
INTRODUCTION
The incidence of pancreatic cancer is on the rise, being projected to become the second cause of cancer-related deaths by 2030 [1]. Patients diagnosed with pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, still face a very poor outcome with a 5-year survival of 5–7%. Surgical resection offers the best chance of cure; however most patients present with locally advanced or metastatic disease and are therefore ineligible for surgery [2]. Novel combination therapies such as nab-paclitaxel with gemcitabine or FOLFIRINOX have improved patient outcomes but improvements are small and toxicity is an issue [2].
Sirtuin1 (SIRT1) inhibitors have shown promising results in preclinical models of PDAC (reviewed in [3]). SIRT1 is an evolutionary conserved protein that senses NAD+ and resides in the nucleus or the cytoplasm where it can deacetylate a panel of proteins that regulate inflammation, lifespan and metabolic homeostasis, among many other processes (reviewed in [3–9]). The tumor suppressor p53 is the best studied protein that is deacetylated and inactivated by SIRT1 [10]. SIRT1 itself is regulated by Cell Cycle and Apoptosis Regulator 2 (CCAR2), an endogenous inhibitor of SIRT1’s deacetylase activity in various organs, including in the pancreas [11].
Not only by its consumption of NAD+ does SIRT1 function as an energy sensor, it also mediates metabolic effects in liver, skeletal muscle, heart and adipocytes by directly deacetylating proteins that regulate gluconeogenesis, fatty acid metabolism and glycolysis [12]. Through effects on Phosphoglycerate 1 (PGAM1) and peroxisome proliferator-activated receptor alpha coactivator 1 alpha (PGC1a), SIRT1 was found a negative regulator of glycolysis in liver and skeletal muscle [12]. Contrary, through inactivation of p53 SIRT1 can stimulate glycolysis [13], underscoring the dependency on the context.
The biological functions of SIRT1 in PDAC are still poorly understood. We recently reported a role for SIRT1 in the development of pancreatic acinar-ductal metaplasia, a process that occurs in acute and chronic pancreatitis and may give rise to neoplastic lesions [14]. We now sought to explore if SIRT1 also impacts the development of (pre)tumoral lesions. In addition, we analyzed a potential metabolic regulation by SIRT1 in pancreatic lesions.
RESULTS
Sirt1 promotes the proliferation of mucinous intraepithelial neoplastic lesions in the murine pancreas
Mice with an activating mutation in Kras (lox-STOP-lox KrasG12D) [15] that is specifically targeted to the pancreas by a Pdx1-Cre driver (abbreviated KC mice) were crossed with a mouse line in which the exon 4 of Sirt1 is flanked by loxP sites to obtain homozygous Sirt1-deficient KC mice (KC;Sirt1-lox) [14, 16]. Only very low expression of the mutant form of SIRT1 protein is found in the pancreas of the KC;Sirt1-lox animals (Figure 1A and [16]). Cheng et al. have previously shown that the resulting short mutant form of SIRT1 lacks SIRT1 deacetylation activity, resulting in a phenotype similar to a full SIRT1 knock out [17]. KC;Sirt1-lox animals were sacrificed at 6 and 12 months of age, in parallel with a cohort of KC control mice. We observed that in the mice that had not been culled earlier because of reaching ethical endpoints (due to PDAC, another tumor or unknown cause), PanIN1-3 lesions progressively developed at 6 and 12 months of age (Figure 1B). The progression of the lesions scored by the grade of the PanINs did not differ when comparing KC controls with KC;Sirt1-lox mice (Figure 1B).
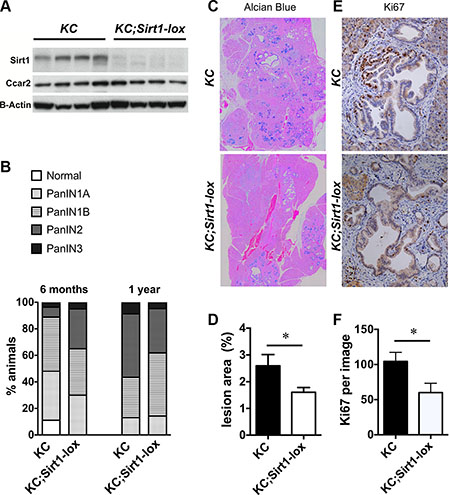
Figure 1: Pancreatic lesions in KC and homozygous KC;Sirt1-lox mice. (A) Western Blot analysis of Sirt1, Ccar2 and Beta-Actin (B-Actin) protein expression in whole pancreas tissue, n = 4 per group. (B) Percentage of animals with the highest lesion in pancreas tissue sections that were classified as normal, PanIN 1,2 or 3. n = 20 KC;Sirt1-lox and n = 27 KC at 6 months of age, n = 21 KC;Sirt1-lox and n = 23 KC at 12 months of age. (C) Representative Alcian Blue staining in pancreas tissue sections of 12 month old animals. (D) Measurement of the percentage Alcian Blue positive area in pancreas tissue sections. n = 9–11. (E) Representative Ki67 immunohistochemistry in pancreas tissue sections of 12 month old animals. (F) Quantification of Ki67 positivity per image of lesion area. n = 9–13. All data are represented as mean +/− SEM, *P < 0.05.
