
INTRODUCTION
Natural Killer (NK) cell neoplasms are a rare and heterogeneous group of disorders characterized by excessive proliferation of cytotoxic CD3− CD16/56+ NK cells. This group comprises two subtypes: aggressive NK leukemia (ANKL) and chronic lymphoproliferative disorder (CLPD) [1, 2]. ANKL is an Epstein Barr virus (EBV)-associated tumor most prevalent among Asian young adults (median age of 42 years). It has a fulminant clinical course, frequently resulting in death within two months. In contrast, CLPD has no demonstrable association with EBV and tends to occur in older adults (median age of 60 years). The clinical course is typically indolent, similarly to T-cell large granular lymphocytes leukemia (T-LGLL) [3–5].
Since no standard therapies for aggressive NK cell neoplasms have been established so far and the overall outcomes are dismal, new therapeutic options are needed.
Fyn, a tyrosine-specific phospho-transferase, is a member of Src family kinases which includes c-Src, Yes, Lck, Lyn, Hck, Fgr and Blk [6–8]. It phosphorylates a variety of target proteins involved in different signaling pathways [6]. Moreover, it regulates several biological functions, including growth factor and cytokine receptor signaling, cell-cell adhesion, integrin-mediated signaling, ion channel function, platelet activation, T and B-cell receptor signaling, axon guidance, mitosis, differentiation of NK cells [9–11].
In the last decade, the implication of Fyn in cancer biology and in hematologic malignancies has become more apparent. In chronic myeloid leukemia (CML) Fyn is up-regulated and its activation seems to be important in imatinib resistance [12, 13]. It is notably involved in the pathogenesis of peripheral T cell lymphomas [14] and in acute myeloid leukemia its higher expression, combined to FLT3-Internal Tandem Duplication (ITD), is correlated with poor prognosis [15].
The tissue-specific pattern of Fyn mRNA indicates that it is more expressed in normal NK and T cells respect to other human tissues [16].
Tintori et al., by a structure-based drug design protocol and following hit-to-lead optimization, found 4c pyrazolo[3,4-d]pyrimidine compound driving inhibition of Fyn phosphorylation with a nanomolar range in an enzymatic cell-free assay. Moreover, the compound showed anti-proliferative activities against different cancer cell lines [17].
Therefore, in the present study we investigated the expression of Fyn in NK leukemic cells and the effect of 4c pyrazolo[3,4-d]pyrimidine compound in NK cell lines and in primary cells from chronic leukemic neoplasms.
RESULTS
Fyn is highly expressed in NK leukemic cells
In order to quantify the presence of Fyn in NK leukemic cells, we firstly assessed its mRNA expression levels in peripheral blood mononuclear cells (PBMCs) from 10 healthy donors (HDs) and 8 CLPD patients by quantitative Real Time-Polymerase Chain Reaction (qRT-PCR). Fyn level was significantly up-regulated in PBMCs from CLPD patients compared to HDs (p < 0.001; Figure 1A). We also analyzed Fyn protein level by western blotting (WB) in PBMCs of 3 HDs, 3 CLPD patients and in two NK cell lines, KHYG1 and NK92. We observed a high level of Fyn protein in PBMCs from chronic patients and in NK leukemic cell lines respect to PBMCs from HDs (Figure 1B).
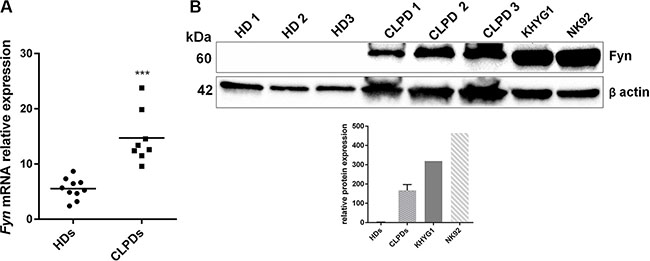
Figure 1: Fyn is over-expressed in NK malignant cells. (A) qRT-PCR of Fyn mRNA in PBMCs from 10 HDs and from 8 patients with CLPD (UPN: 1–8). Statistical significance was determined using an unpaired t test and P-value is indicated by asterisk: ***p < 0.001. (B) WB analysis of Fyn in 3 HDs, 3 CLPD patients (UPN: 6–7) and two NK cell lines, KHYG1 and NK92. Quantification of Fyn protein levels was normalized with β-actin bands.
4c pyrazolo[3,4-d]pyrimidine compound reduced cell viability, induced apoptosis and cell cycle arrest in NK leukemic cells
We treated two cell lines (KHYG1 and NK92), 3 PBMCs from HDs and NK cells isolated from 3 HDs with 4c pyrazolo[3,4-d]pyrimidine compound or with dimethyl sulfoxide (DMSO) vehicle control at different concentrations (2–10 μM) for 24, 48 and 72 hours, cultured with IL2. After 4c treatment, we performed viability test which showed that 4c compound reduced viability of both cell lines in a dose-dependent manner. The effect was observed at 24 hours and remained constant in the other time points (Figure 2A). Importantly, both cell lines needed a significantly lower drug concentration (p < 0.01) to reach 50% reduction of viability (EC50) (Table 1). Interestingly, 4c compound had negligible effect in PBMCs and in purified NK cells from HDs (Figure 2A). Same results were obtained in primary NK cells from HDs treated with 4c compound and cultured without IL2 (Supplementary Figure S1).
