmentalization of SPARC may represent a state of aberrant homeostasis with increased inflammation that might be directly involved in urothelial cell transformation through the persistent release of inflammatory mediators and ROS, ancillary to tumor growth and metastasis (Figure 4).
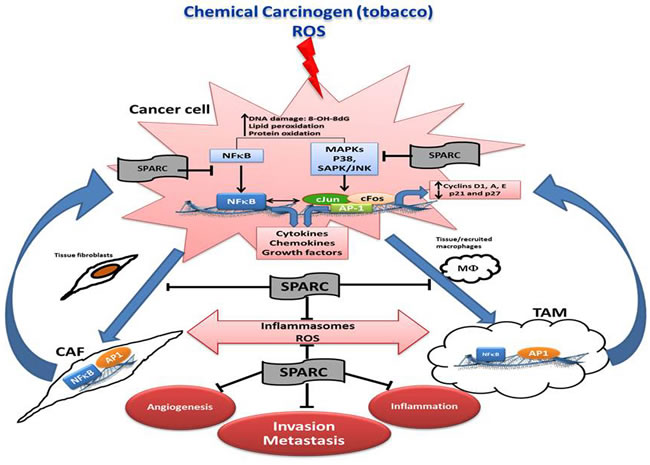
Figure 4: Schematic illustration summarizing the effects of SPARC on the interactions of cancer cells, and stromal cells in the multistep carcinogenesis cascade.
Inhibition of angiogenesis
Tumor angiogenesis is one of the hallmarks of cancer [52]. Initial growth of transformed cells and tumor mass increases the demands for oxygen from blood, thus tumor tissue becomes hypoxic. In turn, hypoxia produces ROS which activate potent angiogenic hypoxia inducible factor (HIF-1α) to increase the transcription of angiogenic growth factors [79]. Angiogenesis is stimulated by growth factors such as VEGF, FGF as well as pro-inflammatory mediators enriched in the TME which induce the proliferation and migration of endothelial cells followed by assembly into patent blood vessels [79, 80]. The anti-angiogenic effects of SPARC have been long recognized as evidenced by its inhibitory effect on endothelial cell proliferation and migration in vitro and in vivo by increased angiogenesis and tumor growth in syngeneic tumors implanted in SP-/- mice and paradoxically, decreased angiogenesis and growth of human tumor xenografts expressing SPARC [7, 9, 19, 21-25, 81-83]. In urothelial cancer, the anti-angiogenic effect of SPARC is attributed to its anti-oxidant, anti-inflammatory effects with subsequent inhibition of angiogenesis at multiple levels [19]. SPARC significantly suppressed carcinogen-induced ROS generation and inflammation while restraining cell cycle progression in cancer cells, thus limiting the increasing demands of the growing tumor cells to new blood vessels. Through modulation of cancer cells’ interactions with stromal cells, SPARC inhibited phenotypic commitment of macrophages and fibroblasts into inflammatory TAM and CAF phenotype. Consequently, the pro-angiogenic factors in the tumor milieu significantly decreased, with subsequent inhibition of endothelial cell recruitments, angiogenesis and vasculogenesis. Furthermore, the observation that SPARC-deficient autochthonous urothelial cancers and in syngeneic tumors growing in SP-/- mice exhibited increased vascularity macroscopically with the increased mean vascular density and area (number and size of blood vessels) strongly implicates a direct effect of host and/or tumor SPARC on endothelial cells in the multistep cascade of tumor angiogenesis and vasculogenesis.