To evaluate the lesion burden, we quantified the area occupied by PanIN lesions using two different methods: Keratin 19 (Krt19) immunohistochemistry and Alcian Blue staining of mucinous lesions. The relative area of Krt19 positive lesions presented no differences between KC controls and KC;Sirt1-lox mice (Supplementary Figure S1). However, the area of mucinous early PanINs, measured by the percentage of Alcian Blue–positive, was significantly lower in the KC;Sirt1-lox mice (1.6 ± 0.2% versus 2.6 ± 0.4% area, n = 9–11, P < 0.05, Figure 1C, 1D).
The reduced area occupied by mucinous neoplastic lesions was attributed to lower proliferation, measured by Ki67 immunohistochemistry (IHC) in the KC;Sirt1-lox background compared to KC controls (60 ± 13 versus 104 ± 13 counts of Ki67 positive cells per field, n = 9–13, P < 0.05, Figure 1E, 1F). There was no statistically significant difference in the proliferation of the neighboring stroma. In addition, all mice analyzed only showed very low apoptosis in the lesions, detected by cleaved caspase-3 staining (not shown). Pancreata from Kras wild type Pdx1-Cre;Sirt1-lox mice did not show alterations in the proliferation of any cell type (acinar, duct, islets or stroma) when compared with Pdx1-Cre controls (Supplementary Figure S2), suggesting the effect on proliferation is specific for the early lesions in the Kras mutant background.
In conclusion, SIRT1 is dispensable for pancreatic lesion development, does not affect the proliferation of normal pancreatic tissue but specifically stimulates the growth of early mucinous PanIN lesions.
SIRT1 regulates the expression of glycolysis genes in mouse pancreas
Ample evidence is available that proliferation of (pancreatic) tumor cells is enabled by an increased rate of intracellular glucose import and a higher rate of aerobic glycolysis (referred to as the Warburg effect) [18]. Moreover, SIRT1 is a known metabolic regulator and deacetylates several proteins that play critical roles in glycolysis such as p53 [13] and MYC [19]. Therefore, we verified if SIRT1’s effect on proliferation of the pancreatic lesions involved changes in glycolysis genes.
We used our SIRT1-deficient mouse model to verify a regulation of glycolysis proteins by SIRT1 in pancreatic lesions in vivo. When comparing immunostainings in the PanIN lesions in KC;Sirt1-lox versus KC controls, we found a weaker staining for GLUT1 (Glucose transporter 1), GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) and PKM2 (Pyruvate kinase isozyme m2) and slightly weaker HK2 (Hexokinase 2) (Figure 2).
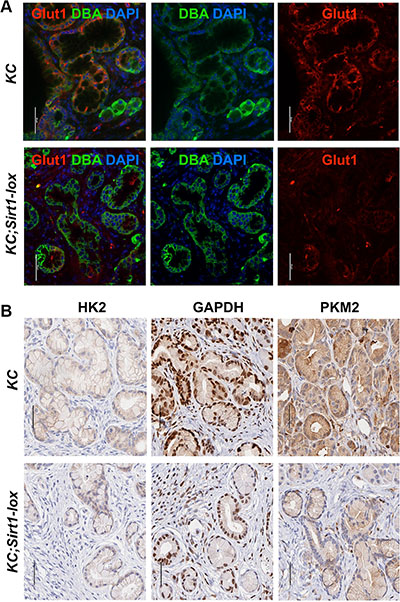
Figure 2: Expression of glycolytic proteins in murine SIRT1-deficient PanIN lesions. (A) Immunofluorescence for GLUT1 (red) and (B) immunohistochemistry for HK2, GAPDH and PKM2 in pancreas tissue sections (bars = 50 micrometer).
In conclusion, SIRT1 stimulates the expression of glycolysis genes in mouse PanIN lesions.
SIRT1 stimulates proliferation and glycolysis gene expression in human PDAC
Complementary to our in vivo observations in the mice, we down-regulated SIRT1 gene expression using siRNA in a human pancreatic tumor cell line. Effective knock down of SIRT1 in Panc1 cells was demonstrated by reduced SIRT1 protein in Western Blotting analysis, along with increased acetylation of SIRT1’s best-characterized substrate p53 [10] and downstream upregulation of p21, a known p53 target gene (Figure 3A and Supplementary Figure S3). We found that knock down of SIRT1 impaired the proliferation of the PDAC cells, an extension of previous reports [14, 20] (Figure 3B). The cells treated with siRNA for SIRT1 had a consistent reduction in the mRNA of SIRT1, GLUT1 (Glucose transporter 1), GAPDH, PKM2 and LDHA (Lactate dehydrogenase A) (Figure 3C).
