
Introduction
Recurrent chromosomal translocations and resultant gene fusions have long been recognized as critical events in the oncogenesis of hematological malignancies and soft-tissue neoplasms. The important role of recurrent gene fusions in epithelial malignancies started to emerge recently with the advent of new genome-wide profiling technologies. It is now recognized that actionable gene fusions are prevalent among carcinomas with over 9,100 documented translocations in solid tumors and over 176 of these described as recurrent events [1]. Although PLAG1 rearrangements characterizing pleomorphic adenoma have been known for almost two decades [2], the role of recurrent chromosomal aberrations in other types of salivary gland tumors had not been understood until now.
Recent advances such as the identification of MECT1-MAML2 fusion in mucoepidermoid carcinoma (MEC), the recognition of a new disease entity (mammary analogue secretory carcinoma [MASC]) characterized by the ETV6-NTRK3 fusion gene and the discovery of the MYB-NFIB oncogene in adenoid cystic carcinoma (ACC), have begun to refine our knowledge of salivary gland carcinogenesis [3]. ACC, one of the most common salivary gland malignancies, represents a significant challenge in head and neck oncology due to its aggressive and unpredictable phenotype. Given the high rate of late local recurrence and distant metastasis, ACC patients require intensive oncological surveillance. Unfortunately, with no effective systemic therapy available, the long-term disease control is poor and overall disease-associated mortality remains high. The discovery of the translocation between chromosome 6q and 9p and the identification of the resultant MYB-NFIB fusion in 2009, led to an important insight into the molecular pathogenesis of this malignancy and highlighted the tumor driving role of the MYB (myeloblastosis) proto-oncogene [4]. Recently, the identification of chromosomal rearrangements that juxtapose super-enhancers to the MYB locus and create a positive feedback elicited by activation of these enhancers by MYB protein, has further enhanced our understanding of the biology of tumors that do not harbor chimeric MYB transcripts [5]. Finally, the identification of a fusion between MYBL1 and NFIB genes in tumors without MYB aberration [6, 7], demonstrates that the pathogenesis of ACC may be driven by genetic alterations in another member of the same transcription factor (TF) gene family. Although the MYB and MYBL1 fusion oncoproteins emerge as attractive diagnostic markers and therapeutic targets to improve clinical management of this lethal disease, the manner by which these and other genetic alterations initiate and drive ACC progression is not yet fully understood, and their impact on clinical outcomes remains to be delineated.
In this review, we summarize the current status of the genomic translocations in ACC, discuss challenges associated with underpinning their role in ACC pathogenesis and focus on possible clinical implications stemming from the current research.
Adenoid cystic carcinoma - enigmatic and challenging malignancy
ACC was first recognized as a distinct head and neck neoplasm over 150 years ago by Robin, Lorain and Laboulbene, who provided its microscopic description [8]. With an incidence of 4.5 cases per million individuals, it is the most common malignant tumor of minor salivary glands and the second most prevalent cancer of parotid and sublingual salivary glands [9, 10]. ACC arises sporadically in other exocrine glands located in breast, lacrimal glands, nasal passages, tracheobronchial tree, prostate, cervix and vulva [9, 11-16]. Interestingly, irrespective of the site of origin, these tumors display similar histological characteristics and share nonrandom cytogenetic anomalies such as copy number alterations involving chromosomes 12q, 6q, 8q, 9p, 1p and 22q [17-19]. Histologically, these neoplasms are composed of two types of cells, inner epithelial/luminal and outer myoepithelial cells, recapitulating the structure of intercalated ducts of secretory glands from which ACC are thought to originate. ACC can be classified into three subtypes: tubular and cribiform variants, which are characterized by the presence of both epithelial and myoepithelial components and display an indolent growth pattern, and the solid phenotype, associated with the loss of myoepithelial cells and more aggressive biology[20].
The clinical behavior of head and neck ACC has been described as a “paradox” [8]. While the primary tumor often manifests itself as a small and inconspicuous nodule with low growth kinetics, the disease displays a relentlessly progressive course. Consequently, although a patient’s short term prognosis is favorable with an expected 5-year survival rate of 77%, rates drop significantly after 10 and 15 years with survival estimated at 60% and 45% respectively, and most patients dying as a result of the disease progression in later decades [21]. Additionally, although total resection with a clean surgical margin is usually possible, late local relapses are likely to occur even after a radical resection and adjuvant radiation therapy [11]. The ACC tumors exhibits a high tendency for neurotropic invasion leading to deep and destructive infiltration of craniofacial region, skull base and intracranial cavity [11]. Distant metastases are frequently observed in lung, liver and bones as a result of hematologic spread, while metastasis to lymph nodes are rare. Owing to these highly aggressive characteristics, it is not surprising that salivary gland ACC has long been recognized as “one of the most biologically destructive and unpredictable tumors of the head and neck” [22]. Interestingly, ACC localized in the breast exhibits favorable clinical characteristics with an excellent prognosis, despite a common growth pattern, histology and overlapping chromosomal alterations [23].
Until recently, little was known about the molecular background of the ACC’s pathogenesis. Non-random chromosomal aberrations were observed and reported in clinical material since the 1980s, with special attention given to the most recurrent t(6;9) rearrangements [24-26]. Unfortunately, past efforts to identify the significance of this anomaly have been largely hampered by the lack of validated ACC cell lines [27]. In 2009, Persson et al. used short-lived primary cultures obtained from fresh tumor specimens to demonstrate that the t(6;9)(q22-23;p23-24) translocation results in a fusion between two TF genes, MYB and NFIB [4]. Although this initial study suggested that MYB-NFIB fusion might constitute a hallmark of all ACC tumors, studies that followed have detected that approximately 50% of the ACC patients do not harbor the MYB-NFIB translocation [15, 16, 28-31]. These studies, however, helped to refine the understanding of MYB-NFIB fusion oncogene as a specific and common driver of ACC pathogenesis in multiple anatomical locations, including breast [15, 30], lacrimal glands [16] and skin [32, 33]. Furthermore, overexpression of the 5’ fragment of MYB was observed in 89-97% [17, 34] of all ACC cases, indicating that MYB-NFIB fusion is not the only mechanism of MYB overexpression and suggests that ACC may also arise from other molecular aberrations involving the MYB transcription factor. Indeed, the discovery of recurrent alternate rearrangements that repose super-enhancers in the NFIB and TGFBR3 loci into proximity of MYB gene, uncovers an additional mechanism that may drive MYB overexpression in ACC tumors that do not express the fusion transcript [5]. Another subset of ACC tumors was found to harbor a novel MYBL1-NFIB fusion, an alteration found to be mutually exclusive of the “classical” MYB-NFIB rearrangement [6, 7]. The extensive homology in the DNA binding domain between MYB and MYBL1 and a common change in the gene expression signature induced by these fusions strongly suggest that pathogenesis of virtually all of ACC tumors is uniquely driven by overexpression of members of the MYB TF gene family [6].
The role of MYB transcription factor family in tumorigenesis
MYB, one of the earliest identified proto-oncogenes, was discovered almost 30 years ago as a cellular homologue of the viral oncogene (v-MYB) carried by two different avian leukemia retroviruses, the avian acute leukemia virus (AMV) and the E26 virus [35]. MYB is a founding member of c-MYB TF family, encompassing structurally related MYBL1 (AMYB) and MYBL2 (BMYB) proteins. It plays a key role in the control of cell proliferation, survival, differentiation and angiogenesis [35, 36]. Over 80 genes are known as MYB cellular targets, including pro-proliferative genes MYC, CCNA1, CCNB1, CCNE1, c-KIT, anti-apoptotic BCL-2, HSPA5, HSP70, pro-inflammatory COX-2 and differentiation regulator genes such as GATA3 [35]. MYB has a vital functional role in the establishment of definitive hematopoiesis, inducing both expansion and differentiation of progenitor cells of erythroid and lymphoid lineages. Additionally, MYB drives renewal of colonic epithelium, regulates airway epithelial cells differentiation and is a critical player of adult brain neurogenesis [35, 37].