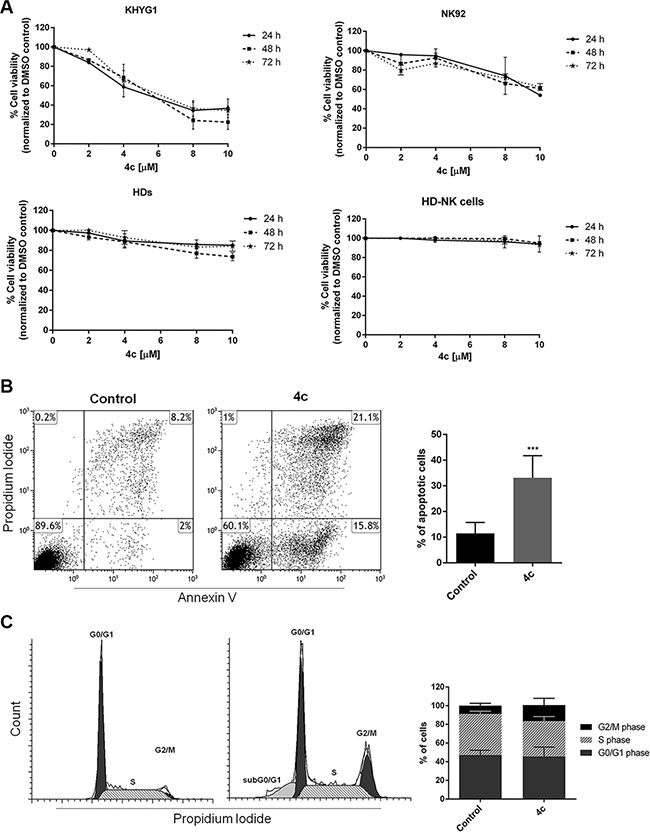
Figure 2: 4c compound reduced cell viability inducing apoptosis and cell cycle arrest in NK leukemic cells. (A) Viability of KHYG1, NK92, 3 PBMCs from HDs (HDs) and NK primary cells sorted from 3 HDs (HD-NK cells) was evaluated by MTS assay after treatment with 4c compound at different concentration (2–10 μM) for 24–72 hours. Results are expressed as percent of cell viability normalized to DMSO-treated control cells. The bar-graphs represent mean with S.D. from three independent experiments. (B) Apoptosis and (C) cell cycle analysis were evaluated by flow cytometer in NK cell line after 4c or DMSO vehicle treatment at 4 μM for 24 hours. Dot plots and cell cycle histograms show a single representative experiment, the bar-graphs represent mean with S.D. from three independent experiments. P-value is indicated by asterisk: ***p < 0.001.
Table 1: EC50 obtained in two cell lines and PBMCs and in purified NK cells from HD samples after 4c compound treatment (P value is < 0.01 for both cell lines vs HDs) (n.c.: not calculable)
EC50 (μM) |
|
|---|---|
KHYG1 |
5.4 |
NK92 |
10.6 |
HDs |
59 |
HD-NK cells |
n.c. |
To further investigate cell death mechanism induced after treatment, we performed apoptosis and cell cycle analysis on KHYG1 cell line by cytometric analysis of Annexin V/propidium iodide (PI) and PI, respectively. After treatment with 4c compound at 4 μM for 24 hours, we observed a significant increase of apoptotic cells (p < 0.001) and cell cycle arrest in G2/M phase in treated KHYG1 respect to their control (Figure 2B–2C).
Fyn phosphorylation is reduced after 4c compound treatment and it decreased Akt and P70 S6 kinase activation
To verify Fyn inhibition we performed its immunoprecipitation in KHYG1 cell line treated with 4c compound or with DMSO vehicle control and we detected its phosphorylation. We observed that Fyn phosphorylation significantly decreased after treatment (p < 0.01; Figure 3A).
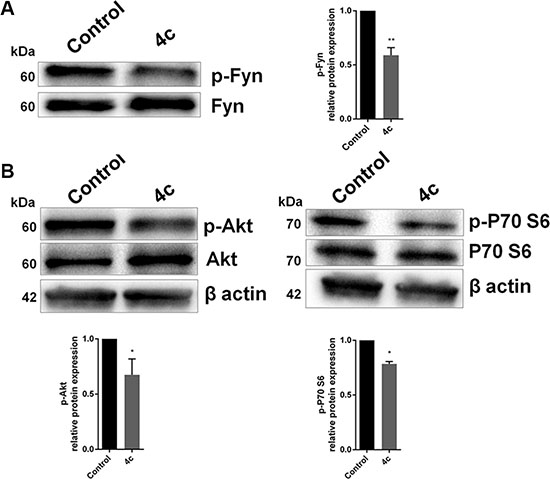
Figure 3: Inhibition on Fyn phosphorylation by 4c compound decreased Akt and P70 phosphorylation. WB analysis of (A) phospho-Fyn and (B) total and phosphorylated Akt and P70 S6 kinase in NK cell line treated with 4c compound compared to control. Protein levels were normalized on β-actin signals. Bar-graphs represent mean with S.D. from three independent experiments. P-values are indicated by asterisk: *p < 0.05, **p < 0.01.
We also explored, by WB, the activation of two protein involved in Fyn pathway, Akt and P70 S6 kinase. Our data showed that there was a decrease of phosphorylation of Akt and P70 S6 kinase after treatment with 4c compound (p < 0.05; Figure 3B).
Gene expression and protein profile of treated NK leukemic cells showed the activation of apoptotic pathways
We performed gene expression profile (GEP) analysis of KHYG1 cell line after 4 μM drug incubation at 24 hours compared to control. A total of 697 genes (297 up-regulated vs 400 down-regulated genes) were differentially expressed. Ingenuity Pathway Analysis (IPA) gene ontology analysis demonstrated that most of genes were involved in cellular growth, death, development and cell cycle (Table 2). Moreover, analysis carried out with DAVID let us to cluster all genes in the same functions indicated by IPA. In fact, the first up-regulated cluster genes [e.g. BCL2-like 13, Caspase-Related Inducer of Apoptosis (CFLAR), DNA Fragmentation Factor Alpha (DFFA) and Fas Ligand] included pro-apoptotic functions (Figure 4A and Supplementary Material, Table S1); conversely, the first down-regulated cluster genes [e.g. Survivin, Cell Division Cycle 34, Aurora Kinase A] included pro-survival functions such as cell cycle progression, cell division, mitosis (Figure 4B and Supplementary Material, Table S2).