Inhibition of invasion and metastasis
The process of metastasis is defined by distinct steps involving local invasion, intravasation into adjacent blood and lymphatic vessels, transit through circulation and evasion of host immune systems, progressive preconditioning of the prospective metastatic sites, extravasation into the parenchyma of metastatic site, and colonization and formation of micro-metastases, followed by proliferation and progression to macro-metastases. This process is largely inefficient due to the many obstacles tumor cells must overcome to successfully metastasize. In this respect SPARC exerts an anti-metastatic effect at multiple levels. In the carcinogen-induced bladder cancer, SP-/- mice not only exhibited accelerated growth that involved the full bladder wall with invasion of the muscle layer and peri-vesical tissues, but also exhibited early onset metastasis and significantly more metastases [19]. Primary SP-/- tumors exhibited significantly higher pro-inflammatory and pro-invasive mediators and were more vascular than SP+/+ tumors. Pairwise examination of matching lungs and bladder tissues with pre-neoplastic and neoplastic lesions revealed progressive increase in the levels of pro-inflammatory mediators with significantly higher levels in SP-/- compared with SP+/+ lungs as a function of disease progression. In addition, macrophage infiltration was significantly higher in SP-/- compared with the SP+/+ lung metastases. These data suggest that the anti-metastatic effect of SPARC is mediated in part through its anti-inflammatory effect on the primary tumor, suppressing the pre-conditioning of the pre-metastatic lung tissues, “pre-metastatic niche” [19, 84-87] (Figure 5).
The contribution of cancer cell- vs. stromal cell- SPARC in bladder cancer metastasis was dissected using multiple independent approaches. Using a syngeneic model of spontaneous metastasis in which SPARC-proficient MB49 cells were injected SC in SP-/- and SP+/+ mice, host-SPARC not only inhibited the in vivo growth, invasiveness, angiogenesis and inflammation of primary tumors, but it also inhibited spontaneous lung metastasis through inhibition of the pre-conditioning of the inflammatory pre-metastatic and metastatic lung niche [19] (Figure 5).
The role of tumor-SPARC in lung colonization was further investigated in a human experimental metastasis model, injecting human bladder cancer cells genetically modified for SPARC expression into nude mice [19]. SPARC expression inversely correlated with the incidence and multiplicity of lung metastasis. Of note, bladder cancer cells depleted of SPARC exhibited a dramatic decrease in early lung colonization implicating a counter-adhesive effect of SPARC inhibiting tumor cell adhesion to pulmonary endothelial cells and early metastatic colonization.
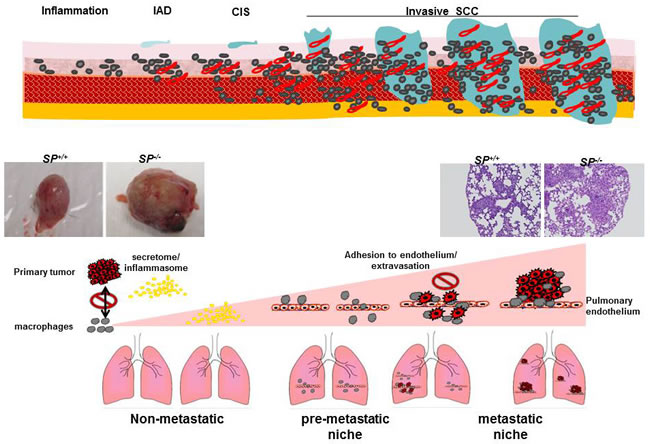
Figure 5: A view of the effect of SPARC on the multistep metastasis cascade and preconditioning pf the metastatic niche.
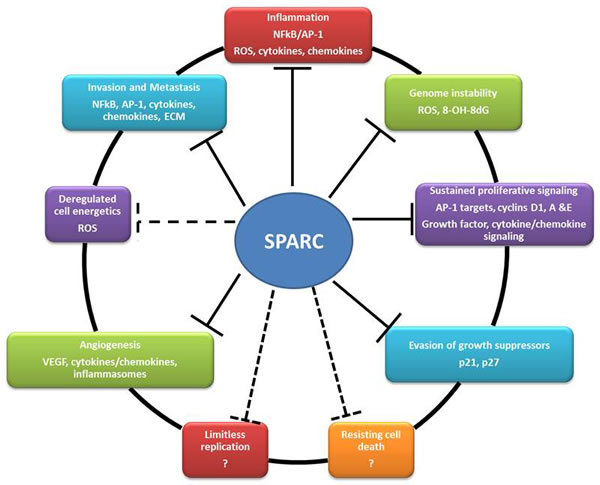
Figure 6: Summary of the reported effects of SPARC on the hallmarks of cancer (solid lines) and the potential effects (dashed lines).