Ample evidence demonstrates that aberrant MYB expression is a potent driver of neoplasia in animal and human malignancies. The first evidence of its oncogenic potential came from the discovery that viral oncogene v-MYB is capable of inducing myeloblastic transformation in chickens and quails [35]. v-MYB represents a truncated version of the MYB genes and its leukemogenic potential has been linked to deletions and mutations in its C-terminal regulatory domain [38]. In humans, MYB overexpression is detected in most myeloid and acute lymphoid leukemia [35]. Its altered activity in hematological malignancies is often linked to the presence of recurrent chromosomal aberrations, such as amplifications, promoter rearrangements or translocations. For example, a transforming MYB-GATA1 fusion gene has been reported in acute basophilic leukemia [39]. Furthermore, high levels of MYB mRNA and protein expression were detected in various solid tumors, such as colorectal and breast cancers [35]. It has been proposed that MYB overexpression commonly seen in these malignancies, may partially result from the disruption of the transcriptional elongation blockade imposed by the stem-loop poly-T structure formed by genomic motifs located in the first intron of MYB transcript. It was suggested that this stem-loop structure may lead to RNA polymerase II stalling, and subsequently result in transcription attenuation [40]. Consistent with this concept, it was reported that intronic mutations in the poly-T motifs, may reverse the transcriptional arrest and lead to increased MYB transcription in colon cancers [41]. In the case of breast cancer, relief of the elongation blockade has been linked to elevated activity of the estrogen receptor α [35]. Moreover, sporadic MYB amplifications, which have been reported in BRCA1 positive tumors, may further contribute to the overall high levels of MYB expression in patients with breast cancer [42].
MYB TF consists of three functional domains: N-terminal DNA-binding domain (DBD), which recognizes a PyAACG/TG consensus sequence, a centrally located transcription activation domain (TAD); and a negative regulatory domain (NRD), located at protein’s C-terminus [40] (Figure 1). Interaction of the TAD with several co-repressor and co-activator proteins, such as CBP/p300, is essential for induction and regulation of MYB transcriptional activity [40, 43]. The post-translational modifications in NRD, such as phosphorylation, acetylation, ubiquitylation and sumoylation were shown to affect MYB activity [44-47]. Studies demonstrate that NRD loss or disruption of its leucine zipper-like and EVES-motifs enhances MYB activity and subsequently drives neoplastic progression in several solid and hematopoietic malignancies [35, 48-50].
Involvement of MYB in ACC has been associated with a spectrum of complex structural rearrangements, of which, the in-frame fusion with the NFIB gene is the most prominent (Figure 2). Although multiple breakpoints ranging from exon 8 to 3’-UTR of the MYB gene have been reported in the fusion positive tumors (Figure 3), the minimal common fragment of MYB yet retained within the MYB-NFIB chimeric transcripts consists of its first 8 exons. As a result, critical functional domains of MYB, DBD and TAD, are always preserved within the fusion oncoprotein and contribute to its transcriptional activity [4]. In addition to the fusion of MYB and NFIB, other MYB translocations have been incidentally identified in ACC, including fusion of MYB exon 14 with intron 3 of the PDCD1LG2 gene on chromosome 9p24 or MYB exon 12 with intron 22 of the EFR3A gene on chromosome 8q24. However, the role of these sporadic events has not yet been elucidated [29].
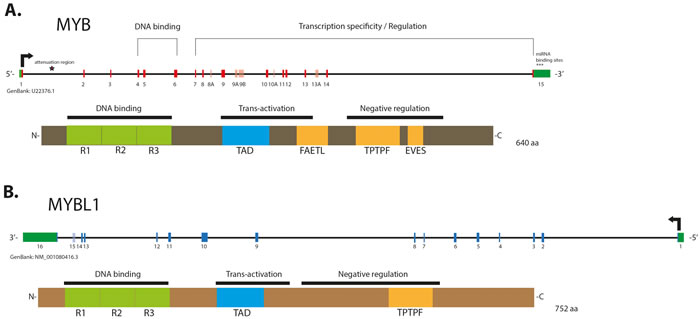
Figure 1: Schematic structure of gene and protein of MYB (A.) and MYBL1 (B). Alternative exons in the genes are shown in a lighter color. The 1st intron of MYB contains the attenuation region, whose polyT motifs may induce formation of energetically stable stem loop which is predicted to block RNA elongation by RNA Polymerase II stalling. MYB contains miRNA binding sites located in its 3’-UTR and involved in repression of its transcriptional activity. MYB and MYBL1 proteins contain evolutionary conserved N-terminal R1, -2, -3 repeats forming the DNA binding domain (DBD), and a centrally located transactivation domain (TAD), both essential for the protein activity. The negative regulatory domains (NRD) are located in the C-terminal elements of the proteins. Labels indicate conserved domains: “FAETL” (which is required for oncogenic activity), “TPTF” motif (conserved in all MYB proteins) and “EVES” domain (involved in the negative regulation).
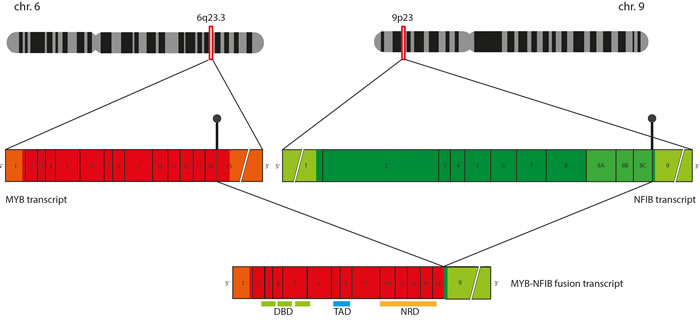
Figure 2: Schematic representation of MYB-NFIB chimeric transcript formation. Translocation between chromosome 6q and 9q results in breakpoints in 3’ termini of MYB and NFIB genes, which often take place in the sequences following exon 14 of MYB and sequences preceding exon 9 of NFIB. When MYB-NFIB “long fusion” is formed, sequences coding the functional MYB domains (DNA binding domain [DBD], Transactivation domain [TAD] and Negative regulatory domain [NRD]) are preserved within the fusion transcript.
MECHANISM OF MYB OVER EXPRESSION IN ACC
MYB-NFIB fusion is a dominating mechanism of 5’ MYB up-regulation in ACC. Persson et al. have postulated that MYB deregulation may be attributed to the miRNA target site loss [4]. The 3’ UTR of MYB, which is lost as a result of t(6;9)(q22-23;p23-24) translocation, contains highly conserved binding sites for certain miRNA molecules, including miR-15a, miR-16 and miR-150. It was demonstrated that transfection with these miRNAs induces a 30% down-regulation of wild-type MYB mRNA level in a T-cell acute lymphoblastic leukemia cell line, whereas this treatment does not decrease levels of the chimeric transcripts in ACC cells [4]. This observation is reminiscent of the mechanism observed in lipoma, where truncation of another oncogene, HMGA2, and its fusion with NFIB results in loss of miRNA target sites, which consequently leads to deregulated HMGA2 expression [51].
Until recently, mechanisms underlying MYB overexpression in cases devoid of MYB structural aberrations remained largely unclear. It has been shown that epigenetic mechanisms such as promoter methylation do not play a role in regulation of MYB expression in ACC [52]. On the other hand, the role of MYB-targeting miRNAs, such as down-regulated in ACC miR-150 [53, 54], has not yet been fully elucidated. The impact of MYB amplification in ACC is probably limited, as copy number gains of MYB has been reported only incidentally [16, 55] and predominantly in cases already harboring “conventional” MYB-NFIB fusion [16]. The most attractive mechanism explaining MYB up-regulation in tumors without MYB-NFIB fusion are other critical translocations bringing regulatory elements into proximity of MYB locus. It has been shown that ACCs may harbor complex rearrangements, either centromeric or telomeric to MYB locus [17]. These structural aberrations may result in translocation of segments of chromosome 9, including sequences of the NFIB gene, placing them from 0.1 Mb to 10 Mb upstream of MYB [6, 17, 29]. This intragenic region between MYB and HBS1L contains multiple long-range enhancer elements, which bring various TFs into the proximity of MYB promoter and its negative regulatory elements, therefore inducing MYB expression [56, 57]. MYB locus is also a common site of retroviral insertion in leukemia, with multiple insertion sites localized upstream and downstream of the gene [58]. Furthermore, it has been observed that translocation of TCRB (T-cell receptor beta) gene to region telomeric of MYB, results in MYB overexpression in childhood T-cell acute lymphoblastic leukemia [58]. The hypothesis suggesting that MYB upregulation in ACC is a result of the regulatory element translocations, has been confirmed by a recent study. It has been reported that rearrangements of enhancers located within the NFIB, TGFBR3 or RAD51B loci and their relocation upstream or downstream of the MYB gene, result in a significant physical interaction of these regulatory element with MYB promoter and subsequently high level of MYB mRNA expression [5]. Furthermore, binding of MYB protein to the translocated enhancers in the NFIB and TGFBR3 loci, creates a positive feedback loop that fuels further expression of MYB. Interestingly, the mechanisms of enhancer-driven overexpression of MYB are not limited to ACC, as an analogous event has been recently described in angiocentric gliomas, which harbor MYB-QKI rearrangements. It has been shown that this structural aberration drives tumorigenesis though three mechanisms: MYB truncation, fusion oncogene overexpression via translocation of enhancer elements and hemizygous loss of the QKI tumor suppressor [59]. Therefore, it is tempting to speculate that this “single event-multiple mechanisms” paradigm may also happen in ACC and that the relocation of NFIB regulatory elements contributes to high expression of the MYB-NFIB or MYBL1-NFIB chimeric transcripts in tumors that harbor the in-frame fusions between these genes.