Table 2: Molecular and cellular functions of up- and down-regulated genes resulting from IPA gene ontology analysis in GEP analysis of 4c compound vs DMSO treated KHYG1 cells
Molecular and Cellular Functions |
p-value range |
Molecules |
|---|---|---|
Cellular Growth and Proliferation |
7.29E-04/5.16E-15 |
259 |
Cellular Function and Maintenance |
6.79E-04/5.82E-14 |
126 |
Cell Death and Survival |
7.81E-04/6.66E-14 |
239 |
Cellular Development |
5.67E-04/6.26E-13 |
227 |
Cell Cycle |
7.64E-04/2.48E-11 |
126 |
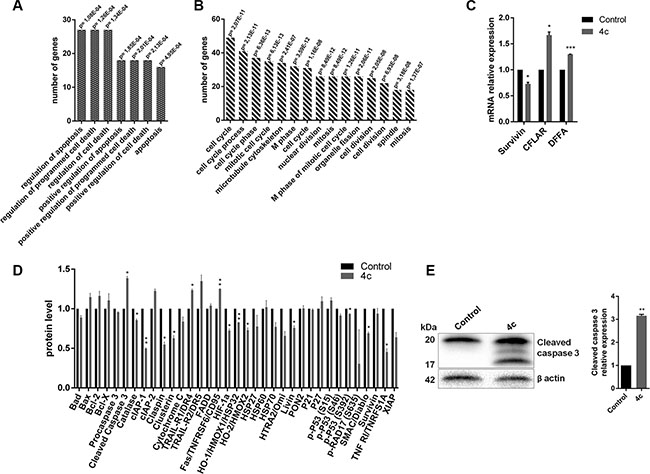
Figure 4: Activation of apoptotic pathways in 4c treated cell line. (A–B) Bar-graphs of the first cluster up-regulated (A) and down-regulated (B) genes resulting by DAVID bioinformatic tool in GEP analysis of KHYG1 treated with 4c compound or with DMSO control. (C) qRT-PCR of Survivin, CFLAR and DFFA in KHYG1 treated vs control. Bar-graphs represent mean with S.D. from three independent experiments. (D) Bar-graphs of protein levels resulted from apoptotic protein array and (E) WB analysis of caspase 3 in KHYG1 treated vs control. P-values are indicated by asterisk: *p < 0.05, **p < 0.01, ***p < 0.001.
We validated GEP data by qRT-PCR confirming the down-regulation of anti-apoptotic gene, Survivin (p < 0.05), and the up-regulation of two pro-apoptotic genes, CFLAR (p < 0.05) and DFFA (p < 0.001), in KHYG1 treated vs control (Figure 4C).
Subsequently, to corroborate apoptosis and GEP data, we also performed an array analysis of multiple apoptotic proteins after treatment. As expected, we observed a low expression of anti-apoptotic proteins as cellular Inhibitors of Apoptosis Protein 1 (cIAP1), Claspin, Clusterin, Heat Shock Protein32 (HSP32), Livin, and high expression of apoptotic ones as cleaved caspase 3, TRAIL receptor 2 (TRAIL-R2) and Fas (Figure 4D).
In order to validate protein array, we showed a significantly increase of cleaved caspase 3 level (p < 0.05) in 4c compound treated cell line by WB (Figure 4E).
4c pyrazolo[3,4-d]pyrimidine compound induced cytotoxic effect and cell cycle arrest in primary NK leukemic cells
In order to evaluate Fyn inhibitor effects on primary cells, we exposed PBMCs from CLPD patients to 4 μM of 4c compound for 24 hours and we evaluated cell viability by trypan blue count. We observed a decrease of 30% of viable cells in treated PBMCs respect control (Figure 5A). Notably, in treated NK cells from leukemic patients there was an increase of active caspase 3/7 level indicating an apoptosis caspase 3-mediated (p < 0.01, Figure 5B).
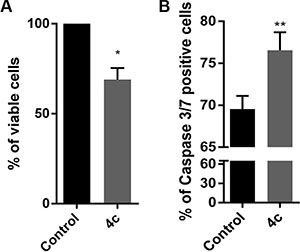
Figure 5: 4c treatment reduced viability and induced caspase 3-mediated apoptosis in PBMC from CLPD patients. (A) Trypan blue count was performed in primary cells, isolated from 3 HDs and 3 CLPD patients (UNP: 6–8) and (B) Caspase 3/7 activity assay on 3 CLPD patients (UNP: 6–8) after 24 hours of treatment with 4c compound at 4 μM concentration. P-values are indicated by asterisk: *p < 0.05, **p < 0.01.
To better elucidate 4c effects on primary NK cells, we sorted CD56+ NK cells from 3 PBMCs of CLPD patients and we exposed them to 4c compound for 24 hours. After incubation time, we evaluated viability by trypan blue count, proliferation by PKH67 labeling, apoptosis and cell cycle by Annexin V/7-Amino-Actinomycin D (7ADD) and PI respectively. Firstly, we observed a decrease of 37% of viable cells (Figure 6A). Furthermore, we analyzed proliferation, apoptosis and caspase 3/7 rate both on CD56+/CD16+ and CD56+/CD16− after 4c treatment. We noted a reduction of proliferation in both CD56+/CD16+ and CD56+/CD16−cells (Figure 6B). Moreover, in both treated populations, we observed a significantly increased apoptosis compared to their respective control (+ 17% in CD56+/CD16+ cells and + 5.4% in CD56+/CD16− cells; p < 0.01 and p < 0.05 respectively) (Figure 6C). The 4c effect on apoptosis was also confirmed by increased expression of caspase3/7 levels in both population (+ 2.5% in CD56+/CD16+ cells and + 8% in CD56+/CD16− cells; p < 0.01 and p < 0.05 respectively) (Figure 6D). Cell cycle analysis after 4c treatment showed that there was an increase of number of NK primary cells in G0/G1 phase (Figure 6E).
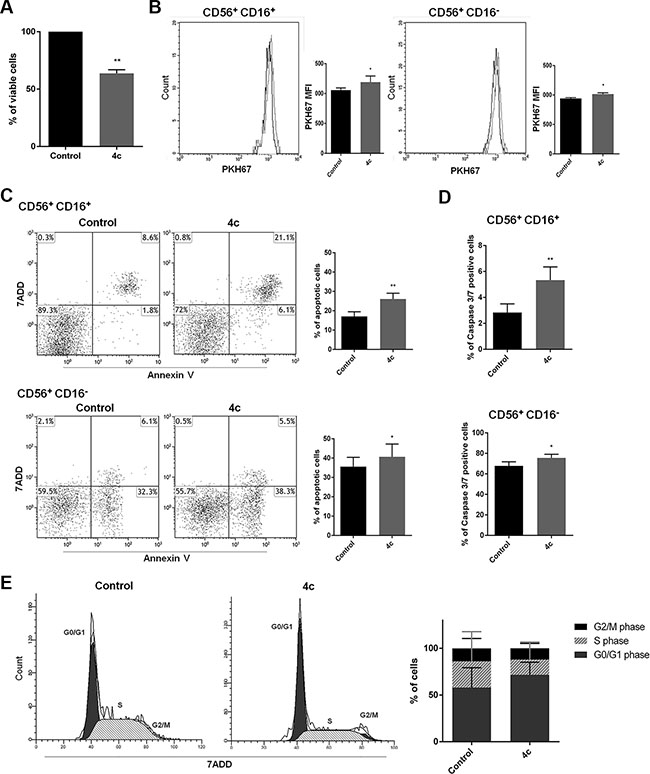
Figure 6: 4c compound treatment reduced cell viability and proliferation, induced caspase 3-mediated apoptosis and cell cycle arrest in primary NK leukemic cells. On 4c treated or not primary NK cells sorted from PBMCs of 3 NK-CLPD patients (UPN: 6–8) (A) Trypan blue count, (B) PKH67 proliferation test, (C) Apoptosis and (D) Caspase 3/7 activity assay, (E) Cell cycle analysis were performed. Proliferation, apoptosis and caspase 3/7 level were analyzed on CD56+/CD16+ and CD56+/CD16− NK cell populations. P-values are indicated by asterisk: *p < 0.05, **p < 0.01. (MFI= mean fluorescent intensity).