Conclusions and Perspective
A large body of research over the last three decades has established SPARC as an ECM protein that plays a substantial role in tissue homeostasis in physiological and pathological contexts characterized by proliferation, differentiation and plasticity of mesenchymal, myeloid, neuronal as well as endothelial and epithelial cells. The role of SPARC in tumorigenesis has been recently recognized and new functions of SPARC in disease, organ and cell-specific contexts are being unveiled. In bladder cancer, our reports strongly suggest a tumor suppressor effect of SPARC modulating the putative hallmarks of cancer (Figure 6). Collectively, findings from our published reports strongly suggest a SPARC as a potential prognostic biomarker in bladder cancer. The identification of the molecular signature associated with SPARC gene/protein expression could serve as prognostic biomarkers to stratify patients who might develop invasive, metastatic, indolent or recurrent disease. In addition to prognostication, in the era of precision and personalized medicine, it is imperative that future investigations develop and test specific pharmacological agents that target SPARC and/or its regulating/regulated signaling pathways in preclinical and clinical settings.
ACKNOWLEDGMENTs
This study was supported by R01 CA193437 to NS.
ConflictS of Interest
There is no conflict of interest.
References
1. Mason IJ, Murphy D, Munke M, Francke U, Elliott RW and Hogan BL. Developmental and transformation-sensitive expression of the Sparc gene on mouse chromosome 11. The EMBO journal. 1986; 5(8):1831-1837.
2. Mason IJ, Taylor A, Williams JG, Sage H and Hogan BL. Evidence from molecular cloning that SPARC, a major product of mouse embryo parietal endoderm, is related to an endothelial cell ‘culture shock’ glycoprotein of Mr 43,000. The EMBO journal. 1986; 5(7):1465-1472.
3. Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML and Martin GR. Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 1986; 26(1):99-105.
4. Mayer U, Aumailley M, Mann K, Timpl R and Engel J. Calcium-dependent binding of basement membrane protein BM-40 (osteonectin, SPARC) to basement membrane collagen type IV. European Journal of Biochemistry. 1991; 198(1):141-150.
5. Chavey C, Boucher J, Monthouel-Kartmann MN, Sage EH, Castan-Laurell I, Valet P, Tartare-Deckert S and Van Obberghen E. Regulation of secreted protein acidic and rich in cysteine during adipose conversion and adipose tissue hyperplasia. Obesity. 2006; 14(11):1890-1897.
6. Nie J and Sage EH. SPARC functions as an inhibitor of adipogenesis. Journal of cell communication and signaling. 2009; 3(3-4):247-254.
7. Said N, Socha MJ, Olearczyk JJ, Elmarakby AA, Imig JD and Motamed K. Normalization of the ovarian cancer microenvironment by SPARC. Molecular cancer research. 2007; 5(10):1015-1030.
8. Chlenski A and Cohn SL. Modulation of matrix remodeling by SPARC in neoplastic progression. Seminars in cell & developmental biology. 2010; 21(1):55-65.
9. Chlenski A, Guerrero LJ, Peddinti R, Spitz JA, Leonhardt PT, Yang Q, Tian Y, Salwen HR and Cohn SL. Anti-angiogenic SPARC peptides inhibit progression of neuroblastoma tumors. Molecular cancer. 2010; 9:138.
10. Chiodoni C, Colombo MP and Sangaletti S. Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer metastasis reviews. 2010; 29(2):295-307.
11. Sangaletti S, Tripodo C, Cappetti B, Casalini P, Chiodoni C, Piconese S, Santangelo A, Parenza M, Arioli I, Miotti S and Colombo MP. SPARC oppositely regulates inflammation and fibrosis in bleomycin-induced lung damage. The American journal of pathology. 2011; 179(6):3000-3010.
12. Bradshaw AD. The role of SPARC in extracellular matrix assembly. Journal of cell communication and signaling. 2009; 3(3-4):239-246.
13. Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Bonnema DD and Zile MR. Age-dependent alterations in fibrillar collagen content and myocardial diastolic function: role of SPARC in post-synthetic procollagen processing. American journal of physiology Heart and circulatory physiology. 2010; 298(2):H614-622.