MYBL1 - a partner in crime
MYBL1 (AMYB), a gene located at chromosome 8q, is another member of the MYB gene family. Although MYBL1 protein and MYB share extensive structural homology in the DNA binding domain (Figure 1), and activate the same reporter genes in vitro, they exhibit distinct biological functions [60, 61]. In a series of deletion and domain swap experiments, Lei et al. have demonstrated that individual functional elements within the TAD, NRD and the C-terminus domains may play a crucial role in the target specificity of different MYB family members, providing a possible explanation for their diverse functionality [62]. MYBL1 has been recently shown to be implicated in the oncogenesis of diffuse astrocytoma, which were discovered to harbor recurrent rearrangements in chromosome 8q, resulting in tandem duplication/truncation of the MYBL1 gene. Consequently, the MYBL1 transcript truncated at exon 9 was shown to have oncogenic properties [63, 64]. The recently discovered MYBL1-NFIB fusion gene, a result of t(8q;9p) translocation, provides another example of MYBL1 neoplastic potential. The structure of the MYBL1-NFIB fusion gene exhibits a striking similarity to the MYB-NFIB fusion, with MYBL1 breakpoints identified in exons 8, 9, 14 and 15, preserving the DNA binding and transactivation domains in all fusion proteins [6] (Figure 3). This fusion is mutually exclusive to the “classical” MYB-NFIB translocation and induces the overexpression of transcriptionally active 5’- MYBL1 fragment [6, 7]. In a subset of ACCs, other MYBL1 rearrangements were also observed, such as fusion with YTHDF3 or MYBL1 3’- truncation, resulting in a similarly abnormal expression profile [6, 7]. Since, in the context of the fusion, both MYB and MYBL1 lose elements responsible for their target specificity, the resulting oncoproteins may induce common expression signatures. Indeed, it was previously demonstrated that all ACC share similar expression profile, regardless of MYB-NFIB fusion status or level of MYB expression [53]. Common transcriptome signatures found in tumors harboring MYB-NFIB and MYBL1-NFIB fusions, further corroborate these findings [6, 7].
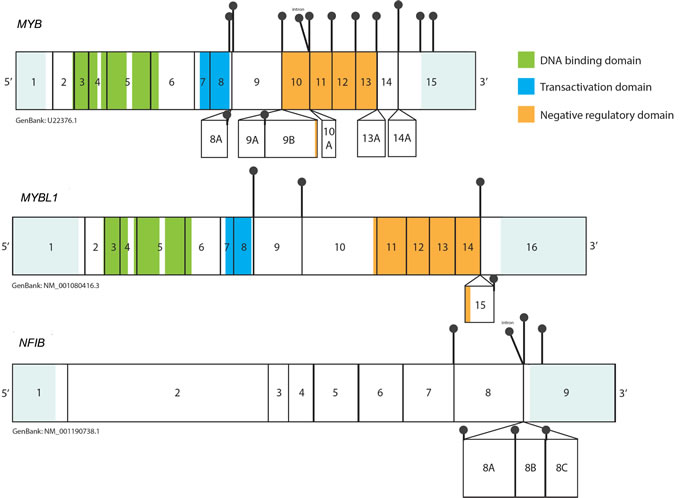
Figure 3: Representation of the breakpoints within the MYB, MYBL1 and NFIB transcripts based on the ‘up-to-date’ literature analysis. Black pins represent the breakpoints observed in the MYB-NFIB and MYBL1-NFIB transcripts. Alternative exons are shown as boxes below the main transcripts. UTRs are shown in light blue (not to scale). The breakpoint in MYB and MYBL1 have been observed in sequences that follow exon 8, thus preserving DBD and TAD within all fusion oncoproteins. The proximal breakpoints (exon 8, exon 9) lead to formation of a “short fusion”, in which sequences encoding for the NRD are lost, while more distal breakpoints preserve the elements of NRD (“long fusion”). In most cases NFIB contributes its terminal exon 9 to the chimeric transcript. In some tumors exons 8A-8C may also be present in MYB-NFIB mRNA as a result of alternative splicing of NFIB fragment. 3’UTR breakpoints in NFIB have also been reported.
Implications of variable STRUCTURE OF MYB AND MYBL1 IN THE FUSION TRANSCRIPTS
Past studies of the transcriptional activity of MYB suggest that alternately spliced RNA forms of MYB may produce proteins with different quantitative and qualitative activities [50]. Indeed, recent observations demonstrate that the structure of MYB fusion transcripts may also implicate varying level of oncogenic potential. Certain MYB-NFIB and MYBL1-NFIB transcripts preserve their respective NRD as a result of distal breakpoint (“long fusion”), whereas other fusion products lose it as a results of a breakpoint located in proximity of exon 8 (“short fusion”). It has been previously reported that the highest expression levels of the fusion mRNA transcripts are commonly observed in cases harboring a breakpoint in exon 8 of MYB [28, 29]. Recent studies suggest that “long” and “short” fusion genes may actually differ in regards to their target specificity and transcriptional activity. Mitani et al. [6] identified that a cohort of ACC harboring MYB or MYBL1 breakpoint after exon 11 exhibits a distinct expression profile, which differs from tumors with fusions at exon 8 or 9. The first group was enriched for expression of 19 gene sets predominantly involved in RNA processing and regulation of translation, while the latter group was enriched for 5 gene sets related to tissue development [6]. Another study demonstrated that transfection with different MYB-NFIB and MYBL1-NFIB fusion genes activates the same synthetic promoters containing MYB binding sites, although the magnitude of activation differs significantly between fusion constructs. The transfection with the wild-type MYB gene or “long fusion” MYB-NFIB constructs containing transformation and negative regulation motifs resulted in 100-fold activation of the 5xMRE-Luc reporter, while overexpression of the “short fusion” MYB-NFIB or 3’-truncated MYB resulted in approximately 200 to 600 fold activation [7]. Although analogous findings were observed in regards to MYBL1 and MYBL1 fusion constructs [7], the clinical implications of these findings are unclear and warrant further investigation.
Role of NFIB gene in ACC pathogenesis
Contrary to MYB, the role of NFIB (Nuclear Factor I B) in normal and cancer cell biology remains obscure. NFIB is a member of the Nuclear Factor I gene family, also known as ‘CAAT box TFs’ (CTF). NFIB binding sites have been identified in the promoter, enhancer and silencer elements of over 100 cellular and viral genes, although the exact function of most of them is poorly understood [65]. Studies suggest that upon dimerization and association with its target, NFIB may modulate transcriptional activation or repression of specific gene promoters in a tissue-specific manner [65]. The diverse cellular functions of NFIB are further corroborated by studies reporting its oncogenic or tumor suppressor roles in different tumor types. For example, NFIB inactivation was shown to contribute to osteosarcoma progression [66] and cutaneous carcinogenesis [67], while other studies demonstrates that NFIB may acts as an oncogene in small cell lung cancer [68]. A study conducted in NFIB-deficient mice model demonstrated that this TF plays a key role in tubule cell differentiation during embryonic development of submandibular glands [69]. Additionally, the loss of NFIB gene leads to fetal lung maturation defects in heterozygous NFIB-deficient mice, indicating possible NFIB haploinsufficiency [70]. Recently, it has been shown that ACC tumors, regardless of the fusion status, overexpress NFIB as compared to normal salivary gland tissue [71]. Although the functional significance of this upregulation is not yet clear, it may be speculated that the elevated NFIB expression is a result of MYB TF interaction with NFIB gene enhancers [5].