4c compound treatment induced phenotype changes in NK primary cells
To evaluate if 4c compound influence NK cell activation, we sorted CD56+ NK cells from 3 PBMCs of CLPD patients and we exposed them to 4c compound for 24 hours to study cell phenotype changes. We analyzed the expression of two markers related to NK activation, CD38 and CD25. We observed that there was a decrease of CD38 expression and similar expression of CD25 after treatment respect to control in NK cells (Figure 7A–7B).
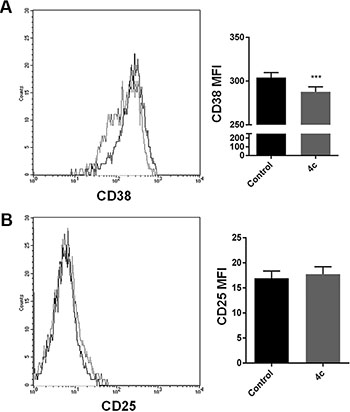
Figure 7: 4c compound treatment induced phenotype changes in NK primary cells. On 4c primary NK cells sorted from PBMCs of 3 CLPD patients (UPN: 6–8) (A) CD38 and (B) CD25 expression were evaluated after 4c treatment. P-values is indicated by asterisk: ***p < 0.001. (MFI = mean fluorescent intensity)
DISCUSSION
NK cell-type large granular lymphocyte leukemia is a rare neoplasm divided in two subtype which could be considered different and independent neoplasms because they have distinct clinical course and management. Chronic lymphoproliferative subtype is characterized by indolent clinical course lacking effective therapeutic approaches [18]; the number of circulating NK cells remains stable for long periods of time and some cases have been reported to show spontaneous regression. On the other hand, aggressive NK leukemia subtype is a fulminant disorder which is not curable.
By tissue-specific pattern of mRNA [16], it was identified Fyn like as a gene more expressed in normal NK cells respect to other tissues. It is required to control cytotoxicity and cytokine production in NK cells [19, 20], and it has a critical role in NK and T-cell development and activation [9, 21].
Since NK leukemia is characterized by an outgrowth of CD3− CD16/56+ cells, Fyn could be a possible target in this hematological disease. In fact, analyzing its expression in NK leukemic cells, we found an increased Fyn transcript level in PBMCs from CLPD patients respect to HDs. Interestingly, Fyn protein levels was higher in CLPD patients and in NK cell lines respect to HDs. In these last samples Fyn protein level was not detectable, perhaps for the lower percent of NK cells in HD-PBMCs (< 10%) respect to CLPD patients (20–40%), to equal protein loaded lysate in western blotting assay. The over-expression of Fyn is documented in different cancers, such as CML, brain, prostate and breast tumors where it facilitates the growth, the migration of tumor cells and genomic instability [22–26]. Recently, FYN mutations were found in peripheral T-cell lymphomas where they impaired DNA damage response and escape from immune surveillance [14].
Our previous data showed that 4c pyrazolo[3,4-d]pyrimidine compound is able to inhibit Fyn kinase in a cell-free assay and to induce apoptosis and cell cycle arrest in CML cell line [17].
In this paper, for the first time, we investigated the effect of this compound on NK cell leukemia.
First of all, we demonstrated that 4c reduced cell viability in NK cell lines. Interestingly, it had no effect on both PBMCs and NK purified cells from HDs.
Since it was discovered previously that exposure to stimulatory factors such as the cytokine IL2 enhanced NK cell potency significantly [27], we cultured primary HD-NK cells with and without IL2 in order to evaluate if 4c compound could have diverse effect on NK cells differently activated. We observed that 4c compound had no effect on HD-NK cell viability in both culture conditions. So the presence of IL2 not influenced response to 4c treatment.
These results allowed us to conclude that 4c compound acted only on NK leukemic cells and not on healthy NK cells. Moreover, it not influenced viability of other PBMC population.
We obtained different percent of viability reduction in cell lines after 4c treatment. More precisely, the major effect was observed in KHYG1 respect to NK92. The diverse response to 4c compound of the two cell lines could be due to their different origin. In particular, KHYG1 was established from peripheral blood of a woman with aggressive NK leukemia at diagnosis, while NK92 was established from a man with non-Hodgkin’s lymphoma with large granular lymphocytes, more corresponding to chronic subtype.
We decided to study in deep the viability reduction in NK aggressive KHYG1 cells and demonstrated that 4c induced a G2/M cell cycle arrest and a significant apoptosis.
Recent studies show the possibility to conduct NK malignant cells to apoptosis thus supporting a possible therapeutic option. Quoc Trung L et al. showed that resveratrol induced G0/G1 cell cycle arrest and apoptosis in NK leukemic cell lines by inhibiting STAT3 signaling [28]; similar results were obtained by Sun X et al. using a combination of drugs, vorinostat and cladribine [29].
To demonstrate that effects on cell viability were due to Fyn inhibition, we confirmed its reduced phosphorylation. These findings are in agreement with Ninio-Many L et al. who revealed that miR-125a-3p causes an arrest of cell cycle at G2/M stage and a decreases cell viability in a Fyn-dependent manner [30]. Other studies, in glioma and prostate cells, revealed similar results after Fyn down modulation [23, 31]. All this confirms the fact that Fyn is considered an important regulator of cell cycle, growth and proliferation [32–34].
To the best our knowledge, this is the first study concerning the Fyn targeting in NK malignancies.
Moreover, we investigated Fyn downstream pathway. Since some studies have shown that Src kinases, including Fyn, may be required for Akt activation by a tyrosine phosphorylation-dependent mechanism [35, 36], we also investigated the Akt behavior after 4c treatment and we observed that Fyn inhibition reduces Akt phosphorylation. Additionally, our data showed a reduction of P70 S6 kinase phosphorylation, an important downstream target of Akt involved in cell survival and in apoptosis inhibition [37, 38].
