

INTRODUCTION
Uveal melanoma is the most common primary cancer of the eye and the second most common form of melanoma. Due to a high rate of metastasis, much research has focused on the development of biomarkers to predict metastatic risk. Previously, we described a gene expression profile that could be performed on a fine needle biopsy of the primary tumor that accurately predicted metastasis [1]. Tumors with the Class 1 profile have a low metastatic risk, whereas those with the Class 2 profile have a high metastatic risk. Consequently, a 15 gene array (12 discriminating genes and 3 control genes) was developed and prospectively validated [2, 3]. This assay is now available commercially as the DecisionDx-UMTM test (Castle Biosciences), which has been independently validated [4] and is widely used to stratify patients for metastatic surveillance and to identify high risk patients for adjuvant therapy trials [5].
While the vast majority of metastatic events in uveal melanoma arise from Class 2 tumors, a small subset of Class 1 tumors also give rise to metastasis. We found that the expression of two of the 12 discriminating genes on the array (CDH1 and RAB31) could be used to identify Class 1 tumors that may have increased metastatic risk. Class 1 tumors with low expression of these genes and very low predicted metastatic risk were called Class “1A,” whereas those with high expression and higher predicted metastatic risk were called Class “1B.” In our efforts to further improve the prognostic accuracy of the gene array platform, we conducted a genome wide search for new biomarkers and found that mRNA expression of the cancer-testis antigen Preferentially Expressed Antigen in Melanoma (PRAME) was an accurate biomarker for metastasis in Class 1 tumors [6]. In that initial study, we found that any detectable mRNA expression of PRAME above baseline was associated with increased metastatic risk. Limitations of that study included a relatively small number of tumor samples that were biased towards larger tumor size.
To date, there have been five common driver mutations identified in uveal melanoma: BAP1, EIF1AX, GNA11, GNAQ and SF3B1 [7–11]. Mutations in BAP1, SF3B1 and EIF1AX are almost mutually exclusive and are associated with high, intermediate and low metastatic risk, respectively [6, 12]. Also, SF3B1 mutations were found to be associated with PRAME expression [6].
The purpose of the present study was to study PRAME expression in a much larger number of Class 1 and, for the first time, in Class 2 uveal melanomas spanning the true range of tumor sizes encountered in clinical practice. We sought to define a threshold value for calling a tumor sample positive for PRAME expression (PRAME+), compare PRAME expression to the 1A/1B designation in Class 1 tumors, identify clinical and molecular factors associated with PRAME expression, evaluate the prognostic value of PRAME expression in Class 2 tumors, and determine whether PRAME expression in uveal melanoma is correlated with promoter hypomethylation.
RESULTS
To evaluate the spectrum of PRAME mRNA expression and to establish a threshold for positive PRAME expression in primary uveal melanoma, we analyzed qPCR data from 678 tumor samples, including 123 of our samples and 555 de-identified samples submitted from a large number of ocular oncology centers to Castle Biosciences. These samples included 454 (67.0%) Class 1 tumors and 224 (33.0%) Class 2 tumors. Class 1 tumors included 317 (69.8%) Class 1A tumors, 131 (28.9%) Class 1B tumors, and 6 (1.3%) tumors for which 1A/1B information was not available. Whereas most samples showed negligible PRAME expression, a subset of samples showed a broad range of PRAME expression (Figure 1A). We previously showed that any PRAME expression above baseline was associated with increased metastasis in Class 1 tumors and consequently defined any expression above baseline as positive PRAME expression (PRAME+) [6]. In this study, we used the same methodology to establish a broadly applicable PRAME+ threshold from qPCR data using a much larger dataset that included both Class 1 and Class 2 tumors, with a majority derived from fine needle biopsy of small and medium sized tumors and a smaller number from large, enucleated specimens that is representative of the actual distribution of tumor sizes encountered in clinical practice (Figure 1B–1C). A similar method was used to determine a PRAME+ threshold using RNA-Seq data from The Cancer Genome Atlas (TCGA) dataset (Figure 1D).