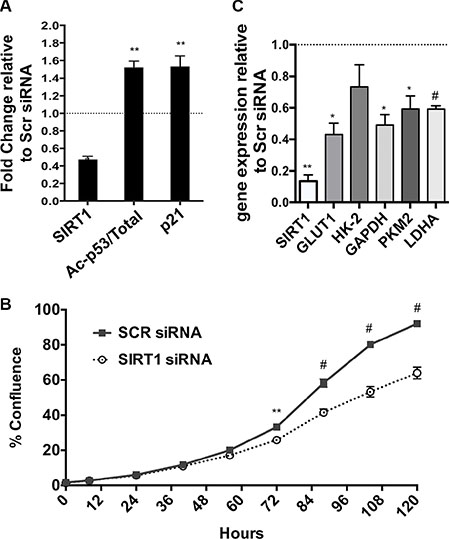
Figure 3: Effects of SIRT1 siRNA in human PDAC cells. (A) Band density quantification by Image J analysis of Western Blot of SIRT1, acetylated and total p53 and p21, using B-ACTIN as a loading control, from Panc1 cells treated with a scrambled siRNA sequence or with siRNA for SIRT1. n = 6 (See representative Western Blot in Supplementary. Figure S3). (B) Real time RT-PCR analysis of the mRNA expression of SIRT1, GLUT1, HK2, GAPDH, PKM2, LDHA and PDK1 relative to HPRT as housekeeping gene (n = 4) in SIRT1 siRNA treated Panc1 cells. (C) IncuCyte graph of the % increase in cell confluence in siRNA treated cells versus scr controls. One of three representative experiments is shown. All data are represented as mean +/− SEM, *P < 0.05, **P < 0.01, #P < 0.001.
High proliferation, high glycolysis gene expression and low expression of a SIRT1 inhibitor corresponds to the poorest patient survival outcomes
We analyzed if SIRT1 expression was prognostic in human PDAC, using a patient cohort of treatment-naive early stage I and II resected PDAC (n = 104) [21]. Staining for SIRT1 protein expression (nuclear, cytoplasmic or total) in the PDAC cells, following the protocol we had previously optimized in another smaller cohort [14], did not directly correlate with survival outcome (Figure 4A and not shown). We verified if instead, expression of the SIRT1 inhibitor CCAR2 was related to clinical outcomes. CCAR2 protein expression is very high in the majority of the samples (H-score > 200, Supplementary Figure S4) and correlated with improved survival; the 25% low CCAR2 expressing tumors corresponded to patients with poorer survival (Figure 4B). The low CCAR2 PDAC also independently correlated with undifferentiated and poorly differentiated tumors (n = 104, P < 0.01). Pathway analysis of the 88 genes that positively correlated with CCAR2 gene expression in microarray data of these human PDAC samples [21] resulted in ‘chromatin modification’, ‘histone acetylation’ and ‘cell cycle’ (Table 1, n = 104), supportive of a regulatory role of CCAR2 in typical SIRT1 activities in PDAC.
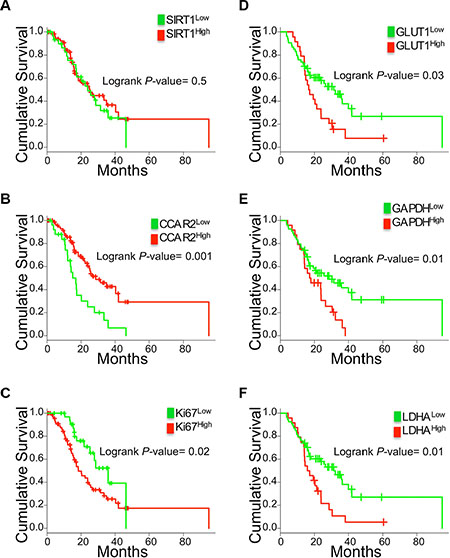
Figure 4: Correlation of SIRT1, CCAR2, Ki67 and glycolysis genes with PDAC patient survival outcomes. Kaplan Meier curves for the protein expression of SIRT1 (A) and CCAR2 (B) and Ki67 (C) scored in the tumor epithelium of PDAC patients (n = 104). Kaplan Meier curves of GLUT1 (D), GAPDH (E) and LDHA (F) gene expression in high cellularity patient samples (21) (n = 78).