Furthermore, GEP analysis of 4c treated KHYG1 cells confirmed the down-regulation of pro-survival gene pathways and the up-regulation of apoptotic functions. In particular, we validated by qRT-PCR the reduction of Survivin, and the increase of CFLAR and DFFA mRNA. Survivin, a member of the inhibitor of apoptosis protein (IAP) family, regulates leukemic NK cell survival via ERK/MAPK signaling [39]. Interestingly, Liu et al. demonstrated that targeting of survivin may be a possible therapeutic approach for NK leukemia because it is highly expressed in both aggressive and chronic leukemic NK cells respect to normal ones [40].
The first up-regulated gene, CFLAR, also known as Casper/cFLIP, is a FADD- and Caspase-Related inducer of apoptosis and its over-expression induces apoptotic cell death [41–43]. It also play a critical role in autophagy, necroptosis and apoptosis in T-lymphocytes [44] and it has prognostic and therapeutic relevance in acute myeloid leukemia (AML) [45] as well as in development of drug resistance [46]. DFFA gene is the substrate for caspase 3 and triggers DNA fragmentation during apoptosis [47, 48].
In addition, the protein array confirmed the down modulation of survivin, of other members of IAP family, such as cIAP-1, XIAP, livin [49, 50] and of cell cycle regulators such as Cluspin [51], and the high modulation of pro-apoptotic ones, such as cleaved caspase 3.
Interestingly, dot blot assay showed a decrease of Cytochrome C and Smac/DIABLO and an increase of Fas levels suggesting the activation of extrinsic apoptotic pathway [52]. We also confirmed, by western blotting (WB), the presence of cleaved caspase 3 after treatment letting us to assert that 4c Fyn inhibitor compound is able to induce apoptotic death caspase 3-mediated in NK malignant cells.
Finally, we demonstrated that 4c pyrazolo[3,4-d]pyrimidine compound reduced viability and induced apoptosis in PBMCs and in NK cells from patients with NK leukemia.
In human PBMCs the NK cells can be divided on the basis of the expression of CD56 and CD16 markers. CD56+/CD16+ and CD56+/CD16− populations show different proliferation, redox state and function. Specifically, CD56+/CD16− cells are responsible for immunoregulatory cytokines production, CD56+/CD16+ cells are responsible for the cytotoxic lysis of target cells [53–54]. On the basis of these considerations, we analyzed the 4c effects on both NK purified population. We demonstrated that there was a reduction of viability after treatment but also a reduction of proliferation and an increase of apoptosis caspase dependent in both population CD56+/CD16+ and CD56+/CD16−. From cell cycle analysis, 4c compound resulted to be cytostatic, in fact there was an increase of G0/G1 phase.
In order to study the 4c effect on NK cell phenotype, we evaluated two important surface molecule involved in NK cells activation, such as CD38 and CD25 [55]. We demonstrated that 4c treatment reduced the expression of CD38, while CD25 expression remained constant. It means that NK treated cells could be slightly active.In summary, the present study demonstrated that Fyn kinase has a role in the pathogenesis of NK leukemia. Moreover, inhibition of Fyn phosphorylation by 4c pyrazolo[3,4-d]pyrimidine compound induces apoptosis and cell cycle arrest in NK leukemic cells.
Fyn targeting by 4c compound could represent a potential and possible strategy for NK leukemia treatment.
MATERIALS AND METHODS
Patients, cell lines and chemical
Peripheral blood samples from HDs and NK-CLPD (patient characteristics are shown in Table 3) were obtained with informed consent. PBMCs were isolated by Ficoll-hypaque gradient separation and NK cells were isolated by cell sorter MoFlo Atrios (Beckman Coulter, Brea, California, USA) by labeling with anti-CD56 phycoerythrin (PE) (Becton Dickinson, BD, Franklin, NJ, USA). Human NK cell lines, KHYG1 and NK92 [56, 57], were purchased from Leibniz-Institut DSMZ – Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH and American Type Culture Collection, respectively. NK primary cells and cell lines were cultured in RPMI 1640 (Gibco, Life technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 1% of penicillin-streptomycin (Gibco) and 10 ng/ml interleukin-2 (IL-2, Miltenyi Biotec, Auburn, CA) at 37°C and 5% CO2.
Table 3: Clinical characteristics of NK-CLPD patients
UPN |
Gender |
Age |
NK cells |
|---|---|---|---|
1 |
F |
65 |
> 60% |
2 |
M |
63 |
> 60% |
3 |
M |
49 |
40–60% |
4 |
M |
49 |
40–60% |
5 |
M |
45 |
40–60% |
6 |
F |
77 |
30–40% |
7 |
M |
45 |
30–40% |
8 |
M |
51 |
20–30% |
Abbreviations: UPN, unique patient number; F, female; M, male.
4c pyrazolo[3,4-d]pyrimidine compound, given by Lead Discovery Siena s.r.l. (patent: WO2016066755), was dissolved in DMSO (Sigma Aldrich, St Louis, MO, USA) and diluted in FBS for cell treatments.
RNA extraction and qRT-PCR
Total RNA was extracted with AllPrep DNA/RNA/miRNA Universal Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. Reverse transcription was performed using Transcriptor First Strand cDNA Synthesis kit (Roche, Indianapolis, IN, USA).
mRNA expression was evaluated by qRT-PCR, performed on Light Cycler 480 II (Roche) with intercalating dye SYBR Green I Master Mix (Roche) using 100 ng cDNA at the following conditions: 95°C for 10 min, 45 cycles at 95°C for 10 sec, 60°C for 10 sec, 72°C for 15 sec. Each sample was analyzed in triplicate. Relative mRNA expression values were normalized using GAPDH as reference gene and Universal Human Reference RNA (Qiagen) as calibrator and calculated on the basis of the E-ΔΔCp method.
The primer sequences of human Fyn, GAPDH, Survivin, CFLAR and DFFA were as follows: Fyn forward 5′-AGATTGCTGACTTCGGATTG-3′, Fyn reverse 5′-CAGACTTGATTGTGAACCTC-3′, GAPDH forward 5′-AGGCTGAGAACGGGAAGC-3′, GAPDH reverse 5′-CCATGGTGGTGAAGACGC-3′, Survivin forward 5′-AGAACTGGCCCTTCTTGGAGG-3’, Survivin reverse 5′-CTTTTTATGTTCCTCTATGGGGTC-3′, CFLAR forward 5′-TGGTAAGGCATGCTTCCAGA-3′, CFLAR reverse 5′-ACAGTATCAGAAGGTGGGGC-3′, DFFA forward 5′-CACTCCAACAGGTGCTTGAC-3′, DFFA reverse 5′-AGTGCAGTAAGGATGTGGCT-3′.