Although the NFIB fragment present within the MYB-NFIB fusion content may differ as a result of alternative splicing, the exon 9 (encoding the last 5 amino acids) is present in virtually all chimeric mRNA transcripts (Figure 3) [4]. It has been suggested that due to its small size, the contribution of this coding fragment to the properties of the fusion oncoprotein is likely very limited [4]. However, it is important to note that exactly the same fragment of NFIB is fused with HMGA2 and HMGIC genes in lipoma [72] and pleomorphic adenoma [73], respectively. HMGA2-NFIB fusion strongly resembles the rearrangement between MYB and NFIB, as in both instances the aberration results in highly deregulated expression of DNA-binding domains of the TF linked to the small C-terminal fragment of NFIB. Hence, although the exact role of NFIB as a fusion partner remains to be discovered, it is possible that NFIB contributes stabilizing or regulatory elements to the fusion protein [34]. On the other hand, it has been reported that in some cases, NFIB fragment within the MYB-NFIB mRNA may be limited to its 3’ UTR only (our unpublished observations in ACC samples further support these findings) [29]. Furthermore, NFIB translocations have been reported in FISH t(6;9)-positive/MYB-NFIB-transcript negative ACCs, resulting in fusions between the 5’-part of NFIB and miscellaneous partner genes, such as XRCC4, PTPRD, NKAIN2 or AIG1 [6, 29]. Additionally, our lab has recently reported presence of NFIB fusions with the RIMS1, MAP3K5, RPS6KA2, MYO6 genes, all of which are located on chromosome 6q [71]. It is uncertain whether these fusions produce functional proteins contributing to oncogenesis per se. However, it was noted that cases harboring these alternative NFIB gene rearrangements, have NFIB segments relocated into the proximity of the MYB locus and concurrently overexpressed the intact MYB transcript [6]. As discussed above, it is possible that these rearrangements relocate NFIB-associated super-enhancers, resulting in their physical interaction with MYB promoter and augmented expression of MYB mRNA [5].
Incidence of the structural aberrations in ACC
The incidence of MYB-NFIB fusion varies across studies with reported rates ranging from 23% to 86% (Table 1). Several possible explanations for the disparities across studies may be suggested, e.g. different anatomical location of tumors tested, variable quality/origin of material studied (archival FFPE vs. frozen tumors), or different analytical methods used (RT-PCR, FISH, RNA-sequencing, WGS). For example, when assessed with RT-PCR, a higher incidence of MYB-NFIB fusions was reported in fresh-frozen material than in FFPE samples (86% vs. 44% respectively) [34]. Furthermore, it has been reported that positive FISH status may not always be associated with the chimeric MYB-NFIB transcript formation. In a subset of these ‘nontranscript forming’ tumors, breakpoints at the flanking sites of MYB have been identified (in many instances involving the NFIB segments) [5, 29]. Similarly, NFIB fusions with genes other than MYB that are located on chromosome 6q can also account for this discrepancy [71]. Consequently, our summary, which includes all up-to-date studies investigating the incidence of the MYB-NFIB fusion in ACC, indicates that t(6;9) rearrangement was observed in 57% (127/223) of all ACC tumors analyzed using in situ hybridization techniques, and the chimeric mRNA transcript was detectable in 51.1% (162/317) of the tumors (Table 1).
Table 1: Reported incidence of MYB and MYBL1 rearrangements in ACC categorized by the detection methodology used.
MYB-NFIB fusion |
||||||
Paper |
ACC source |
MYB break apart (FISH) |
t(6;9) |
MYB-NFIB fusion in WGS |
MYB-NFIB Transcript |
Other structural aberrations observed in MYB or NFIB |
Persson et. al 2009 [4] |
Salivary, other Head and Neck, Breast |
|
6/6 |
|
11/11 |
|
Salivary gland, other Head and Neck, Respiratory Tract |
|
54/102 |
|
39/102 (RT-PCR and 3' RACE PCR) |
MYB-PDCDILG2; MYB-EFR3A; NFIB-AIG1; NFIB-XRCC4; NFIB-NKAIN2; NFIB-PTPRD |
|
Brill et. al 2011 [34] |
Salivary, Respiratory Tract, Breast, Vulva |
|
|
|
39/61 |
|
West et. al 2011 [31] |
Salivary gland |
24/37 |
18/37 |
|
|
NFIB translocation into proximity of intact MYB; 5' MYB copy gain without association with NFIB; MYB translocation without involvement of NFIB |
Persson et. al 2012 [17] |
Salivary, Lacrimal gland, other Head and Neck, Respiratory Tract, Breast, Distant mets |
|
|
|
30/35 |
NFIB translocation upstream of MYB; breakpoints distal to MYB |
Ho et. al 2013 [74] |
Salivary, Lacrimal gland, other Head and Neck, Respiratory Tract |
|
34/60 |
|
|
|
Costa et. al 2014 [104] |
Salivary gland |
|
3/5 |
|
|
|
Hudson et. al 2014 [55] |
Salivary gland, Respiratory Tract, Breast, Distant mets. |
4/10 |
|
|
|
MYB trisomy without translocation in 1 case |
Rettig et. al 2015 [82] |
Salivary gland; other Head and Neck |
59/91 |
|
|
|
|
Brayer et. al 2015 [7] |
Salivary gland |
|
|
|
8/20 (RNA-seq, RT-PCR) |
|
Tian et. al. 2015 [105] |
Cribiform salivary ACC |
9/20 |
|
|
|
|
Argyris et. al. 2016 [106] |
Salivary gland |
5/5 |
|
|
|
|
Rettig et. al 2016 [71] |
Salivary gland; other Head and Neck |
|
|
11/25 |
|
RIMS1-NFIB; RPS6A2-NFIB, MAP3K5-NFIB, MYO6-NFIB |
Wetterskog et. al 2012 [30] |
Breast |
12/13 |
12/13 |
|
4/13 (all negative sampes low RNA quality) |
|
D'Alfonso et. al 2014 [107] |
Breast |
7/31 |
|
|
6/29 |
|
Martoletto et. al 2015 [15] |
Breast |
10/12 |
|
|
10/12 |
|
Von Holstein et. al 2013 [16] |
Lacrimal gland |
8/13 |
|
|
7/14 |
MYB copy number gain in 1 case |
Bishop et. al 2015 [108] |
Prostate basal cell carcinoma |
2/12 (2/7 ACC-like hist.) |
|
|
|
|
Fehr et. al 2011 [32] |
Dermal cylindroma |
|
|
|
6/11 |
|
North et. al 2015 [33] |
Primary cutaneous ACC |
6/11 |
|
|
2/9 |
|
Drier et. al. 2016 [5] |
Primary tumors (as in Ho 2013 and Stephens 2013) and Primagrafts: Salivary gland ACC, Respiratory Tract, other Head and Neck, Distant Mets |
|
|
12/20 (6 cases with loss of MYB 3'-UTR, |
|
MYB-TGFBR3; MYB-RAD51B; super-enhancer translocations in NFIB, TGFBR3 and RAD51B loci act as drivers of MYB activation |
TOTAL |
|
146/250 (58.4%) |
127/223 (57.0%) |
23/45 (51.1%) |
162/317 (51.1%) |
|
|
MYBL1-NFIB fusion |
||||||
Paper |
ACC source |
t(8;9) |
MYBL1-NFIB fusion in WGS |
MYBL1-NFIB fusion transcript |
Other structural aberrations in MYBL1 |
|
Brayer et. al 2015 [7] |
Salivary gland |
|
|
2/20 (RNA-seq, RT-PCR) |
MYBL1 truncation - result of MYBL1-RAD51B (1/20) |
|
Mitani et. al 2015 [6] |
Salivary gland, other Head and Neck, Respiratory Tract |
14/102 |
4/21 (samples preselected with known MYB-NFIB status) |
12/102 |
MYBL1-YTHDF3 (2/102) |
|
Drier et. al. 2016 [5] |
Primary tumors (as in Ho 2013 and Stephens 2013) and Primagrafts: Salivary gland ACC, Respiratory Tract, other Head and Neck, Distant Mets |
|
0/20 |
|
||
Rettig et al. 2016 [71] |
Salivary gland; other Head and Neck |
|
2/25 |
2/25 (RNA-seq, RT-PCR) |
||
TOTAL |
|
14/102 (13.7%) |
6/66 (9.1%) |
16/147 (10.9%) |
6/122 (4.9%) |
The second known fusion, MYBL1-NFIB, has been reported independently by three studies to be present in 11% of the ACC cases [6, 7, 71]. Our team has detected a similar incidence of this novel fusion, with 7/56 (12.5%) of ACCs identified as MYBL1-NFIB positive (unpublished data; manuscript in preparation). MYBL1-NFIB fusion was found to characterize 19% of all cases that do not harbor any MYB fusions. Additional structural aberrations involving MYBL1, such as fusion with YTHDF3, or MYBL1 transcript truncations, have been identified in 6% of ACC tumors [6]. Taken together, the MYB and MYBL1 gene rearrangements are observed in approximately two-thirds of all ACC cases.
Another subset of ACCs with no structural aberrations involving MYB or MYBL1 genes, commonly overexpresses the intact MYB mRNA transcript. Collectively, this group contains tumors which harbor t(6;9) that do not result in formation of the MYB-NFIB transcript, and ACCs with no known t(6;9) translocations [6]. In the light of the recent discoveries, it appears that enhancer rearrangements in the vicinity of the MYB locus may account for MYB upregulation in the majority of these cases, however further studies are warranted to unravel alternative events that may also lead to MYB overexpression.