14. Card L, Henderson N, Zhang Y, Bornstein P and Bradshaw AD. Expression in SPARC-null mice of collagen type I lacking the globular domain of the alpha1(I) N-propeptide results in abdominal hernias and loss of dermal collagen. Matrix biology. 2010; 29(7):559-564.
15. Goldsmith EC, Bradshaw AD and Spinale FG. Cellular mechanisms of tissue fibrosis. 2. Contributory pathways leading to myocardial fibrosis: moving beyond collagen expression. American journal of physiology Cell physiology. 2013; 304(5):C393-402.
16. McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD and Lindsey ML. SPARC mediates early extracellular matrix remodeling following myocardial infarction. American journal of physiology Heart and circulatory physiology. 2011; 301(2):H497-505.
17. Nie J, Bradshaw AD, Delany AM and Sage EH. Inactivation of SPARC enhances high-fat diet-induced obesity in mice. Connective tissue research. 2011; 52(2):99-108.
18. Rentz TJ, Poobalarahi F, Bornstein P, Sage EH and Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. The Journal of biological chemistry. 2007; 282(30):22062-22071.
19. Said N, Frierson HF, Sanchez-Carbayo M, Brekken RA and Theodorescu D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. The Journal of clinical investigation. 2013; 123(2):751-766.
20. Sangaletti S, Stoppacciaro A, Guiducci C, Torrisi MR and Colombo MP. Leukocyte, rather than tumor-produced SPARC, determines stroma and collagen type IV deposition in mammary carcinoma. The Journal of experimental medicine. 2003; 198(10):1475-1485.
21. Said N, Frierson HF, Jr., Chernauskas D, Conaway M, Motamed K and Theodorescu D. The role of SPARC in the TRAMP model of prostate carcinogenesis and progression. Oncogene. 2009; 28(39):3487-3498.
22. Said N and Motamed K. Absence of host-secreted protein acidic and rich in cysteine (SPARC) augments peritoneal ovarian carcinomatosis. The American journal of pathology. 2005; 167(6):1739-1752.
23. Said N, Najwer I and Motamed K. Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. The American journal of pathology. 2007; 170(3):1054-1063.
24. Said NA, Elmarakby AA, Imig JD, Fulton DJ and Motamed K. SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia. 2008; 10(10):1092-1104.
25. Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, Castrillon DH, Sage EH, Puolakkainen P, Bradshaw AD and Brekken RA. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech. 2010; 3(1-2):57-72.
26. Pan MR, Chang HC, Chuang LY and Hung WC. The nonsteroidal anti-inflammatory drug NS398 reactivates SPARC expression via promoter demethylation to attenuate invasiveness of lung cancer cells. Experimental biology and medicine. 2008; 233(4):456-462.
27. Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, Rumio C, Brekken RA, Chiodoni C and Colombo MP. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer research. 2008; 68(21):9050-9059.
28. Socha MJ, Said N, Dai Y, Kwong J, Ramalingam P, Trieu V, Desai N, Mok SC and Motamed K. Aberrant promoter methylation of SPARC in ovarian cancer. Neoplasia. 2009; 11(2):126-135.
29. Arnold SA and Brekken RA. SPARC: a matricellular regulator of tumorigenesis. Journal of cell communication and signaling. 2009; 3(3-4):255-273.
30. Arnold SA, Rivera LB, Carbon JG, Toombs JE, Chang CL, Bradshaw AD and Brekken RA. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFbeta activation. PloS one. 2012; 7(2):e31384.
31. Mok SC, Chan WY, Wong KK, Muto MG and Berkowitz RS. SPARC, an extracellular matrix protein with tumor-suppressing activity in human ovarian epithelial cells. Oncogene. 1996; 12(9):1895-1901.
32. Yiu GK, Chan WY, Ng SW, Chan PS, Cheung KK, Berkowitz RS and Mok SC. SPARC (secreted protein acidic and rich in cysteine) induces apoptosis in ovarian cancer cells. The American journal of pathology. 2001; 159(2):609-622.
33. Dhanesuan N, Sharp JA, Blick T, Price JT and Thompson EW. Doxycycline-inducible expression of SPARC/Osteonectin/BM40 in MDA-MB-231 human breast cancer cells results in growth inhibition. Breast cancer research and treatment. 2002; 75(1):73-85.