Gene expression profile analysis
Total RNA was quantified with a NanoDrop 2000c spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and its quality was assessed by capillary electrophoresis on an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc, Santa Clara, CA) using RNA 6000 Nano Assay Kit (Agilent). Only samples with RNA integrity number (RIN) > 7 were used.
Samples preparation, hybridization, staining and scanning of Illumina HumanHT12 v4.0 Expression BeadChip array (Illumina Inc., San Diego, CA, USA) on HiScanSQ system (Illumina Inc.) was performed as described before [58].
The intensity files were loaded into the Illumina Genome Studio software for quality control and gene expression analysis. Quantile normalization algorithm was applied on the data set to correct systematic errors, values below a detection score of 0.05 were filtered out and missing values were imputed. Differently expressed genes (DEGs) were selected with differential score (DiffScore) cutoff set at ± 13 (p < 0.05). Microarray data were submitted to Array Express under accession number E-MTAB-4536. DEGs list was used to evaluate the functional behavior in terms of Biological Processes performing an enrichment analysis with IPA (Ingenuity Systems; Mountain View, CA, USA). DEGs list was uploaded also into DAVID bioinformatic tool [59] and functional annotation clustering was evaluated separately for up- and down-regulated genes.
Western blotting analysis
Cells were lysed as previously reported [60]. 80 μg was subjected to sodium dodecyl sulfate polyacylamide gel electrophoresis on a 10% gel under reducing conditions and then electrotransferred onto a polyvinylidene difluoride membranes using Trans Blot Turbo Transfer System (BioRad, Hercules, CA, USA). Membranes were probed with primary antibodies directed against Fyn, phospho-Src Family (Tyr416), phospsho-AKT (Ser473), Akt, phospho-P70 S6 kinase (Thr389), P70 S6 kinase, β actin (Cell Signaling, Beverly, MA, USA), active caspase 3 (Abcam, Cambridge, UK) and then incubated with secondary antibody (horseradish peroxidise-conjugated goat anti-mouse or anti-rabbit; Cell Signaling). Immune complexes were detected by ECL chemiluminescence system (Bio-Rad Laboratories), as recommended by the manufacturer. Densiometric analysis was performed using BioRad Image Lab 4.1 software. The intensity of bands of all proteins was normalized to the β actin signal.
Immunoprecipitation
Protein immunoprecipitation was carried out starting from 1 mg of total protein extracts. Lysates were pre-cleared by incubating with protein A/G-Agarose (Santa Cruz Biotechnology) for 1 hour, subsequently incubated with anti-Fyn antibody for 18 hours and then with fresh A/G-Agarose for 2 hours. All incubation were conducted at 4°C. The product was collected by centrifugation and washed twice. WB analysis was performed as previously reported.
Proteome profiler array
Protein extract of KHYG1 cell line, treated with 4c compound at 4 μM or with DMSO vehicle control for 24 hours, were subjected to the human apoptosis array following the manufacturer’s instructions (Human Apoptosis Array kit, R&D Systems, Abingdon, UK).
Cell viability
KHYG1 and NK92 cell lines, PBMCs and NK cells from HDs were seeded into 96-well plates (3 × 104 cells/100 μl) and incubated with 4c compound at increasing concentrations (2–10 μM) for 24, 48 and 72 hours. Cells treated with DMSO vehicle were used as control. Cell viability was determined using the CellTiter 96 Aqueous One Solution assay kit (MTS, Promega, Madison, WI, USA). The optical density was measured at 492 nm. Cellular viability was calculated as percentage of viable cells compared with control. All experiments were conducted in triplicate. EC50 values were obtain by GraphPad Prism (GraphPad Prism, San Diego, CA). Viability of cell lines and primary NK cells from NK-CLPD was assessed by Trypan blue dye count method.
Functional tests
KHYG1 and NK cells from NK-CLPD samples were treated with 4 μM of 4c compound or with DMSO vehicle control for 24 hours (cell density 3 × 105 cells/ml) and used in:
Apoptosis assay
Apoptosis of KHYG1 was evaluated by cytometric analysis of Annexin V and Propidium Iodide (PI)-stained cells using fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection kit I (BD). NK cells isolated from NK-CLPD samples were firstly labeled with anti CD56 PE and CD16 allophycocyanin (APC), than labeled with Annexin V and 7-Amino-Actinomycin D (7ADD, BD) to evaluate apoptosis. Stained cells were acquired using FACSCalibur flow cytometer and analyzed by CellQuest Pro software (BD). Single positive for Annexin V and double positive for Annexin V and PI/7ADD cells were interpreted as signs of early and late phases of apoptosis respectively.
Cell cycle analysis
After treatment KHYG1 cells were fixed in cold ethanol 70% for 1 hour, then labeled with PI/RNase staining solution for 30 minutes. Instead, after treatment, NK cells from NK-CLPD samples were labeled with anti-CD56 PE and anti-CD16 APC, fixed and permeabilzed by Intracell kit (Immunostep, S.L. Avda), then labeled with 7ADD/RNase staining solution for 30 minutes. Samples were acquired by FACSCalibur (BD). Data were analyzed by ModFit LT Software (Verity Software House).
Caspase 3/7 activity assay
NK primary cells, treated with 4c compound or with DMSO vehicle for 24 hours (cell density 1 × 106/ml), were labeled with anti-CD56 PE and anti-CD16 APC, then incubated with 1 μM of CellEvent caspase3/7 Green Detection Reagent (Life technologies, Carlsbad, CA, USA) in complete medium at 37°C for 30 minutes as manufacturer’s protocol. Stained cells were analyzed by FACSCalibur cytometer (BD).
Proliferation assay
NK primary cells were stained with PKH67 (PKH67 Green Fluorescent Cell Linker Kit for General Cell Membrane Labeling, Sigma Aldrich) as manufacturer’s protocol, treated with 4c compound or with DMSO vehicle control for 24 hours and analyzed by FACSCalibur cytometer (BD).
Analysis of NK phenotype
NK primary cells, treated with 4c compound or with DMSO vehicle control for 24 hours, were labeled with anti-CD25 FITC, anti CD38-APC (BD) and analyzed by FACSCalibur cytometer (BD).