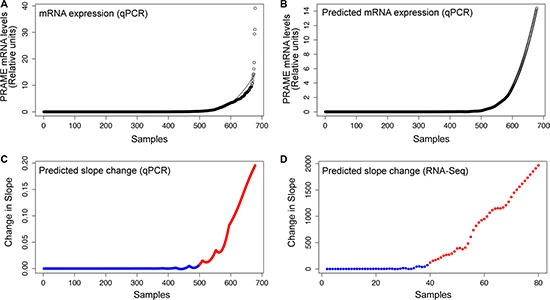
Figure 1: Defining the threshold for PRAME+ expression status. (A) PRAME mRNA expression plotted from lowest to highest expression for 678 uveal melanoma samples measured by qPCR with a LOESS model (second degree, family = ”Gaussian”, spanning 0.4, fitting by least-squares). (B) Predicted PRAME mRNA expression for an additional “hypothetical” 678 samples based on the LOESS model. (C) Predicted slope change between each of these predicted points. (D) The same process depicted in panels A–C was repeated separately for the RNA-Seq data from 80 TCGA uveal melanoma samples in order to generate a predicted slope change plot. For both datasets, the threshold for PRAME+ (red) was defined as the point where the slope sustainably rose above baseline (blue).
Overall, 498 (73.5%) tumors were PRAME− and 180 (26.5%) were PRAME+. Class 1 tumors were more likely to be PRAME−, whereas Class 2 tumors were more likely to be PRAME+ (Fisher exact test, P < 0.0001) (Figure 2A). Among Class 1 tumors, 357 (78.6%) were PRAME− and 97 (21.4%) were PRAME+. Among Class 1A tumors, 261 (82.3%) were PRAME− and 56 (17.7%) were PRAME+. Among Class 1B tumors, 93 (71.0%) were PRAME− and 38 (29.0%) were PRAME+. Class 1A tumors were more likely to be PRAME−, whereas Class 1B tumors were more likely to be PRAME+ (Fisher exact test, P = 0.01) (Figure 2B). Among Class 2 tumors, 141 (62.9%) were PRAME− and 83 (37.1%) were PRAME+. Additionally, we determined PRAME mRNA status in commonly used UM cell lines: Mel202 and MP41 are PRAME+, whereas 92.1, Mel270, Mel290, and MP46 are PRAME−.
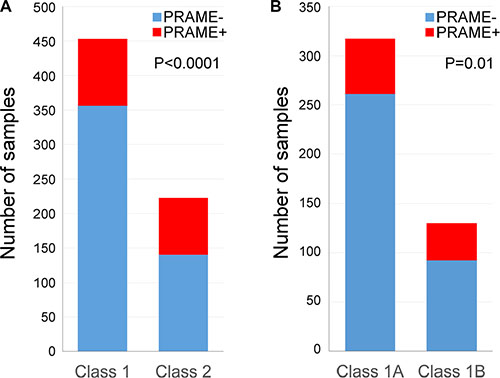
Figure 2: Summary of PRAME expression status measured by qPCR. (A) PRAME expression status with respect to gene expression profile classification in 678 uveal melanomas. (B) PRAME expression status in 454 Class 1 uveal melanomas with respect to 1A/1B sub-classification.
Association between PRAME and clinical features
Clinical annotations and PRAME expression were available for 123 of our uveal melanoma samples (Supplementary Table S1). The only features that were significantly associated with PRAME+ status were larger tumor diameter and thickness (Mann-Whitney test, P = 0.01 and P = 0.02, respectively). To expand this analysis, we examined the TCGA Research Network dataset which consists of an independent cohort of 80 primary uveal melanoma samples (http://cancergenome.nih.gov/) (Supplementary Table S2). Since PRAME expression data were obtained from RNA-Seq analysis in the TCGA dataset, we established a threshold for PRAME+ expression using the same procedure as for qPCR data (Figure 1D). Consistent with our original dataset, PRAME+ status in the TCGA dataset was associated with larger tumor diameter (P = 0.02). In both datasets, well-known risk factors for metastasis such as increased patient age, ciliary body involvement, and extrascleral tumor extension were not associated with PRAME expression status.