Table 1: Reactome pathways enriched among the genes that positively correlated (Pearson and Spermann correlation coefficient ≥ 0.4) with CCAR2 in cDNA microarray analysis of PDAC (n = 104)
#Term |
Corrected P-Value |
|---|---|
Chromatin modifying enzymes |
0, 0008 |
Chromatin organization |
0, 0008 |
RNA transport |
0, 0014 |
HATs acetylate histones |
0, 0030 |
mRNA surveillance pathway |
0, 0056 |
G2/M Transition |
0, 0069 |
Mitotic G2-G2/M phases |
0, 0069 |
Centrosome maturation |
0, 0082 |
Recruitment of mitotic centrosome proteins and complexes |
0, 0082 |
Then we assessed the clinical significance of increased epithelial proliferation and glycolysis gene expression, the latter confined to high cellularity samples (n = 78) to enrich for the epithelial gene expression. Higher percentage of Ki67-stained epithelium correlated with poor outcomes (Figure 4C). Also high SLC2A1/GLUT1, GAPDH and LDHA but not PKM2 correlated with worse patient survival (Figure 4D–4F and not shown).
In conclusion, CCAR2 protein but not SIRT1 is prognostic in PDAC. In addition, reduced patient survival time is seen in tumors that proliferate more and have higher expression of glycolysis genes.
DISCUSSION
SIRT1 has been attributed roles in tumor formation [7, 22–24] and is a regulator of metabolism, a process that interconnects with cancer [4, 25]. Previously we had shown a role of SIRT1 in metaplasia of pancreatic cells [14, 16]. Because of the link between glycolytic metabolism, tumor formation and tumor cell growth, we set out to explore whether these mechanisms were regulated by SIRT1 in the pancreas.
The development of pancreatic lesions is dependent on the activation of KRAS [15, 26, 27]. In the vast majority of PDAC patients, the KRAS gene presents an activating mutation [21]. Here we investigated the effects of SIRT1 inactivation in a KRAS mutant background using genetically engineered mice [15]. We are the first to demonstrate that a pancreas-specific inactivation of SIRT1 limited the proliferation of early mucinous PanIN lesions. Similar to our observations, SIRT1-deficiency reduced polyp area and size in the APC+/min model of colorectal cancer [23] and reduced the number and the size of tumors in a colitis-induced colorectal cancer model [24]. We extended our findings on tumor development to tumor progression, where we also found that SIRT1 stimulated the proliferation of established pancreatic tumor cells, in line with previous reports (reviewed in [3]).
Aerobic glycolysis plays a central role in cellular transformation and the proliferation of tumors [18]. In PDAC, aerobic glycolysis is a source of intermediates for the non-oxidative pentose phosphate pathway [30] [31] . Our analyses demonstrated that SIRT1 positively regulated the expression of key genes in the glycolysis pathway. This new finding was made, both in the Kras mutant mouse model and in KRAS mutant human PDAC cells. A recent paper by Daemen et al [32] found that pancreatic tumor cell lines can be sub-typed according to their gene expression signature and metabolic profile with the quasi-mesenchymal sub-type having a glycolytic nature. Our patient samples were categorized by this gene expression signature into exocrine, quasi-mesenchymal and classical subtype (as also reported in [33]) but did not show sub-type specific differences in SIRT1 expression (not shown).
Several genes may be mediating the SIRT1 dependent regulation of glycolytic gene expression; SIRT1 deacetylates and inactivates p53 which is a negatively regulator of glycolysis [13]. We detected consistent changes in acetylation of p53 upon manipulation of SIRT1 in a PDAC cell line. Despite our attempts, we did not succeed in detecting acetylated P53 in tissue sections of human PDAC. More in-depth study and suitable antibodies would be required to decipher if indeed p53 is the critical mediator. Other genes may be accountable such as MYC, a transcriptional activator of glycolysis genes, since SIRT1 is reported to increase MYC’s transcriptional program [19, 22]. Alternatively, SIRT1 protects HIF1a from acetylation and degradation and thereby can increase GLUT1 expression, a mechanism reported before [34]. Although not specific for SIRT1, HDAC inhibitors were found to down-regulate GLUT1 expression and to inhibit hexokinase 1 enzymatic activity in multiple myeloma cells [35], underscoring that acetylation plays a key role in regulation of glycolytic metabolism. The opposite regulation has also been reported, e.g. inhibition of SIRT1 leads to acetylation and degradation of PKM2 while stimulating tumor proliferation [36]. Apart from glycolysis, other energy generating and biosynthetic pathways need to be controlled by the tumor cells in order to promote their cell proliferation [2–4]. SIRT1 deacetylates and thereby stimulates PGC1α. PGC1α is a negative regulator of glycolysis but promotes tumor progression through oxidative phosphorylation and lipogenesis [37] and by increasing mitochondrial gene expression that protects against pancreatic tumour cell apoptosis [38]. Therefore, SIRT1’s effect on PGC1α may also have contributed to our observations. We note that SIRT1 was reported to deacetylate KRAS thereby promoting its downstream signaling [39]. Knowing that oncogenic KRAS signaling upregulates many key enzymes in glycolysis (extensively reviewed in [40]), this may be another mechanism by which SIRT1 indirectly promotes glycolysis.