Comprehensive genetic studies identified low rates of somatic mutations in ACC tumors (0.3 somatic mutations per 1 Mb) with wide mutational diversity scattered among genes involved in chromatin remodeling, DNA damage/checkpoint, FGF-IGF-PI3K, Rho family, axonal guidance, Notch and MYB-MYC signaling pathways [15, 71, 74, 75]. Mutations in NOTCH1 and SPEN, a negative NOTCH signaling regulator, were reported to be preferentially found in tumors with no MYB or MYBL1 fusions [6]. Interestingly, another study has shown that MYB signaling cooperates with distinct pathways in eliciting biphenotypical differentiation of ACC cells, with myoepithelial cells enriched for TP63 signaling, abd Notch signal orchestrating expression program in luminal cells [5]. Absence of myopithelial component in solid histology high grade tumors has been found to be associated with activation of Notch signaling by gain-of-function mutation in NOTCH1 or loss-of-function aberrations in SPEN [5]. Therefore, while dependency on Notch signaling has been associated with more aggressive phenotype, it implies that this group of ACC patients may respond to the targeted therapy with NOTCH1 inhibitors, as confirmed by a recent tumor xenograft study [76].
It is also possible that a number of cases which do not demonstrate aberrant expression of MYB or MYBL1 may represent other types of salivary gland tumors with an ACC-like morphology, such as polymorphous low-grade adenocarcinoma (PLGA) or basal cell adenocarcinoma [77]. The problem of histological overlap between different salivary gland tumors may be exemplified by a report which identified an instance of MYB-NFIB fusion in a tumor diagnosed as PLGA [78]. However, it was recently discovered that PLGAs are characterized by a highly recurrent and pathognomonic hotspot mutation in the PRKD1 gene [79], demonstrating that PGLA and ACC are genetically distinct entities. Future comprehensive studies may shed light on whether precise diagnosis and classification of salivary gland tumors based solely on molecular characteristics will be possible.
Impact of MYB and MYBL1 ABERRATIONS ON CLINICAL OUTCOMES
High recurrence of MYB-NFIB fusions and their ACC-specificity in the context of other types of head and neck neoplasms [4, 28] have encouraged various attempts to assess the diagnostic relevance of these genetic aberrations. Hudson et al. utilized FISH to detect MYB structural aberrations in cytological material obtained via fine-needle aspiration biopsy of primary and metastatic ACC tumors. Using this approach, they were able to successfully identify 5 out of 10 ACCs and distinguish them from pleomorphic adenomas (PA), which did not demonstrate MYB abnormalities in any of the studied cases [55]. The clinical utility for the detection of MYB protein overexpression in ACCs has also been evaluated in similar cytological material, with fine-needle aspiration biopsy specimens from ACCs found positive for MYB in most of the studied cases, successfully discriminating them from pleomorphic adenomas [80, 81].
Despite its potential diagnostic value, there is no consensus on the utility of MYB and MYBL1 fusions as prognostic markers. It has been reported that MYB-NFIB fusion status is not significantly associated with overall survival [28, 82], although a trend toward higher likelihood of local recurrence, perineural invasion and decrease in disease-free survival (DFS) has been observed [6, 31, 82]. On the other hand, MYB overexpression regardless of the fusion status has been significantly associated with a poor patient survival [29]. Furthermore, combined MYB and MYBL1 expression was shown to be associated with a higher disease stage and poor clinical outcome [7]. Intriguingly, a difference in the outcomes of patients with MYB and MYBL1 alterations has been recently revealed, with the former group showing a significantly shorter survival [6], although studies conducted in larger cohorts of ACC patients are essential to delineate clinical impact of this finding.
MYB transcription factor as a therapeutic target
Treatment options available to ACC patients remain limited to surgery and/or radiation and efficiency of currently available chemotherapeutic agents is extremely low [83]. Recurrent MYB alterations observed in ACC may therefore present an attractive target for potential therapeutic interventions. Unfortunately, inhibition of TF activity has been historically proven to be challenging. With no kinase activity, ligand binding sites or hydrophobic pockets that may be targeted by small molecule inhibitors (with exception of nuclear hormone receptors), inhibition of TF activity requires disruption of complex protein-DNA or protein-protein interactions [84]. Nonetheless, success in blocking protein-protein interactions, illustrated by the development of a direct NOTCH1 inhibitor [85], are encouraging and suggest that this strategy might be viable. Furthermore, it was shown that direct inhibition of other aberrant transcription factors, such as CBFβ-SMMHC fusion protein in AML, appears to be feasible [86].
The possibility of targeting MYB protein has predominantly been explored in leukemia studies, and several molecular approaches have been proposed (reviewed by Pattabiraman et al. [40]). One of these approaches suggests the use of antisense nucleotides or RNA interference to suppress MYB expression. Although this method proved to be successful in a mouse model of AML [87], use of RNA inference in clinical trials has been associated with substantial toxicity and problems with delivery [40]. Another approach aiming at suppression of the MYB protein expression proposes to target protein complexes that relieve the elongation blockade imposed by the poly-T motifs located in the first intron of MYB (see above). Deregulation of the interaction between these complexes (involving NF-kB and c-Jun) [88, 89] and the intronic region of MYB might constitute an attractive therapeutic strategy [40]. As MYB activity is regulated by an interplay with various partner proteins such as CBP/p300 co-activator, disruption of their physical associations might pose as another promising approach to reduce MYB activity in cancer. Interaction between the MYB transactivating domain and the KIX domain of CPB/p300 has been evaluated in depth by nuclear magnetic resonance studies [90], providing important information which may guide the development of inhibitors that could specifically bind to the protein surfaces. This strategy has been recently successfully utilized for establishment of two small molecule inhibitors of the MYB/p300 interaction, Naphthol AS-E phosphate and triterpenoid Celastrol, which were shown to impose the inhibitory effect on MYB [91, 92]. Another study was utilizing screens of small-molecule libraries and identified a promising specific inhibitor of MYB activity, natural sesquiterpene lactone mexicanin-I [93]. Although subsequent studies revealed even more potent inhibitors of MYB in the sesquiterpene lactone group, the exact mechanism of their action and their clinical utility remain to be delineated [94]. Finally, dependence of MYB expression on the activity of super-enhancers with strong bromodomain protein occupancy, suggests that use of BET bromodomain inhibitors may render potential anti-tumorigenic response in ACC [5]. BET inhibitors were shown to have oncostatic effect on ACC xenografts by disrupting MYB circuitry, as suggested by a modest decrease in MYB level and MYB target gene expression. This effect was however restricted to grade 2 tumors, as solid phenotype tumors (grade 3) exhibited resistance to BET inhibition, possibly reflecting their stronger dependency on NOTCH signaling, which in turn may be potentially circumvented by Notch inhibitors [5].
Targeting the downstream effectors of MYB, such as c-KIT, could pose an alternative approach to reduce MYB activity. c-KIT is a strong oncogene involved in leukemia, gastrointestinal stromal tumors (GIST) and melanoma [95-97], and its inhibition with imatinib has proven to be a highly successful strategy for management of these diseases. Unfortunately, although c-KIT overexpression is observed in 95% of ACCs [98], clinical trials with imatinib or second generation c-KIT inhibitors, such as dasatinib, produced no objective responses in ACC patients [83]. This has been attributed to the lack of c-KIT gain-of-functions mutations (which are vital for c-KIT overexpression in GIST) among the ACC tumors [20, 99]. BCL-2, a key pro-survival molecule and a MYB target, is also overexpressed in a vast majority of ACCs [98]. The development of selective BCL-2 inhibitors [100, 101] may open up a fertile avenue for novel therapeutic opportunities, but their utility in ACC has not yet been evaluated. Although inhibitors of other MYB downstream targets, such as COX-2, are currently available [98], given the broad and complex transcriptional activity of MYB, it is unlikely that targeting a single effector molecule will emerge as a successful strategy.
Conclusions
The identification of recurring t(6;9) and t(8;9) chromosomal translocations resulting in MYB-NFIB and MYBL1-NFIB fusion oncogenes and the unravel of the role of super-enhancer translocations in the oncogene activation, dramatically extend our understanding of the role of the MYB transcription factors in the pathogenesis of ACC. New findings solidify our knowledge on the involvement of the complex structural alterations in head and neck neoplasias and indicate that disruption of regulatory mechanisms may play vital roles in overexpression of these potent oncogenes. Although the exact biological consequences of these events are not yet entirely clear, the technical progress in genomic profiling and new experiment models of ACC, such as a recently developed cell line [102] and patient-derived xenografts [103], may significantly aid in uncovering the impact of these genetic events on ACC pathogenesis. Furthermore, advancements in pharmacogenetics indicate that the MYB protein, previously regarded as an “undruggable” target, may be potently inhibited with novel promising agents. In this context, there is growing hope that intensified efforts will become fruitful with the emergence of new diagnostic and therapeutic avenues that will improve clinical outcomes for ACC patients.