We previously showed that PRAME expression was associated with increased metastatic risk in Class 1 uveal melanomas [6]. Here, we extend that analysis to determine whether PRAME expression is also a biomarker for metastasis in Class 2 tumors. For this analysis, we combined our 123 cases with the 80 cases from the TCGA. Using Kaplan-Meier survival analysis, PRAME+ status was associated with shorter time to metastasis for both Class 1 and Class 2 tumors together (P = 0.0002; 22 metastatic events; median follow-up of 19 months, range 0–125 months) and for Class 2 tumors alone (P = 0.01; 15 metastatic events; median follow-up of 18 months, range 0–89 months)(Figure 3A–3B). Similarly, PRAME+ status was associated with shorter time to melanoma-specific mortality for both Class 1 and Class 2 tumors together (P = 0.001; 32 melanoma-specific mortality events; median follow-up of 19 months, range 0–142 months) and for Class 2 tumors alone (P = 0.02; 28 melanoma-specific mortality events, median follow-up of 18 months, range 0–89 months) (Figure 3C–3D).
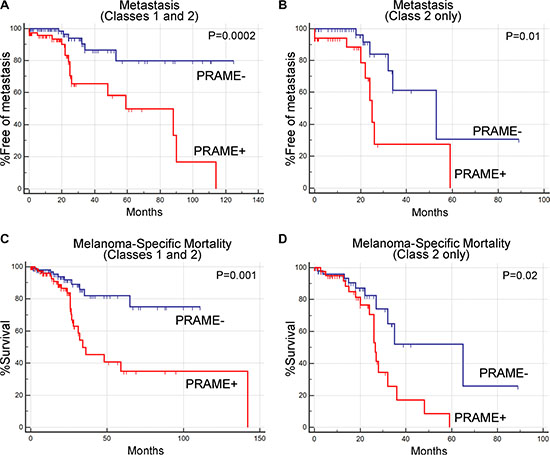
Figure 3: Prognostic significance of PRAME expression status in uveal melanoma. (A) Kaplan-Meier survival plot showing metastasis-free survival for Class 1 and Class 2 tumors combined, with respect to PRAME expression status. (B) Kaplan-Meier survival plot showing metastasis-free survival for Class 2 tumors only, with respect to PRAME expression status. (C) Kaplan-Meier plot showing melanoma-specific mortality for Class 1 and Class 2 tumors combined, with respect to PRAME expression status. (D) Kaplan-Meier survival plot showing melanoma-specific mortality for Class 2 tumors only, with respect to PRAME expression status.
Association between PRAME and chromosomal alterations
To identify chromosomal copy number changes that may be associated with PRAME expression, we analyzed 26 of our cases and 80 TCGA cases for which chromosomal copy number and PRAME expression data were available (Supplementary Table S3). Overall, PRAME+ tumors were strongly associated with 6q loss (P < 0.0001), 8p loss (P = 0.04), 8q gain (P < 0.0001) and 16q loss (P < 0.0001) (Figure 4). Notably, PRAME expression status was not associated with monosomy 3 (P = 0.3), the chromosomal alteration most strongly associated with metastasis in uveal melanoma, highlighting the potential benefit of including PRAME expression status in a prognostic test.
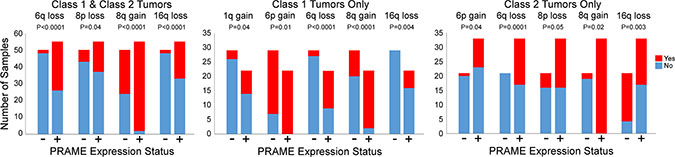
Figure 4: Association of PRAME expression with chromosomal gains and losses. The bar graphs depict chromosomal gains and losses that were significantly associated with PRAME+ tumors when Class 1 and Class 2 tumors were analyzed together, and when each class was analyzed separately. PRAME+ (red), PRAME− (blue).