34. Feng J, Shi Z and Ye Z. Effects of metabolites of the lignans enterolactone and enterodiol on osteoblastic differentiation of MG-63 cells. Biological & pharmaceutical bulletin. 2008; 31(6):1067-1070.
35. Sato N, Fukushima N, Matsubayashi H, Iacobuzio-Donahue CA, Yeo CJ and Goggins M. Aberrant methylation of Reprimo correlates with genetic instability and predicts poor prognosis in pancreatic ductal adenocarcinoma. Cancer. 2006; 107(2):251-257.
36. Cheetham S, Tang MJ, Mesak F, Kennecke H, Owen D and Tai IT. SPARC promoter hypermethylation in colorectal cancers can be reversed by 5-Aza-2’deoxycytidine to increase SPARC expression and improve therapy response. British journal of cancer. 2008; 98(11):1810-1819.
37. Yang E, Kang HJ, Koh KH, Rhee H, Kim NK and Kim H. Frequent inactivation of SPARC by promoter hypermethylation in colon cancers. International journal of cancer. 2007; 121(3):567-575.
38. Kahn SL, Ronnett BM, Gravitt PE and Gustafson KS. Quantitative methylation-specific PCR for the detection of aberrant DNA methylation in liquid-based Pap tests. Cancer. 2008; 114(1):57-64.
39. Rodriguez-Jimenez FJ, Caldes T, Iniesta P, Vidart JA, Garcia-Asenjo JL and Benito M. Overexpression of SPARC protein contrasts with its transcriptional silencing by aberrant hypermethylation of SPARC CpG-rich region in endometrial carcinoma. Oncology reports. 2007; 17(6):1301-1307.
40. Wang Y, Yu Q, Cho AH, Rondeau G, Welsh J, Adamson E, Mercola D and McClelland M. Survey of differentially methylated promoters in prostate cancer cell lines. Neoplasia. 2005; 7(8):748-760.
41. Kwabi-Addo B, Wang S, Chung W, Jelinek J, Patierno SR, Wang BD, Andrawis R, Lee NH, Apprey V, Issa JP and Ittmann M. Identification of differentially methylated genes in normal prostate tissues from African American and Caucasian men. Clinical cancer research. 2010; 16(14):3539-3547.
42. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66(1):7-30.
43. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65(1):5-29.
44. Knowles MA and Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015; 15(1):25-41.
45. Wasco MJ, Daignault S, Zhang Y, Kunju LP, Kinnaman M, Braun T, Lee CT and Shah RB. Urothelial Carcinoma with Divergent Histologic Differentiation (Mixed Histologic Features) Predicts the Presence of Locally Advanced Bladder Cancer When Detected at Transurethral Resection. Urology. 2007; 70(1):69-74.
46. Morris DS, Weizer AZ, Ye Z, Dunn RL, Montie JE and Hollenbeck BK. Understanding bladder cancer death: tumor biology versus physician practice. Cancer. 2009; 115(5):1011-1020.
47. Hollingsworth JM, Zhang Y, Krein SL, Ye Z and Hollenbeck BK. Understanding the variation in treatment intensity among patients with early stage bladder cancer. Cancer. 2010; 116(15):3587-3594.
48. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507(7492):315-322.
49. Thomas R, True LD, Bassuk JA, Lange PH and Vessella RL. Differential Expression of Osteonectin/SPARC during Human Prostate Cancer Progression. Clinical Cancer Research. 2000; 6(3):1140-1149.
50. Kosman J, Carmean N, Leaf EM, Dyamenahalli K and Bassuk JA. The motif of SPARC that inhibits DNA synthesis is not a nuclear localization signal. J Mol Biol. 2007; 371(4):883-901.
51. Larson J, Yasmin T, Sens DA, Zhou XD, Sens MA, Garrett SH, Dunlevy JR, Cao L and Somji S. SPARC gene expression is repressed in human urothelial cells (UROtsa) exposed to or malignantly transformed by cadmium or arsenite. Toxicol Lett. 2010; 199(2):166-172.