Despite the difference in area occupied by mucinous Alcian Blue positive lesions and the reduced expression of proteins in the glycolytic pathway, total area of lesions, as stained by Krt19 showed no differences between controls and SIRT1 deficient mice. Our observation suggests that only the early PanIN stage is affected in the SIRT1 loss of function model. We speculate that a subset of cells have started to compensate for the lower proliferation/reduced glycolysis of the early PanIN lesions. These cells must have progressed faster or have skipped the early PanIN stage. It has been reported that pancreatic tumor cells, upon genetic or pharmacological ablation of the bulk of glycolysis-dependent cells can become more aggressive and can lead to full recurrence of the tumor. This involves the acquisition of stem cell features and metabolic reprogramming with a shift from glycolysis to oxidative phosphorylation and is notably dependent on PCG1a [28], a protein well known for its regulation by SIRT1 [29], and important in tumor growth, as outlined above.
We verified the significance of our findings in clinical samples. In a cohort of human early stage tumors, high proliferation and high expression of glycolysis genes corresponded, not unexpectedly, to poorer patient outcomes. This inferred that high expression of SIRT1 might also lead to worse outcome. However, expression of SIRT1 in our analysis did not directly correlate with the patient survival outcomes, in discrepancy with other studies reporting that SIRT1 in PDAC correlated with poor histological differentiation and poor postsurgical survival [41] and with increased size, lymph node burden and hepatic metastasis [20]. Nevertheless, we found that CCAR2, an inhibitor of SIRT1’s deacetylase activity, was prognostic in our cohort, with the lower CCAR2 expressing tumors corresponding to poorly differentiated high mortality tumors. Based on literature [11, 42], the lowest CCAR2 expressing tumors would have the highest SIRT1 activity. Microarray data analysis of the patient samples also further suggested that CCAR2 regulates typical SIRT1 activities such as histone deacetylation but more research and the development of suitable antibodies would be needed before CCAR2 could be definitely used as a surrogate marker of low SIRT1 activity. Indeed, CCAR2 may also exert other effects independent of SIRT1 inhibition; for example, a recent report showed that CCAR2 is a tumor suppressor that stabilizes p53 with effects independent of SIRT1 [43].
In conclusion, our new results underscore a novel oncogenic function of SIRT1 during pancreatic cancer development where it stimulates the proliferation and expression of glycolytic proteins. SIRT1 is an interesting druggable target to halt tumor cell proliferation with successes in chronic myeloid leukemia [44] and with an inhibitor found to be safe in a clinical trial [45, 46]. Several groups have found promising effects of SIRT1 drugs in preclinical models of PDAC [3]. Our data do not only reinforce the idea to explore SIRT1 as a druggable target in early stage pancreatic cancer, we also deciphered a relation with tumor metabolism. We found that SIRT1 is a stimulus for glycolytic enzymes. Interestingly, specific drugs for these glycolytic enzymes are currently being tested, e.g. the LDH-A antagonist FX11 in PDAC [47] or the Hexokinase II inhibitor, 3-BrPA in breast cancer [48]. One could envision a combination therapy of a SIRT1 inhibitor and one of these drugs that target the metabolism of the pancreatic tumor or one could potentially use low CCAR2 expression, likely corresponding to high SIRT1 activity, as a biomarker to select a population of early stage PDAC patients for therapy with drugs that target the glycolytic metabolism. Our study paves the way for further investigation into these new therapeutic angles.
MATERIALS AND METHODS
Animals and in vivo experimentation
B6;129-Sirt1tm1Ygu/J (Sirt1-lox) strains were obtained from Jackson Laboratories and bred with the B6.FVB-Tg(Ipf1-cre)6Tuv/J (Pdx1-Cre, abbreviated as C) [15] and the lox-STOP-lox-KrasG12D line (abbreviated as KC) [15] to create KC;Sirt1-lox mice. The Sirt1-lox mice [49] have a loxP-flanked neomycin cassette just upstream of exon 4, and a third loxP site just downstream of exon 4 (encoding a conserved Sirt1 motif) of the targeted gene. The resulting offspring have exon 4 deleted in the pancreas. In all experiments, only homozygous Sirt1-lox mice were analyzed and the respective Sirt1 wild-type line was used as control. All animal experiments were approved by the Garvan Animal Ethical Committee (AEC approval #12-52).
Patient derived tissue collection
Biospecimens (tissue microarrays of paraffin embedded material and microarray gene expression datasets) and the clinico-pathological data were provided by the Australian Pancreatic Cancer Genome Initiative (APGI) [21, 50, 51] (ethical approval:HREC/11/RPAH/329 – The Molecular Pathology of Pancreatic Cancer, incorporating the APGI, approved by Sydney Local Health District –RPA Zone, protocol x11-0220).