We then analyzed Class 1 and Class 2 tumors separately. Among Class 1 tumors, PRAME+ status was associated with 1q gain (P = 0.04), 6p gain (P = 0.01), 6q loss (P < 0.0001), 8q gain (P < 0.0001), and 16q loss (P = 0.004). PRAME+ status in Class 2 tumors was associated with 6p gain (P = 0.04), 6q loss (P = 0.0001), 8p loss (P = 0.05), 8q gain (P = 0.02) and 16q loss (P = 0.003). PRAME expression was not associated with monosomy 3 in either comparison (Supplementary Table S3).
Association between PRAME and driver gene mutations
To identify common driver mutations that may be associated with PRAME+ status, we analyzed 59 of our cases for which mutation data were available, as well as the 80 TCGA cases, for mutations in EIF1AX, BAP1, GNA11, GNAQ and SF3B1 (Supplementary Table S4). When Class 1 and Class 2 tumors were considered together, PRAME+ status was associated with BAP1 mutations (P = 0.02). However, this association is likely due to BAP1 mutations occurring almost exclusively in Class 2 tumors [9], which we show here to be associated with PRAME+ status. When Class 1 tumors were analyzed separately, PRAME expression was directly associated with SF3B1 mutations (P < 0.0001) and inversely associated with EIF1AX mutations (P = 0.004). There were no mutations associated with PRAME expression in Class 2 tumors when analyzed separately.
PRAME expression is associated with aberrant promoter hypomethylation
Testes is the only normal adult tissue that expresses PRAME mRNA at appreciable levels (Figure 5A), which strongly suggests that the expression of PRAME in uveal melanoma is anomalous. Consequently, we hypothesized that PRAME may become aberrantly activated in uveal melanoma by hypomethylation of the promoter region. Consistent with this hypothesis, 12 CpG sites within and near the PRAME promoter were significantly hypomethylated (FDR < 0.05 for all probes) in PRAME+ tumors compared to PRAME− tumors (Figure 5B). We validated these findings using bisulfite conversion followed by Sanger sequencing in a subset of cases (Supplementary Table S5). Strikingly, there was a highly significant correlation between the level of hypomethylation of all 12 CpG sites and the level of mRNA expression (P < 0.0001) (Figure 5C and Supplementary Figure S1). The most differentially methylated CpG site (recognized by probe cg27303185) is hypermethylated in all adult tissues except placenta and sperm (Figure 5D). These data indicate that the PRAME promoter region is normally hypermethylated and silenced in virtually all normal adult tissues, but it is targeted for hypomethylation and aberrant transcriptional activation during uveal melanoma progression.
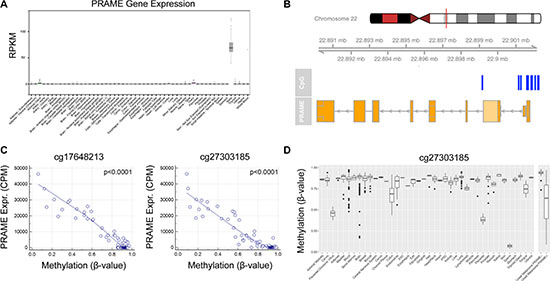
Figure 5: Transcriptional activation of PRAME is associated with hypomethylation of the PRAME promoter in uveal melanoma. (A) The only normal adult human tissue that expresses high levels of PRAME mRNA is testis. Data were obtained through the GTEx Portal [41]. (B) Locations of 12 CpG sites (blue bars) within or near the PRAME promoter that exhibited significantly decreased methylation in PRAME+ uveal melanomas (n = 41) compared to PRAME- samples (n = 39) at a significance level of FDR < 0.05. (C) Scatter plots showing the relationship between PRAME mRNA expression levels (obtained from TCGA RNA-Seq data) and PRAME promoter methylation (obtained from TCGA Infinium HumanMethylation450 BeadChip data) using two representative methylation probes (cg17648213 and cg27303185). Spearman’s rank correlation coefficient was used to determine P-values. Graphs depicting the other 10 differentially methylated probes are in Supplementary Figure S1. (D) Methylation data for the cg27303185 methylation probe was plotted for normal tissues obtained from Marmal-aid [40]. A separate panel (right) depicts PRAME+ and PRAME- uveal melanomas samples for comparison. RPKM, reads per kilobase of transcript per million mapped reads; CPM, counts per million.