52. Hanahan D and Weinberg RA. The Hallmarks of Cancer. Cell. 2000; 100(1):57-70.
53. Shariat SF, Ashfaq R, Sagalowsky AI and Lotan Y. Correlation of cyclin D1 and E1 expression with bladder cancer presence, invasion, progression, and metastasis. Human Pathology. 2006; 37(12):1568-1576.
54. Galmozzi F, Rubagotti A, Romagnoli A, Carmignani G, Perdelli L, Gatteschi B and Boccardo F. Prognostic value of cell cycle regulatory proteins in muscle-infiltrating bladder cancer. Journal of Cancer Research and Clinical Oncology. 2006; 132(12):757-764.
55. Chatterjee SJ, Datar R, Youssefzadeh D, George B, Goebell PJ, Stein JP, Young L, Shi SR, Gee C, Groshen S, Skinner DG and Cote RJ. Combined effects of p53, p21, and pRb expression in the progression of bladder transitional cell carcinoma. J Clin Oncol. 2004; 22(6):1007-1013.
56. Gao L, Fang YQ, Zhang TY, Ge B, Xu B, Huang JF, Zhang ZF and Tan N. GSTP1 arrests bladder cancer T24 cells in G0/G1 phase and up-regulates p21 expression. Int J Clin Exp Med. 2014; 7(9):2984-2991.
57. Ioachim E, Michael M, Stavropoulos NE, Kitsiou E, Hastazeris K, Salmas M, Stefanaki S and Agnantis NJ. Expression patterns of cyclins D1, E and cyclin-dependent kinase inhibitors p21(Waf1/Cip1) and p27(Kip1) in urothelial carcinoma: correlation with other cell-cycle-related proteins (Rb, p53, Ki-67 and PCNA) and clinicopathological features. Urol Int. 2004; 73(1):65-73.
58. Jia ZM, Ai X, Teng JF, Wang YP, Wang BJ and Zhang X. p21 and CK2 interaction-mediated HDAC2 phosphorylation modulates KLF4 acetylation to regulate bladder cancer cell proliferation. Tumour Biol. 2016.
59. Lashinger LM, Zhu K, Williams SA, Shrader M, Dinney CP and McConkey DJ. Bortezomib abolishes tumor necrosis factor-related apoptosis-inducing ligand resistance via a p21-dependent mechanism in human bladder and prostate cancer cells. Cancer Res. 2005; 65(11):4902-4908.
60. Li TM, Chen GW, Su CC, Lin JG, Yeh CC, Cheng KC and Chung JG. Ellagic acid induced p53/p21 expression, G1 arrest and apoptosis in human bladder cancer T24 cells. Anticancer Res. 2005; 25(2A):971-979.
61. Shariat SF, Ashfaq R, Sagalowsky AI and Lotan Y. Predictive value of cell cycle biomarkers in nonmuscle invasive bladder transitional cell carcinoma. J Urol. 2007; 177(2):481-487; discussion 487.
62. Shariat SF, Tokunaga H, Zhou J, Kim J, Ayala GE, Benedict WF and Lerner SP. p53, p21, pRB, and p16 expression predict clinical outcome in cystectomy with bladder cancer. J Clin Oncol. 2004; 22(6):1014-1024.
63. Tang K, Wang C, Chen Z, Xu H and Ye Z. Clinicopathologic and prognostic significance of p21 (Cip1/Waf1) expression in bladder cancer. Int J Clin Exp Pathol. 2015; 8(5):4999-5007.
64. Vallmanya Llena FR, Laborda Rodriguez A, Lloreta Trull J, Cortadellas Angel R, Placer Santos J and Mas Gelabert A. [Immunohistochemical expression of p53, p21, p16, and cyclin D1 in superficial bladder cancer. A tissue microarray study [Article in Spanish]. Actas Urol Esp. 2006; 30(8):754-762.
65. Wang C, Chen Z, Ge Q, Hu J, Li F, Hu J, Xu H, Ye Z and Li LC. Up-regulation of p21(WAF1/CIP1) by miRNAs and its implications in bladder cancer cells. FEBS Lett. 2014; 588(24):4654-4664.
66. Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, Frierson HF, Conaway MR and Theodorescu D. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002; 62(22):6418-6423.