Statistical analysis
Statistical significance was determined using a paired or unpaired t test by GraphPad Prism. All error bars represent the standard deviation (SD) of the mean.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by Italian Ministry of Health, Current Research Founds for IRCCS, CUP E66J12000230001. The authors acknowledge Lead Discovery Siena s.r.l. that holds intellectual property of patent [WO2016066755].
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
REFERENCES
1. Liang X, Graham DK. Natural killer cell neoplasms. Cancer. 2008; 112:1425–36.
2. Shi M, Savage NM, Salman H, Morice WG. A case of lymphoproliferative disorder of NK-cells: aggressive immunophenotype but indolent behavior. Clin Case Rep. 2015; 3:740–3.
3. Zambello R, Teramo A, Gattazzo C, Semenzato G. Are T-LGL Leukemia and NK-Chronic Lymphoproliferative Disorder really two distinct diseases? Transl Med UniSa. 2014; 8:4–11.
4. Zhang Q, Jing W, Ouyang J, Zeng H, George SK, Liu Z. Six cases of aggressive natural killer-cell leukemia in a Chinese population. Int J Clin Exp Pathol. 2014; 7:3423–31.
5. Poullot E, Zambello R, Leblanc F, Bareau B, De March E, Roussel M, Boulland ML, Houot R, Renault A, Fest T, Semenzato G, Loughran T, Lamy T, et al. Chronic natural killer lymphoproliferative disorders: characteristics of an international cohort of 70 patients. Ann Oncol. 2014; 25:2030–5.
6. Elias D, Ditzel HJ. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharm Res. 2015; 100:250–4.
7. Kinoshita T, Matsubara M, Ishiguro H, Okita K, Tada T. Structure of human Fyn kinase domain complexed with staurosporine. Biochem Biophys Res Commun. 2006; 346:840–4.
8. Tintori C, Laurenzana I, La Rocca F, Falchi F, Carraro F, Ruiz A, Esté JA, Kissova M, Crespan E, Maga G, Biava M, Brullo C, Schenone S, et al. Identification of Hck inhibitors as hits for the development of antileukemia and anti-HIV agents. ChemMedChem. 2013; 8:1353–60.
9. Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004; 23:7990–8000.
10. Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009; 228:9–22.
11. Saito YD, Jensen AR, Salgia R, Posadas EM. Fyn: a novel molecular target in cancer. Cancer. 2010; 116 :1629–37.
12. Singh MM, Howard A, Irwin ME, Gao Y, Lu X, Multani A, Chandra J. Expression and activity of Fyn mediate proliferation and blastic features of chronic myelogenous leukemia. PLoS One. 2012; 7:e51611.
13. Fenouille N, Puissant A, Dufies M, Robert G, Jacquel A, Ohanna M, Deckert M, Pasquet JM, Mahon FX, Cassuto JP, Raynaud S, Tartare-Deckert S, Auberger P. Persistent activation of the Fyn/ERK kinase signaling axis mediates imatinib resistance in chronic myelogenous leukemia cells through upregulation of intracellular SPARC. Cancer Res. 2010; 70 :9659–70.
14. Palomero T, Couronné L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, Jiang X, Lossos IS, et al. Recurrent mutations in epigenetic regulators, RHOA, FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014; 46:166–70.
15. Chougule RA, Kazi JU, Rönnstrand L. FYN expression potentiates FLT3-ITD induced STAT5 signaling in acute myeloid leukemia. Oncotarget. 2016; 7:9964–74. doi: 10.18632/oncotarget.7128.
16. http://biogps.org/#goto=generepo&id=2534
17. Tintori C, La Sala G, Vignaroli G, Botta L, Fallacara AL, Falchi F, Radi M, Zamperini C, Dreassi E, Dello Iacono L, Orioli D, Biamonti G, Garbelli M, et al. Studies on the ATP Binding Site of Fyn Kinase for the Identification of New Inhibitors and Their Evaluation as Potential Agents against Tauopathies and Tumors. J Med Chem. 2015; 58:4590–609.
18. Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M, Gomez-Segui I, Guinta K, Durkin L, Hsi ED, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012; 120:3048–57.
19. Lowin-Kropf B, Kunz B, Schneider P, Held W. A role for the src family kinase Fyn in NK cell activation and the formation of the repertoire of Ly49 receptors. Eur J Immunol. 2002; 32:773–82.
20. Gerbec ZJ, Thakar MS, Malarkannan S. The Fyn–ADAP axis: cytotoxicity versus cytokine production in killer cells. Front Immunol. 2015; 6:472.
21. Gadue P, Yin L, Jain S, Stein PL. Restoration of NK T cell development in fyn-mutant mice by a TCR reveals a requirement for Fyn during early NK T cell ontogeny. J Immunol. 2004; 172:6093–100.
22. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR–ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001; 344:1031–7.
23. Jensen AR, David SY, Liao C, Dai J, Keller ET, Al-Ahmadie H, Dakin-Haché K, Usatyuk P, Sievert MF, Paner GP, Yala S, Cervantes GM, Natarajan V, et al. Fyn is downstream of the HGF/MET signaling axis and affects cellular shape andtropism in PC3 cells. Clin Cancer Res. 2011; 17:3112–22.
24. Elias D, Vever H, Laenkholm AV, Gjerstorff MF, Yde CW, Lykkesfeldt AE, Ditzel HJ. Gene expression profiling identifies FYN as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene. 2015; 34: 1919–2727.
25. Tabariès S, Annis MG, Hsu BE, Tam CE, Savage P, Park M, Siegel PM. Lynmodulates Claudin-2 expression and is a therapeutic target for breast cancer liver metastasis. Oncotarget. 2015; 6:9476–87. doi: 10.18632/oncotarget.3269.
26. Zhang Q, Qi CB, Chan W, Zhou J, Chen HR, Luo HR, Appin C, Brat DJ, Ye K. Fyn-phosphorylated PIKE-A binds and inhibits AMPK signaling, blocking itstumor suppressive activity. Cell Death Differ. 2015; 23:52–63.
27. Becker PSA, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, Tonn T, Seidl C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016; 65:477–484.
28. Quoc Trung L, Espinoza JL, Takami A, Nakao S. Resveratrol induces cell cycle arrest and apoptosis in malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS One. 2013; 8:e55183.
29. Sun X, Hasanali ZS, Chen A, Zhang D, Liu X, Wang HG, Feith DJ, Loughran TPJr, Xu K. Suberoylanilide hydroxamic acid (SAHA) and cladribine synergistically induce apoptosis in NK-LGL leukaemia. Br J Haematol. 2015; 168:371–83.