Acknowledgments
This work was funded by the Adenoid Cystic Carcinoma Research Foundation (ACCRF) and the United States National Institutes of Health/National Institute of Dental and Craniofacial Research (NIDCR) P50DE019032 Spore in Head & Neck Cancer.
Conflicts of Interest
The authors declare no potential conflict of interest.
References
1. Mertens F, Johansson B, Fioretos T and Mitelman F. The emerging complexity of gene fusions in cancer. Nature reviews Cancer. 2015; 15(6):371-381.
2. Kas K, Voz ML, Roijer E, Astrom AK, Meyen E, Stenman G and Van de Ven WJ. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nature genetics. 1997; 15(2):170-174.
3. Stenman G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head and neck pathology. 2013; 7 Suppl 1:S12-19.
4. Persson M, Andren Y, Mark J, Horlings HM, Persson F and Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(44):18740-18744.
5. Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJ, Kluk MJ, Carey CD, Rodig SJ, Sholl LM, Afrogheh AH, Faquin WC, Queimado L, Qi J, Wick MJ, El-Naggar AK, Bradner JE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nature genetics. 2016.
6. Mitani Y, Liu B, Rao P, Borra V, Zafereo M, Weber RS, Kies MS, Lozano G, Futreal A, Caulin C and El-Naggar A. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clinical cancer research. 2015.
7. Brayer KJ, Frerich CA, Kang H and Ness SA. Recurrent Fusions in MYB and MYBL1 Define a Common, Transcription Factor-Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer discovery. 2015.
8. Bradley PJ. Adenoid cystic carcinoma of the head and neck: a review. Current opinion in otolaryngology & head and neck surgery. 2004; 12(2):127-132.
9. Moskaluk CA. Adenoid cystic carcinoma: clinical and molecular features. Head and neck pathology. 2013; 7(1):17-22.
10. Lee RJ, Tan AP, Tong EL, Satyadev N and Christensen RE. Epidemiology, Prognostic Factors, and Treatment of Malignant Submandibular Gland Tumors: A Population-Based Cohort Analysis. JAMA Otolaryngol Head Neck Surg. 2015.
11. Dillon PM, Chakraborty S, Moskaluk CA, Joshi PJ and Thomas CY. Adenoid cystic carcinoma: A review of recent advances, molecular targets, and clinical trials. Head & neck. 2016; 38(4):620-627.
12. Di Palma S, Fehr A, Danford M, Smith C and Stenman G. Primary sinonasal adenoid cystic carcinoma presenting with skin metastases--genomic profile and expression of the MYB-NFIB fusion biomarker. Histopathology. 2014; 64(3):453-455.
13. Roden AC, Greipp PT, Knutson DL, Kloft-Nelson SM, Jenkins SM, Marks RS, Aubry MC and Garcia JJ. Histopathologic and Cytogenetic Features of Pulmonary Adenoid Cystic Carcinoma. J Thorac Oncol. 2015; 10(11):1570-1575.
14. Halat SK and MacLennan GT. Adenoid cystic/basal cell carcinoma of the prostate. J Urol. 2008; 179(4):1576.
15. Martelotto LG, De Filippo MR, Ng CK, Natrajan R, Fuhrmann L, Cyrta J, Piscuoglio S, Wen HC, Lim RS, Shen R, Schultheis AM, Wen YH, Edelweiss M, Mariani O, Stenman G, Chan TA, et al. Genomic landscape of adenoid cystic carcinoma of the breast. The Journal of pathology. 2015; 237(2):179-189.
16. von Holstein SL, Fehr A, Persson M, Therkildsen MH, Prause JU, Heegaard S and Stenman G. Adenoid cystic carcinoma of the lacrimal gland: MYB gene activation, genomic imbalances, and clinical characteristics. Ophthalmology. 2013; 120(10):2130-2138.
17. Persson M, Andren Y, Moskaluk CA, Frierson HF, Jr., Cooke SL, Futreal PA, Kling T, Nelander S, Nordkvist A, Persson F and Stenman G. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes, chromosomes & cancer. 2012; 51(8):805-817.
18. Bernheim A, Toujani S, Saulnier P, Robert T, Casiraghi O, Validire P, Temam S, Menard P, Dessen P and Fouret P. High-resolution array comparative genomic hybridization analysis of human bronchial and salivary adenoid cystic carcinoma. Laboratory investigation; a journal of technical methods and pathology. 2008; 88(5):464-473.
19. Rao PH, Roberts D, Zhao YJ, Bell D, Harris CP, Weber RS and El-Naggar AK. Deletion of 1p32-p36 is the most frequent genetic change and poor prognostic marker in adenoid cystic carcinoma of the salivary glands. Clinical cancer research. 2008; 14(16):5181-5187.
20. Bell D and Hanna EY. Head and neck adenoid cystic carcinoma: what is new in biological markers and treatment? Current opinion in otolaryngology & head and neck surgery. 2013; 21(2):124-129.
21. Lloyd S, Yu JB, Wilson LD and Decker RH. Determinants and patterns of survival in adenoid cystic carcinoma of the head and neck, including an analysis of adjuvant radiation therapy. American journal of clinical oncology. 2011; 34(1):76-81.
22. Conley J and Dingman DL. Adenoid cystic carcinoma in the head and neck (cylindroma). Archives of otolaryngology (Chicago, Ill : 1960). 1974; 100(2):81-90.
23. Marchio C, Weigelt B and Reis-Filho JS. Adenoid cystic carcinomas of the breast and salivary glands (or ‘The strange case of Dr Jekyll and Mr Hyde’ of exocrine gland carcinomas). Journal of clinical pathology. 2010; 63(3):220-228.
24. Nordkvist A, Mark J, Gustafsson H, Bang G and Stenman G. Non-random chromosome rearrangements in adenoid cystic carcinoma of the salivary glands. Genes, chromosomes & cancer. 1994; 10(2):115-121.
25. Higashi K, Jin Y, Johansson M, Heim S, Mandahl N, Biorklund A, Wennerberg J, Hambraeus G, Johansson L and Mitelman F. Rearrangement of 9p13 as the primary chromosomal aberration in adenoid cystic carcinoma of the respiratory tract. Genes, chromosomes & cancer. 1991; 3(1):21-23.
26. Stenman G, Sandros J, Dahlenfors R, Juberg-Ode M and Mark J. 6q- and loss of the Y chromosome--two common deviations in malignant human salivary gland tumors. Cancer Genet Cytogenet. 1986; 22(4):283-293.
27. Phuchareon J, Ohta Y, Woo JM, Eisele DW and Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PloS one. 2009; 4(6):e6040.
28. Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM, Weber RS, Caulin C and El-Naggar AK. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clinical cancer research. 2010; 16(19):4722-4731.
29. Mitani Y, Rao PH, Futreal PA, Roberts DB, Stephens PJ, Zhao YJ, Zhang L, Mitani M, Weber RS, Lippman SM, Caulin C and El-Naggar AK. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clinical cancer research. 2011; 17(22):7003-7014.
30. Wetterskog D, Lopez-Garcia MA, Lambros MB, A’Hern R, Geyer FC, Milanezi F, Cabral MC, Natrajan R, Gauthier A, Shiu KK, Orr N, Shousha S, Gatalica Z, Mackay A, Palacios J, Reis-Filho JS, et al. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. The Journal of pathology. 2012; 226(1):84-96.
31. West RB, Kong C, Clarke N, Gilks T, Lipsick JS, Cao H, Kwok S, Montgomery KD, Varma S and Le QT. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. The American journal of surgical pathology. 2011; 35(1):92-99.
32. Fehr A, Kovacs A, Loning T, Frierson H, Jr., van den Oord J and Stenman G. The MYB-NFIB gene fusion-a novel genetic link between adenoid cystic carcinoma and dermal cylindroma. The Journal of pathology. 2011; 224(3):322-327.
33. North JP, McCalmont TH, Fehr A, van Zante A, Stenman G and LeBoit PE. Detection of MYB Alterations and Other Immunohistochemical Markers in Primary Cutaneous Adenoid Cystic Carcinoma. The American journal of surgical pathology. 2015; 39(10):1347-1356.
34. Brill LB, 2nd, Kanner WA, Fehr A, Andren Y, Moskaluk CA, Loning T, Stenman G and Frierson HF, Jr. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Modern pathology. 2011; 24(9):1169-1176.
35. Ramsay RG and Gonda TJ. MYB function in normal and cancer cells. Nature reviews Cancer. 2008; 8(7):523-534.
36. Drabsch Y, Robert RG and Gonda TJ. MYB suppresses differentiation and apoptosis of human breast cancer cells. Breast cancer research : BCR. 2010; 12(4):R55.