Immunostaining
Mouse pancreas tissues were fixed in 4% formalin and embedded in paraffin. All immunohistochemistry was performed using the Dako Autostainer, Universal Staining System Model# LV-1 and the Leica BOND RX, according to supplier procedures. After deparaffinization and antigen retrieval (S1699 Target Retrieval Solution, Dako), slides were incubated with diluted primary antibody (Supplementary. Table S1) for 60 minutes and a secondary EnVision antibody (Dako) (Supplementary. Table S1) for 30 minutes. Finally the slides were treated with 3,3′-Diaminobenzidine (DAB+ Substrate Chrmogen system, Dako) for 10 minutes and counterstained with haematoxylin. All immunohistochemistry images were taken using the Aperio Scanscope CS system (Leica Biosystems). For immunofluorescence of GLUT-1, mouse pancreas was treated with EDTA-NaOH antigen retrieval (boiling 20 minutes in 1 mM pH 8). Sections were incubated with primary antibody overnight at 4°C and subsequently with anti-mouse-594, DBA and Dapi. Sections were viewed on a Nikon Eclipse TE300 epifluorescence microscope (Nikon Instruments Inc, Melville, NY) equipped with a DS-5M Standard charge-coupled device camera (Nikon Instruments Inc) and acquired with NISElements (Nikon Instruments Inc).
Human PDAC samples were assembled by APGI into tissue microarrays containing 3 samples of each patient tumor. SIRT1 and CCAR2 immunohistochemistry (IHC) on the human tissue microarrays was performed as reported before [14] and was scored blindly as absolute intensity (arbitrary values 0–3) and an estimated percentage of positive nuclei (H-score).
Alcian blue staining
Tissue slides were manually stained with Clinipure ALCBLUE1 03 (1% alcian blue, 3% acetic acid solution) for 30 minutes and counterstained with eosin. Alcian Blue quantification was performed by analyzing the stained slides with the Leica DM6000 Power Mosaic microscope and software. Twenty-five pictures per sample were manually selected to discard any images with empty areas or non-pancreatic tissue like lymphoid tissue, blood vessels or fat. The stained area was measured and the percentage of stained area over total tissue area was calculated.
siRNA knockdown of SIRT1
Panc-1 cells were obtained from the American Type Culture Collection (ATCC) Cell Biology Collection and used within 6 months of revival. The cells were tested for Mycoplasma contamination. The cells were plated in DMEM +10% Fetal Bovine Serum. At 24 hours, cells were transfected with 10 nM siRNA / Hyperfect complexes according to the manufacturers instructions (Qiagen) and incubated 37C, 5% CO2. One of two successful siRNAs (Invitrogen) was used for further analysis. Panc1 seeded in 96 well plates at 2 × 103 cells were monitored for growth (percent confluence) for up to 5 days following siRNA treatment using the IncuCyteZOOM live cell imager (Essen Bioscience). Replicate plates were also analysed for cell proliferation via BRDU ELISA (Roche, 11647229001) with a 2 hour Bromodeoxyuridine pulse at 96 hours per manufacturers instructions.
Western blotting
Cells were lysed in Normal Lysis Buffer (Hepes 50 mM, NaCl 150 mM, Glycerol 10%, Triton X100 1%, MgCl2 1.5 mM, EGTA 1 mM, Pyrophosphate 10 mM, NaF 100 mM) complemented with protease and phosphatase inhibitors (Roche) and MG132 (10 ug/ml). Frozen tissue specimens were homogenized in RIPA Buffer (Tris-Hcl 50 mM, NaCl 150 mM, EDTA 5 mM, NP40 0.5%, 0.1%, SDS 0.1%) complemented with protease and phosphatase inhibitors and MG132. Protein extracts were subjected to electrophoresis on 4–12% gradient polyacrylamide gels (SDS-PAGE) and transferred onto a PVDF membrane (Biorad). The membrane was incubated overnight at 4°C with primary antibodies (Supplementary. Table S1) diluted in TBS/BSA (Tris pH 7.4 10 mM, NaCl 150 mM, 5% BSA, 0.05% Sodium Azide and phenol red). The membrane was then incubated with HRP-conjugated secondary antibodies at a 1/2000 dilution. The signals were visualized with ECL (Perkin Elmer) and autoradiography, and band densities of the gene of interest relative to the housekeeping gene were measured with ImageJ software.
Real time PCR analysis
Total RNA from Panc1 cells was isolated using the Purelink RNA mini kit (Ambion, Life Technologies) and subjected to DNase1 treatment (Ambion, Life Technologies). First strand cDNA synthesis was performed with a random hexamer / poly-A primer mix (Roche) according to manufacturers instructions. Real time PCR products were amplified with Fast Start SYBR Green (Roche) using the primers listed in Supplementary. Table S2 using the 7900HT Fast Real Time PCR System (Applied Biosystems). PCR product specificity was confirmed with dissociation curve analysis and 2% agarose gel electrophoresis of products. Expression levels were normalized to house keeping gene expression (HPRT).