DISCUSSION
We previously reported that PRAME mRNA expression is a significant risk factor for metastasis in Class 1 uveal melanomas, and we developed a general method for establishing a PRAME+ threshold in various datasets [6]. In that article, our analysis included a much greater proportion of large tumors treated by enucleation than are encountered in actual clinical practice. However, since we show here that PRAME expression is strongly associated with larger tumor size, a study composed primarily of large tumors may not accurately reflect the true range of PRAME expression. To pursue the development of PRAME as a clinical biomarker, we sought here to rigorously establish a standard method for defining the PRAME+ expression threshold using a standardized and widely used qPCR platform. To achieve a widely applicable threshold and avoid potential systematic biases arising from a single center study, we analyzed a large number of samples obtained from many different ocular oncology centers representing a wide range of tumor sizes and both Class 1 and Class 2 tumors in proportions representative of actual clinical practice. From this analysis, we established a PRAME+ threshold and used it to identify clinical, chromosomal and mutational features associated with PRAME expression. We also established a PRAME+ threshold for RNA-Seq using the TCGA dataset, but this threshold must be considered provisional since that dataset was composed primarily of very large tumors treated by enucleation. Indeed, 43% of the TCGA samples were PRAME+, compared to only 27% of our samples.
Across all samples, larger tumor size was the only clinical feature that correlated with PRAME+ status, suggesting that PRAME becomes transcriptionally activated later during tumor progression. Interestingly, even though PRAME+ status was a stronger predictor of metastasis in Class 1 tumors, it was also associated with metastasis in Class 2 tumors. In our earlier study that was much smaller and biased towards larger tumors treated by enucleation, we did not find a correlation between PRAME expression status and the “1A/1B” system that the clinical test currently uses to indicate low (1A) versus moderate (1B) metastatic risk [6]. In the present study that included a much larger number of samples that better represented the full spectrum of uveal melanomas, we observed a highly significant correlation between PRAME+ status and “1B” status. Nevertheless, since there were a number of discordant cases, we are preparing to start a multi-center prospective study to determine the relative prognostic value of PRAME expression status versus the 1A/1B designation in Class 1 tumors in order to determine the optimal biomarker for increased metastatic risk in Class 1 tumors. A limitation of this analysis is the limited follow-up, particularly from the TCGA dataset, which results in a large number of censored data points. Our planned prospective multicenter study with long follow-up is the appropriate study design to validate these findings. Since we found that PRAME+ correlates significantly with tumor size, this multi-center study will also evaluate whether there is a minimum threshold tumor size at which point PRAME becomes prognostic.