37. Pan J-H, Adair-Kirk TL, Patel AC, Huang T, Yozamp NS, Xu J, Reddy EP, Byers DE, Pierce RA, Holtzman MJ and Brody SL. Myb Permits Multilineage Airway Epithelial Cell Differentiation. STEM CELLS. 2014; 32(12):3245-3256.
38. Dubendorff JW and Lipsick JS. Transcriptional regulation by the carboxyl terminus of c-Myb depends upon both the Myb DNA-binding domain and the DNA recognition site. Oncogene. 1999; 18(23):3452-3460.
39. Quelen C, Lippert E, Struski S, Demur C, Soler G, Prade N, Delabesse E, Broccardo C, Dastugue N, Mahon FX and Brousset P. Identification of a transforming MYB-GATA1 fusion gene in acute basophilic leukemia: a new entity in male infants. Blood. 2011; 117(21):5719-5722.
40. Pattabiraman DR and Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia. 2013; 27(2):269-277.
41. Hugo H, Cures A, Suraweera N, Drabsch Y, Purcell D, Mantamadiotis T, Phillips W, Dobrovic A, Zupi G, Gonda TJ, Iacopetta B and Ramsay RG. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes, Chromosomes and Cancer. 2006; 45(12):1143-1154.
42. Kauraniemi P, Hedenfalk I, Persson K, Duggan DJ, Tanner M, Johannsson O, Olsson H, Trent JM, Isola J and Borg A. MYB oncogene amplification in hereditary BRCA1 breast cancer. Cancer Res. 2000; 60(19):5323-5328.
43. Pattabiraman DR, McGirr C, Shakhbazov K, Barbier V, Krishnan K, Mukhopadhyay P, Hawthorne P, Trezise A, Ding J, Grimmond SM, Papathanasiou P, Alexander WS, Perkins AC, Levesque J-P, Winkler IG and Gonda TJ. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood. 2014; 123(17):2682-2690.
44. Aziz N, Miglarese MR, Hendrickson RC, Shabanowitz J, Sturgill TW, Hunt DF and Bender TP. Modulation of c-Myb-induced transcription activation by a phosphorylation site near the negative regulatory domain. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92(14):6429-6433.
45. Miglarese MR, Richardson AF, Aziz N and Bender TP. Differential regulation of c-Myb-induced transcription activation by a phosphorylation site in the negative regulatory domain. The Journal of biological chemistry. 1996; 271(37):22697-22705.
46. Bies J, Markus J and Wolff L. Covalent attachment of the SUMO-1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. The Journal of biological chemistry. 2002; 277(11):8999-9009.
47. Sano Y and Ishii S. Increased affinity of c-Myb for CREB-binding protein (CBP) after CBP-induced acetylation. The Journal of biological chemistry. 2001; 276(5):3674-3682.
48. Press RD, Reddy EP and Ewert DL. Overexpression of C-terminally but not N-terminally truncated Myb induces fibrosarcomas: a novel nonhematopoietic target cell for the myb oncogene. Molecular and cellular biology. 1994; 14(4):2278-2290.
49. Press RD, Wisner TW and Ewert DL. Induction of B cell lymphomas by overexpression of a Myb oncogene truncated at either terminus. Oncogene. 1995; 11(3):525-535.
50. O’Rourke JP and Ness SA. Alternative RNA splicing produces multiple forms of c-Myb with unique transcriptional activities. Molecular and cellular biology. 2008; 28(6):2091-2101.
51. Mayr C, Hemann MT and Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007; 315(5818):1576-1579.
52. Shao C, Bai W, Junn JC, Uemura M, Hennessey PT, Zaboli D, Sidransky D, Califano JA and Ha PK. Evaluation of MYB promoter methylation in salivary adenoid cystic carcinoma. Oral oncology. 2011; 47(4):251-255.
53. Gao R, Cao C, Zhang M, Lopez MC, Yan Y, Chen Z, Mitani Y, Zhang L, Zajac-Kaye M, Liu B, Wu L, Renne R, Baker HV, El-Naggar A and Kaye FJ. A unifying gene signature for adenoid cystic cancer identifies parallel MYB-dependent and MYB-independent therapeutic targets. Oncotarget. 2014; 5(24):12528-12542. doi: 10.18632/oncotarget.2985.
54. Mitani Y, Roberts DB, Fatani H, Weber RS, Kies MS, Lippman SM and El-Naggar AK. MicroRNA profiling of salivary adenoid cystic carcinoma: association of miR-17-92 upregulation with poor outcome. PloS one. 2013; 8(6):e66778.
55. Hudson JB and Collins BT. MYB gene abnormalities t(6;9) in adenoid cystic carcinoma fine-needle aspiration biopsy using fluorescence in situ hybridization. Archives of pathology & laboratory medicine. 2014; 138(3):403-409.
56. Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra R-J, Bryne JC, van den Heuvel A, Stevens M, de Boer E, Kockx C, van der Sloot A, van den Hout M, van Ijcken W, Eick D, Lenhard B, Grosveld F and Soler E. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. The EMBO Journal. 2012; 31(4):986-999.
57. Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van Ijcken W, Lenhard B, Rooks H, Best S, Menzel S, Grosveld F, Thein SL and Soler E. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. The Journal of clinical investigation. 2014; 124(4):1699-1710.
58. Clappier E, Cuccuini W, Kalota A, Crinquette A, Cayuela J-M, Dik WA, Langerak AW, Montpellier B, Nadel B, Walrafen P, Delattre O, Aurias A, Leblanc T, Dombret H, Gewirtz AM, Baruchel A, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. 2007; 110(4):1251-1261.
59. Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, Schumacher SE, Urbanski L, O’Rourke R, Gibson WJ, Pelton K, Ramkissoon SH, Han HJ, Zhu Y, Choudhari N, Silva A, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nature genetics. 2016.
60. Rushton JJ, Davis LM, Lei W, Mo X, Leutz A and Ness SA. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene. 2003; 22(2):308-313.
61. Bolcun-Filas E, Bannister LA, Barash A, Schimenti KJ, Hartford SA, Eppig JJ, Handel MA, Shen L and Schimenti JC. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development (Cambridge, England). 2011; 138(15):3319-3330.
62. Lei W, Rushton JJ, Davis LM, Liu F and Ness SA. Positive and negative determinants of target gene specificity in myb transcription factors. The Journal of biological chemistry. 2004; 279(28):29519-29527.
63. Ramkissoon LA, Horowitz PM, Craig JM, Ramkissoon SH, Rich BE, Schumacher SE, McKenna A, Lawrence MS, Bergthold G, Brastianos PK, Tabak B, Ducar MD, Van Hummelen P, MacConaill LE, Pouissant-Young T, Cho YJ, et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110(20):8188-8193.
64. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, Boop FA, Lu C, Kandoth C, Ding L, Lee R, Huether R, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nature genetics. 2013; 45(6):602-612.
65. Gronostajski RM. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000; 249(1–2):31-45.
66. Mirabello L, Koster R, Moriarity BS, Spector LG, Meltzer PS, Gary J, Machiela MJ, Pankratz N, Panagiotou OA, Largaespada D, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, de Toledo SR, Petrilli AS, et al. A Genome-Wide Scan Identifies Variants in NFIB Associated with Metastasis in Patients with Osteosarcoma. Cancer Discov. 2015; 5(9):920-931.
67. Zhou M, Zhou L, Zheng L, Guo L, Wang Y, Liu H, Ou C and Ding Z. miR-365 Promotes Cutaneous Squamous Cell Carcinoma (CSCC) through Targeting Nuclear Factor I/B (NFIB). PloS one. 2014; 9(6):e100620.
68. Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, Crowley D, Barretina J, Garraway L, Meyerson M and Jacks T. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes & development. 2011; 25(14):1470-1475.
69. Mellas RE, Kim H, Osinski J, Sadibasic S, Gronostajski RM, Cho M and Baker OJ. NFIB regulates embryonic development of submandibular glands. Journal of dental research. 2015; 94(2):312-319.
70. Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, Litwack ED, Richards LJ and Gronostajski RM. The transcription factor gene Nfib is essential for both lung maturation and brain development. Molecular and cellular biology. 2005; 25(2):685-698.
71. Rettig EM, Talbot C, Sausen M, Jones S, Bishop JA, Wood LD, Tokheim C, Niknafs N, Karchin R, Fertig E, Wheelan S, Marchionni L, Considine M, Fakhry C, Papadopoulos N, Kinzler KW, et al. Whole-Genome Sequencing of Salivary Gland Adenoid Cystic Carcinoma. Cancer prevention research (Philadelphia, Pa). 2016.