67. Huang YJ, Zhang BB, Ma N, Murata M, Tang AZ and Huang GW. Nitrative and oxidative DNA damage as potential survival biomarkers for nasopharyngeal carcinoma. Med Oncol. 2010; 28(1):377-384.
68. Tang S-W, Yang T-C, Lin W-C, Chang W-H, Wang C-C, Lai M-K and Lin J-Y. Nicotinamide N-methyltransferase induces cellular invasion through activating matrix metalloproteinase-2 expression in clear cell renal cell carcinoma cells. Carcinogenesis. 2010; 32(2):138-145.
69. Gao J, Song J, Huang H, Li Z, Du Y, Cao J, Li M, Lv S, Lin H and Gong Y. Methylation of the SPARC gene promoter and its clinical implication in pancreatic cancer. J Exp Clin Cancer Res. 2010; 29:28.
70. Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H and Lleonart ME. Oxidative stress and cancer: An overview. Ageing Research Reviews. 2013; 12(1):376-390.
71. Valavanidis A, Vlachogianni T and Fiotakis C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009; 27(2):120-139.
72. Sharma V, Collins LB, Chen T-h, Herr N, Takeda S, Sun W, Swenberg JA and Nakamura J. (2016). Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget. 2016; 7: 25377-90. doi: 10.18632/oncotarget.8298.
73. Tafani M, Sansone L, Limana F, Arcangeli T, De Santis E, Polese M, Fini M and Russo MA. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Medicine and Cellular Longevity. 2016; 2016:18.
74. Gatenby RA and Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004; 4(11):891-899.
75. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC and Levy DE. Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Science. 2009; 324(5935):1713-1716.
76. Ghirelli C and Hagemann T. Targeting immunosuppression for cancer therapy. The Journal of Clinical Investigation. 123(6):2355-2357.
77. Bhoopathi P, Chetty C, Dontula R, Gujrati M, Dinh DH, Rao JS and Lakka SS. SPARC stimulates neuronal differentiation of medulloblastoma cells via the Notch1/STAT3 pathway. Cancer research. 2011; 71(14):4908-4919.
78. Piconese S, Costanza M, Tripodo C, Sangaletti S, Musio S, Pittoni P, Poliani PL, Burocchi A, Passafaro AL, Gorzanelli A, Vitali C, Chiodoni C, Barnaba V, Pedotti R and Colombo MP. The matricellular protein SPARC supports follicular dendritic cell networking toward Th17 responses. Journal of autoimmunity. 2011; 37(4):300-310.
79. Ferrara N, Hillan KJ, Gerber H-P and Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004; 3(5):391-400.
80. Pickup MW, Mouw JK and Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO reports. 2014; 15(12):1243-1253.
81. Bhoopathi P, Chetty C, Gujrati M, Dinh DH, Rao JS and Lakka SS. The role of MMP-9 in the anti-angiogenic effect of secreted protein acidic and rich in cysteine. British journal of cancer. 2010; 102(3):530-540.
82. Chlenski A, Liu S, Baker LJ, Yang Q, Tian Y, Salwen HR and Cohn SL. Neuroblastoma angiogenesis is inhibited with a folded synthetic molecule corresponding to the epidermal growth factor-like module of the follistatin domain of SPARC. Cancer research. 2004; 64(20):7420-7425.
83. Said NA, Najwer I, Socha MJ, Fulton DJ, Mok SC and Motamed K. SPARC inhibits LPA-mediated mesothelial-ovarian cancer cell crosstalk. Neoplasia. 2007; 9(1):23-35.
84. Said N, Sanchez-Carbayo M, Smith SC and Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. The Journal of clinical investigation. 2012; 122(4):1503-1518.
85. Said N, Smith S, Sanchez-Carbayo M and Theodorescu D. Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. The Journal of clinical investigation. 2011; 121(1):132-147.
86. Said N and Theodorescu D. RhoGDI2 suppresses bladder cancer metastasis via reduction of inflammation in the tumor microenvironment. Oncoimmunology. 2012; 1(7):1175-1177.
87. Said N and Theodorescu D. Permissive role of endothelin receptors in tumor metastasis. Life sciences. 2012; 91(13-14):522-527.