PRAME expression was associated with specific chromosomal gains and losses, some of which were specific to either Class 1 or Class 2 tumors. Changes that were associated with PRAME+ status in both Class 1 and Class 2 tumors included 6p gain, 6q loss, 8q gain and 16q loss. 6p gain and 6q loss were frequently found in the same tumor samples, likely representing the formation of an isochromosome 6p [13, 14]. A previous study identified 16q loss in 16% of uveal melanomas, but no prognostic significance was found [14]. Our study using a larger number of samples and more accurate molecular analytical methods indicates that 16q loss may indeed have prognostic significance. 1q gain was associated with PRAME+ status only in Class 1 tumors, which confirms our previous observation [6]. 1q gain has only rarely been mentioned in the uveal melanoma literature [15], but our findings suggest the need for further studies to determine whether 1q gain has pathogenic as well as prognostic significance. 8p loss was associated with PRAME+ status only in Class 2 tumors, whereas 8q gain was associated with PRAME+ status in both tumor classes. 8q gain is prevalent in both Class 1 and Class 2 tumors, but the mechanism leading to 8q gain tends to be different between the two tumor classes [16]. In Class 1 tumors, 8q gain often occurs through gain of an entire copy of chromosome 8 or by simple gain of the q arm, whereas in Class 2 tumors, 8q gain frequently occurs through formation of an isochromosome 8q, which is accompanied by loss of 8p [17]. The common association of PRAME expression and isochromosome formation on chromosomes 6 and 8 is of interest and may provide new insight into uveal melanoma tumorigenesis. We previously showed that genes which become aberrantly up-regulated in PRAME+ tumors are enriched for functions related to chromosome maintenance, meiotic recombination and telomere maintenance [6]. In addition, the PRAME protein has been shown to associate at transcriptional target sites on chromatin with the KEOPS/EKC complex [18], which is involved in chromosome segregation, telomere maintenance and other highly conserved functions [19]. Hence, aberrant expression of PRAME may predispose tumor cells to isochromosome formation, as well as other forms of aneuploidy that promote tumor progression.
Our finding that PRAME becomes aberrantly hypomethylated and transcriptionally activated during uveal melanoma progression is similar to findings in other cancers [20, 21] and may have therapeutic implications. Since PRAME is not normally expressed in most normal adult tissues, targeted molecular inhibition of the PRAME protein or immunotherapy directed against PRAME−expressing tumor cells may be well tolerated. Indeed, there is growing evidence that PRAME may be a good target for immunotherapy [22–25]. Since the PRAME protein is not normally expressed on the cell surface, one strategy is to target PRAME using a T-cell receptor mimic (TCRm) monoclonal antibody that recognizes the PRAME300–309 peptide presented by HLA*A02:01 on the cell surface [26]. Others have developed PRAME−specific cytotoxic T lymphocytes that have shown effective responses against PRAME-expressing tumor cells, including progenitor populations that are notoriously resistant to current cancer therapeutic strategies [24, 27]. Furthest along in development are vaccines against PRAME that are currently undergoing clinical trials in cutaneous melanoma and other cancers (Trial numbers NCT01149343, NCT01853878 and NCT00423254) [28]. Interestingly, we evaluated PRAME expression of two matched primary and metastatic UM samples analyzed by the Illumina HumanRef-8 v1.0 expression microarray in our previously published dataset (GEO accession number GSE39717) [29], and we found that both the primary and metastatic samples were PRAME+ (data not shown), supporting a mechanistic role for PRAME expression in UM metastasis. Since no effective therapies currently exist for metastatic uveal melanoma [30], our center and others are preparing to undertake clinical trials to assess the efficacy of PRAME-directed immunotherapy in appropriately selected patients.
In summary, we have provided a threshold for PRAME+ expression from qPCR data for primary uveal melanomas across a wide spectrum of tumor sizes and in both tumor classes representative of actual clinical practice. We previously identified PRAME expression as a biomarker for increased metastatic risk in Class 1 tumors [6], and here we showed for the first time that PRAME expression is also associated with worse prognosis among Class 2 tumors. We demonstrated that specific chromosomal gains and losses, as well as specific driver mutations, are found preferentially in PRAME+ tumors. Finally, we showed that specific CpG sites around the PRAME promoter are differentially hypomethylated in PRAME+ tumors, suggesting that the aberrant transcriptional activation of PRAME in uveal melanoma is the result of epigenetic reprogramming during tumor progression. In addition to its prognostic value, PRAME expression status may potentially be useful in the future for guiding the use of PRAME-directed immunotherapy, which would make PRAME the first true “companion prognostic” biomarker in uveal melanoma.