Statistics
Results are presented as mean ± SEM. The number of independent experiments is represented as n. Data were analyzed by Prism 6.0 using Student t test (unpaired t test) or two-way ANOVA and results considered significant when P < 0.05.
ACKNOWLEDGMENTS
We thank J. Pettitt and M. Rees for technical assistance.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
GRANT SUPPORT
AVP and IR hold/held fellowships from Cancer Institute NSW (13ECF104 and 10FRL203). IR is supported by an Odysseus fellowship from the Fund for Scientific Research Flanders (FWO). SMRC and EK were supported by University of Zurich.
REFERENCES
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913–2921.
2. Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015; 12:319–334.
3. Giry-Laterriere M, Pinho AV, Eling N, Chantrill L, Rooman I. Emerging Drug Target In Pancreatic Cancer: Placing Sirtuin 1 on the Canvas. Curr Cancer Drug Targets. 2015; 15:463–468.
4. Simmons GE, Jr., Pruitt WM, Pruitt K. Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int J Mol Sci. 2015; 16:950–965.
5. Boutant M, Canto C. SIRT1 metabolic actions: Integrating recent advances from mouse models. Mol Metab. 2014; 3:5–18.
6. Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther. 2013; 6:1399–1416.
7. Song NY, Surh YJ. Janus-faced role of SIRT1 in tumorigenesis. Ann N Y Acad Sci. 2012; 1271:10–19.
8. Martinez-Redondo P, Vaquero A. The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer. 2013; 4:148–163. doi: 10.1177/1947601913483767.
9. Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007; 404:1–13.
10. Lee JT, Gu W. SIRT1: Regulator of p53 Deacetylation. Genes Cancer. 2013; 4:112–117. doi: 10.1177/1947601913484496.
11. Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z, Chini EN. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest. 2010; 120:545–558.
12. Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014; 25:138–145.
13. Zhang C, Liu J, Wu R, Liang Y, Lin M, Chan CS, Hu W, Feng Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014; 5:5535–5546. doi: 10.18632/oncotarget.2137.
14. Wauters E, Sanchez-Arevalo Lobo VJ, Pinho AV, Mawson A, Herranz D, Wu J, Cowley MJ, Colvin EK, Ngawi Njicop E, Sutherland RL, Liu T, Serrano M, Bouwens L, et al. Sirtuin-1 regulates acinar to ductal metaplasia and supports cancer cell viability in pancreatic cancer. Cancer Research. 2013; 73:2357–2367.
15. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003; 4:437–450.
16. Pinho AV, Bensellam M, Wauters E, Rees M, Giry-Laterriere M, Mawson A, Ly le Q, Biankin AV, Wu J, Laybutt DR, Rooman I. Pancreas-Specific Sirt1-Deficiency in Mice Compromises Beta-Cell Function without Development of Hyperglycemia. PLoS One. 2015; 10:e0128012.
17. Cheng H-L, Mostoslavsky R, Saito Si, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:10794–10799.
18. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21:297–308.
19. Menssen A, Hermeking H. c-MYC, SIRT1 locked in a vicious cycle. Oncotarget. 2012; 3:112–113. doi: 10.18632/oncotarget.440.
20. Zhao G, Cui J, Zhang JG, Qin Q, Chen Q, Yin T, Deng SC, Liu Y, Liu L, Wang B, Tian K, Wang GB, Wang CY. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther. 2011; 18:920–928.
21. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012; 491:399–405.
22. Herranz D, Maraver A, Canamero M, Gomez-Lopez G, Inglada-Perez L, Robledo M, Castelblanco E, Matias-Guiu X, Serrano M. SIRT1 promotes thyroid carcinogenesis driven by PTEN deficiency. Oncogene. 2013; 32:4052–4056.
23. Leko V, Park GJ, Lao U, Simon JA, Bedalov A. Enterocyte-specific inactivation of SIRT1 reduces tumor load in the APC(+/min) mouse model. PLoS One. 2013; 8:e66283.
24. Lo Sasso G, Ryu D, Mouchiroud L, Fernando SC, Anderson CL, Katsyuba E, Piersigilli A, Hottiger MO, Schoonjans K, Auwerx J. Loss of Sirt1 function improves intestinal anti-bacterial defense and protects from colitis-induced colorectal cancer. PLoS One. 2014; 9:e102495.
25. Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009; 9:123–128.
26. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H, KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005; 7:469–483.
27. Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012; 122:639–653.
28. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014; 514:628–632.
29. Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009; 20:98–105.
30. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012; 149:656–670.
31. Sousa CM, Kimmelman AC. The complex landscape of pancreatic cancer metabolism. Carcinogenesis. 2014; 35:1441–1450.
32. Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, Boudreau A, Mroue R, Corson L, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A. 2015; 112:E4410–4417.