72. Nilsson M, Panagopoulos I, Mertens F and Mandahl N. Fusion of the HMGA2 and NFIB genes in lipoma. Virchows Archiv. 2005; 447(5):855-858.
73. Geurts JM, Schoenmakers EF, Roijer E, Astrom AK, Stenman G and van de Ven WJ. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene. 1998; 16(7):865-872.
74. Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, Zhang J, Dolgalev I, Huberman K, Heguy A, Viale A, Drobnjak M, et al. The mutational landscape of adenoid cystic carcinoma. Nature genetics. 2013; 45(7):791-798.
75. Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, Gamble J, Gamble S, Hardy C, Hinton J, Jia M, Jayakumar A, et al. Whole exome sequencing of adenoid cystic carcinoma. The Journal of clinical investigation. 2013; 123(7):2965-2968.
76. Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, Yuan J, Ware C, MacLean J, Garrett-Engele PW, Kluk M, Laskey J, Haines BB, Moskaluk C, Zawel L, Fawell S, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014; 4(10):1154-1167.
77. Stenman G, Persson F and Andersson MK. Diagnostic and therapeutic implications of new molecular biomarkers in salivary gland cancers. Oral oncology. 2014; 50(8):683-690.
78. Persson F, Fehr A, Sundelin K, Schulte B, Loning T and Stenman G. Studies of genomic imbalances and the MYB-NFIB gene fusion in polymorphous low-grade adenocarcinoma of the head and neck. International journal of oncology. 2012; 40(1):80-84.
79. Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CK, Perez-Ordonez B, Harding NJ, Alfaro J, Chu KC, Viale A, Fusco N, da Cruz Paula A, Marchio C, Sakr RA, Lim R, Thompson LD, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nature genetics. 2014; 46(11):1166-1169.
80. Pusztaszeri MP, Sadow PM, Ushiku A, Bordignon P, McKee TA and Faquin WC. MYB immunostaining is a useful ancillary test for distinguishing adenoid cystic carcinoma from pleomorphic adenoma in fine-needle aspiration biopsy specimens. Cancer cytopathology. 2014; 122(4):257-265.
81. Foo WC, Jo VY and Krane JF. Usefulness of translocation-associated immunohistochemical stains in the fine-needle aspiration diagnosis of salivary gland neoplasms. Cancer cytopathology. 2016.
82. Rettig EM, Tan M, Ling S, Yonescu R, Bishop JA, Fakhry C and Ha PK. MYB rearrangement and clinicopathologic characteristics in head and neck adenoid cystic carcinoma. The Laryngoscope. 2015; 125(9):E292-299.
83. Chae YK, Chung SY, Davis AA, Carneiro BA, Chandra S, Kaplan J, Kalyan A and Giles FJ. Adenoid cystic carcinoma: current therapy and potential therapeutic advances based on genomic profiling. Oncotarget. 2015; 6(35):37117-34. doi: 10.18632/oncotarget.5076.
84. Patel MN, Halling-Brown MD, Tym JE, Workman P and Al-Lazikani B. Objective assessment of cancer genes for drug discovery. Nat Rev Drug Discov. 2013; 12(1):35-50.
85. Moellering RE, Cornejo M, Davis TN, Bianco CD, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL and Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009; 462(7270):182-188.
86. Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S, Zhou Y, Boulton A, Kuntimaddi A, Gao Y, Rajewski RA, Guzman ML, Castilla LH and Bushweller JH. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science. 2015; 347(6223):779-784.
87. Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, Weissenboeck M, Graeber TG, Kogan SC, Vakoc CR and Lowe SW. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes & development. 2011; 25(15):1628-1640.
88. Pereira LA, Hugo HJ, Malaterre J, Huiling X, Sonza S, Cures A, Purcell DFJ, Ramsland PA, Gerondakis S, Gonda TJ and Ramsay RG. MYB Elongation Is Regulated by the Nucleic Acid Binding of NFκB p50 to the Intronic Stem-Loop Region. PloS one. 2015; 10(4):e0122919.
89. Dooley S, Seib T, Welter C and Blin N. c-myb intron I protein binding and association with transcriptional activity in leukemic cells. Leukemia research. 1996; 20(5):429-439.
90. Zor T, De Guzman RN, Dyson HJ and Wright PE. Solution Structure of the KIX Domain of CBP Bound to the Transactivation Domain of c-Myb. Journal of Molecular Biology. 2004; 337(3):521-534.
91. Uttarkar S, Dukare S, Bopp B, Goblirsch M, Jose J and Klempnauer KH. Naphthol AS-E Phosphate Inhibits the Activity of the Transcription Factor Myb by Blocking the Interaction with the KIX Domain of the Coactivator p300. Molecular cancer therapeutics. 2015; 14(6):1276-1285.
92. Uttarkar S, Dassé E, Coulibaly A, Steinmann S, Jakobs A, Schomburg C, Trentmann A, Jose J, Schlenke P, Berdel WE, Schmidt TJ, Müller-Tidow C, Frampton J and Klempnauer K-H. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood. 2015.
93. Bujnicki T, Wilczek C, Schomburg C, Feldmann F, Schlenke P, Muller-Tidow C, Schmidt TJ and Klempnauer KH. Inhibition of Myb-dependent gene expression by the sesquiterpene lactone mexicanin-I. Leukemia. 2012; 26(4):615-622.
94. Schomburg C, Schuehly W, Da Costa FB, Klempnauer KH and Schmidt TJ. Natural sesquiterpene lactones as inhibitors of Myb-dependent gene expression: structure-activity relationships. European journal of medicinal chemistry. 2013; 63:313-320.
95. Ikeda H, Kanakura Y, Tamaki T, Kuriu A, Kitayama H, Ishikawa J, Kanayama Y, Yonezawa T, Tarui S and Griffin JD. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood. 1991; 78(11):2962-2968.
96. Lyle M and Long GV. Diagnosis and treatment of KIT-mutant metastatic melanoma. J Clin Oncol. 2013; 31(26):3176-3181.
97. Fletcher JA and Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev. 2007; 17(1):3-7.
98. Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS and El-Naggar AK. Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer biology & therapy. 2011; 12(7):569-573.
99. Wetterskog D, Wilkerson PM, Rodrigues DN, Lambros MB, Fritchie K, Andersson MK, Natrajan R, Gauthier A, Di Palma S, Shousha S, Gatalica Z, Topfer C, Vukovic V, A’Hern R, Weigelt B, Vincent-Salomon A, et al. Mutation profiling of adenoid cystic carcinomas from multiple anatomical sites identifies mutations in the RAS pathway, but no KIT mutations. Histopathology. 2013; 62(4):543-550.
100. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19(2):202-208.
101. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, Jin S, Smith M, Xue J, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Science Translational Medicine. 2015; 7(279):279ra240-279ra240.
102. Li J, Perlaky L, Rao P, Weber RS and El-Naggar AK. Development and characterization of salivary adenoid cystic carcinoma cell line. Oral oncology. 2014; 50(10):991-999.
103. Moskaluk CA, Baras AS, Mancuso SA, Fan H, Davidson RJ, Dirks DC, Golden WL and Frierson HF, Jr. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Laboratory investigation; a journal of technical methods and pathology. 2011; 91(10):1480-1490.
104. Costa AF, Altemani A, Garcia-Inclan C, Fresno F, Suarez C, Llorente JL and Hermsen M. Analysis of MYB oncogene in transformed adenoid cystic carcinomas reveals distinct pathways of tumor progression. Laboratory investigation; a journal of technical methods and pathology. 2014; 94(6):692-702.
105. Tian Z, Li L, Zhang CY, Gu T and Li J. Differences in MYB expression and gene abnormalities further confirm that salivary cribriform basal cell tumors and adenoid cystic carcinoma are two distinct tumor entities. Journal of oral pathology & medicine. 2015.
106. Argyris PP, Wetzel SL, Greipp P, Wehrs RN, Knutson DL, Kloft-Nelson SM, Garcia JJ and Koutlas IG. Clinical utility of myb rearrangement detection and p63/p40 immunophenotyping in the diagnosis of adenoid cystic carcinoma of minor salivary glands: a pilot study. Oral surgery, oral medicine, oral pathology and oral radiology. 2016; 121(3):282-289.
107. D’Alfonso TM, Mosquera JM, MacDonald TY, Padilla J, Liu YF, Rubin MA and Shin SJ. MYB-NFIB gene fusion in adenoid cystic carcinoma of the breast with special focus paid to the solid variant with basaloid features. Human pathology. 2014; 45(11):2270-2280.
108. Bishop JA, Yonescu R, Epstein JI and Westra WH. A subset of prostatic basal cell carcinomas harbor the MYB rearrangement of adenoid cystic carcinoma. Human pathology. 2015; 46(8):1204-1208.