30. Ninio-Many L, Grossman H, Shomron N, Chuderland D, Shalgi R. microRNA-125a-3p reduces cell proliferation and migration by targeting Fyn. J Cell Sci. 2013; 126:2867–76.
31. Lewis-Tuffin LJ, Feathers R, Hari P, Durand N, Li Z, Rodriguez FJ, Bakken K, Carlson B, Schroeder M, Sarkaria JN, Anastasiadis PZ. Src family kinases differentially influence glioma growth and motility. Mol Oncol. 2015; 9:1783–98.
32. Ban K, Gao Y, Amin HM, Howard A, Miller C, Lin Q, Leng X, Munsell M, Bar-Eli M, Arlinghaus RB, Chandra J. BCR-ABL1 mediates up-regulation of Fyn in chronic myelogenous leukemia. Blood. 2008; 111:2904–8.
33. Cowden Dahl KD, Dahl R, Kruichak JN, Hudson LG. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia. 2009; 11:1208–1215.
34. Okamoto M, Nakayama Y, Kakihana A, Yuki R, Yamaguchi N. Fyn Accelerates M Phase Progression by Promoting the Assembly of Mitotic Spindle Microtubules. J Cell Biochem. 2016; 117:894–903.
35. Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001; 276:31858–62.
36. Tang X, Feng Y, Ye K. Src-family tyrosine kinase fyn phosphorylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ. 2007; 14:368–77.
37. Kawauchi K, Ihjima K, Yamada O. IL-2 increases human telomerase reverse transcriptase activity transcriptionally and post translationally through phosphatidylinositol 3′-kinase/Akt, heat shock protein 90, and mammalian target of rapamycin in transformed NK cells. J Immunol. 2005; 174:5261–9.
38. Kawada J, Ito Y, Iwata S, Suzuki M, Kawano Y, Kanazawa T, Siddiquey MN, Kimura H. mTOR inhibitors induce cell-cycle arrest and inhibit tumor growth in Epstein-Barr virus-associated T and natural killer cell lymphoma cells. Clin Cancer Res. 2014; 20:5412–22.
39. Sun L, Zhao Y, Shi H, Ma C, Wei L. LMP-1 induces surviving expression to inhibit cell apoptosis through the NF-κB, PI3K/Akt signaling pathways in nasal NK/T-cell lymphoma. Oncol Rep. 2015; 33:2253–60.
40. Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, Tan SF, Kaiser J, Shanmugavelandy SS, Rogers A, Loughran K, Petersen B, Yuen J, et al. Targeting of surviving by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood. 2010; 116:4192–201.
41. Shu HB, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997; 6:751–63.
42. Inohara N, Koseki T, Hu Y, Chen S, Núñez G. CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc Natl Acad Sci USA. 1997; 94:10717–22.
43. Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem. 2000; 275:10838–44.
44. He MX, He YW. CFLAR/c-FLIPL: a star in the autophagy, apoptosis and necroptosis alliance. Autophagy. 2013; 9:791–3.
45. McLornan D, Hay J, McLaughlin K, Holohan C, Burnett AK, Hills RK, Johnston PG, Mills KI, McMullin MF, Longley DB, Gilkes A. Prognostic and therapeutic relevance of c-FLIP in acute myeloid leukaemia. Br J Haematol. 2013; 160:188–98.
46. Lee S, Yoon CY, Byun SS, Lee E, Lee SE. The role of c-FLIP in cisplatin resistance of human bladder cancer cells. J Urol. 2013; 189:2327–34.
47. Wolf BB, Schuler M, Echeverri F, Green DR. Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation Factor-45/inhibitor of caspase-activated DNase inactivation. J Biol Chem. 1999; 274:30651–6.
48. Ninios YP, Sekeri-Pataryas KE, Sourlingas TG. Differential sensitivity of human leukemic cell lines to the histone deacetylase inhibitor, trichostatin A. Leuk Res. 2010; 34:786–92.
49. Lau R, Pratt MA. The Opposing Roles of Cellular Inhibitor of Apoptosis Proteins in Cancer. ISRN Oncol. 2012; 2012: 928120.
50. Gu J, Ren L, Wang X, Qu C, Zhang Y. Expression of livin, survivin and caspase-3 in prostatic cancer and their clinical significance. Int J Clin Exp Pathol. 2015; 8:14034–9.
51. Oakes V, Wang W, Harrington B, Lee WJ, Beamish H, Chia KM, Pinder A, Goto H, Inagaki M, Pavey S, Gabrielli B. Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle. 2014; 13:3302–11.
52. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000; 1:489 – 495.
53. Huth TK, Staines D, Marshall-Gradisnik S. ERK1/2, MEK1/2 and p38 downstream signalling molecules impaired in CD56dimCD16+ and CD56brightCD16dim/− natural killer cells in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. J Transl Med. 2016; 14: 97.
54. Harlin H , Hanson M, Johansson CC, Sakurai D, Poschke I, Norell H, Malmberg KJ, Kiessling R. The CD16−CD56bright NK Cell Subset Is Resistant to Reactive Oxygen Species Produced by Activated Granulocytes and Has Higher Antioxidative Capacity Than the CD16+CD56dim Subset. J Immunol. 2007; 179:4513–9.
55. Mallone R, Funaro A, Zubiaur M, Baj G, Ausiello CM, Tacchetti C, Sancho J, Grossi C, Malavasi F. Signaling through CD38 induces NK cell activation. Int Immunol. 2001; 13:397–409.
56. Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, Konaka Y, Takatsuki K. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000; 14 :922–30.
57. Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S, Keating A. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol. 2005; 33:1160–71.
58. De Luca L, Trino S, Laurenzana I, Simeon V, Calice G, Raimondo S, Podestà M, Santodirocco M, Di Mauro L, La Rocca F, Caivano A, Morano A, Frassoni F, et al. MiRNAs and piRNAs from bone marrow mesenchymal stem cell extracellular vesicles induce cell survival and inhibit cell differentiation of cord blood hematopoietic stem cells: a new insight in transplantation. Oncotarget. 2016; 7:6676–92. doi: 10.18632/oncotarget.6791.
59. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.
60. Trino S, Iacobucci I, Erriquez D, Laurenzana I, De Luca L, Ferrari A, Rorà AG, Papayannidis C, Derenzini E, Simonetti G, Lonetti A, Venturi C, Cattina F, et al. Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: a new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget. 2016; 7:12951–61. doi: 10.18632/oncotarget.7339.