33. Grimont A, Pinho AV, Cowley MJ, Augereau C, Mawson A, Giry-Laterriere M, Van den Steen G, Waddell N, Pajic M, Sempoux C, Wu J, Grimmond SM, Biankin AV, et al. SOX9 regulates ERBB signalling in pancreatic cancer development. Gut. 2015; 64:1790–1799.
34. Joo HY, Yun M, Jeong J, Park ER, Shin HJ, Woo SR, Jung JK, Kim YM, Park JJ, Kim J, Lee KH. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1alpha (HIF-1alpha) via direct interactions during hypoxia. Biochem Biophys Res Commun. 2015; 462:294–300.
35. Wardell SE, Ilkayeva OR, Wieman HL, Frigo DE, Rathmell JC, Newgard CB, McDonnell DP. Glucose metabolism as a target of histone deacetylase inhibitors. Mol Endocrinol. 2009; 23:388–401.
36. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011; 42:719–730.
37. Bhalla K, Hwang BJ, Dewi RE, Ou L, Twaddel W, Fang HB, Vafai SB, Vazquez F, Puigserver P, Boros L, Girnun GD. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011; 71:6888–6898.
38. Huang B, Cheng X, Wang D, Peng M, Xue Z, Da Y, Zhang N, Yao Z, Li M, Xu A, Zhang R. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1alpha signaling. Oncotarget. 2014; 5:4732–4745. doi: 10.18632/oncotarget.1963.
39. Cheng D, Zhao L, Xu Y, Ou R, Li G, Yang H, Li W. K-Ras promotes the non-small lung cancer cells survival by cooperating with sirtuin 1 and p27 under ROS stimulation. Tumour Biol. 2015; 36:7221–7232.
40. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014; 39:91–100.
41. Stenzinger A, Endris V, Klauschen F, Sinn B, Lorenz K, Warth A, Goeppert B, Ehemann V, Muckenhuber A, Kamphues C, Bahra M, Neuhaus P, Weichert W. High SIRT1 expression is a negative prognosticator in pancreatic ductal adenocarcinoma. BMC Cancer. 2013; 13:450.
42. Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, Lou Z, Chini EN. Role of Deleted in Breast Cancer 1 (DBC1) Protein in SIRT1 Deacetylase Activation Induced by Protein Kinase A, AMP-activated Protein Kinase. J Biol Chem. 2012; 287:23489–23501.
43. Qin B, Minter-Dykhouse K, Yu J, Zhang J, Liu T, Zhang H, Lee S, Kim J, Wang L, Lou Z. DBC1 functions as a tumor suppressor by regulating p53 stability. Cell Rep. 2015; 10:1324–1334.
44. Li L, Wang L, Wang Z, Ho Y, McDonald T, Holyoake TL, Chen W, Bhatia R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell. 2012; 21:266–281.
45. Arrowsmith CH, Bountra C Fau - Fish PV, Fish Pv Fau - Lee K, Lee K Fau -Schapira M, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012; 11:384–400.
46. Sussmuth SD, Haider S, Landwehrmeyer GB, Farmer R, Frost C, Tripepi G, Andersen CA, Di Bacco M, Lamanna C, Diodato E, Massai L, Diamanti D, Mori E, et al. An Exploratory Double blind, Randomised Clinical Trial with Selisistat, a SirT1 Inhibitor, in Patients with Huntington’s Disease. Br J Clin Pharmacol. 2014.
47. Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, Kamphorst JJ, Rabinowitz JD, Jain SK, Hidalgo M, Dang CV, Gillies RJ, Maitra A. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of p53 Function. Cancer Res. 2015; 75:3355–3364.
48. Zhang Q, Zhang Y, Zhang P, Chao Z, Xia F, Jiang C, Zhang X, Jiang Z, Liu H. Hexokinase II inhibitor, 3-BrPA induced autophagy by stimulating ROS formation in human breast cancer cells. Genes Cancer. 2014; 5:100–112. doi: 10.18632/genesandcancer.9.
49. Li H, Rajendran GK, Liu N, Ware C, Rubin BP, Gu Y. SirT1 modulates the estrogen-insulin-like growth factor-1 signaling for postnatal development of mammary gland in mice. Breast Cancer Res. 2007; 9:R1.
50. Biankin AV, Kench JG, Colvin EK, Segara D, Scarlett CJ, Nguyen NQ, Chang DK, Morey AL, Lee CS, Pinese M, Kuo SC, Susanto JM, Cosman PH, et al. Expression of S100A2 calcium-binding protein predicts response to pancreatectomy for pancreatic cancer. Gastroenterology. 2009; 137:558–568, 568 e551–511.
51. Nones K, Waddell N, Song S, Patch AM, Miller D, Johns A, Wu J, Kassahn KS, Wood D, Bailey P, Fink L, Manning S, Christ AN, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014; 135:1110–1118.