MATERIALS AND METHODS
The sources of all uveal melanoma samples used in this study are summarized in Supplementary Table S6. Tumor samples were obtained from 123 primary uveal melanomas from the practice of one of the authors (JWH), including 64 samples that were included in a previous publication [6]. The research was conducted in a HIPAA-compliant manner in accordance with the tenets of the Declaration of Helsinki. Approval was obtained from the Institutional Review Board of the University of Miami. Written informed consent was obtained from each patient from our center. Baseline clinical information and patient outcomes were recorded. De-identified PRAME expression and GEP Class data were obtained from 555 uveal melanoma samples from Castle Biosciences that had been collected between July 21, 2015, and March 2, 2016, as part of internal PRAME qPCR method development. These samples were obtained as formalin-fixed paraffin-embedded tissue from enucleations in 55 (9.9%) cases and as fresh-frozen samples from fine needle aspirate biopsies in 500 (90.1%) cases. The data available for these cases included GEP class, 1A versus 1B subtype for Class 1 tumors, and PRAME mRNA expression. Additionally we analyzed clinical, whole exome sequencing, RNA sequencing, SNP 6.0 array data, and DNA methylation data from 80 uveal melanoma samples generated by the TCGA Research Network: http://cancergenome.nih.gov/.
PRAME mRNA expression analysis
For the RNA samples from our center and from Castle Biosciences, PRAME mRNA expression was analyzed by qPCR using the Applied Biosystems 7900 HT Real-Time PCR System with TaqMan primers and Gene Expression Master Mix following the manufacturer’s protocol as previously described [6]. Ct values were calculated using the manufacturer’s software and ΔCt values were calculated by subtracting the geometric mean of the Ct values of the endogenous control genes from the mean Ct values for PRAME, as previously described [6]. Relative normal expression was calculated using the equation 2^-ΔCT. For the 80 samples from the TCGA, raw RNA-Seq datasets were aligned to the hg19 genome using STAR [31], which was also used to generate count files. Count files were then normalized using DeSeq2 [32]. Next-generation sequencing analysis was conducted on Pegasus, the supercomputer administered by the Advanced Computing Group of the Center for Computational Science at the University of Miami. For PRAME mRNA expression in normal tissues, RNA-Seq data was obtained from the Genotype-Tissue Expression (GTEx) project [33].
Estimating class status from RNA-sequencing data
For research purposes of this study, we estimated the gene expression profile class assignment for the 80 TCGA samples, which were analyzed by RNA-Seq. Raw RNA-Seq datasets were prepared using the pipeline described in the previous section. The top 20% most variable genes were selected, analyzed by principal component analysis, and plotted using the stats, matrixstats, and rgl packages, respectively, in R (version 3.2.3). This analysis grouped the samples into two clusters, as we have previously described for Class 1 and Class 2 tumors [34]. The identity of each cluster was determined to be most consistent with Class 1 versus Class 2 based on the expression of genes previously known to be differentially up-regulated in each Class. The DecisionDx-UM test results were available for 11 of these samples, and there was 100% concordance with our class assignment. This method was used solely for research purposes and is not meant for actual clinical testing, as it has not been prospectively validated in a manner analogous to the DecisionDx-UM test.
Determining PRAME+ expression threshold
qPCR and RNA-Seq samples were separately ordered from lowest to highest relative and normalized PRAME expression, respectively, and each was plotted with a line representing the best-fitting LOESS model (second degree, family = ”Gaussian”, spanning 0.4 for qPCR and 0.45 for RNA-Seq, fitting by least-squares) (Figure 1A). Based on the LOESS model, a predicted dataset fitting the LOESS model was generated (Figure 1B) and the slope between each predicted point was calculated and plotted (Figure 1C) to represent the change in slope. The point of inflection where the slope sustainably rose above baseline was defined as the cut-off for PRAME+ and PRAME− (Figure 1C–1D).
Exome sequencing and chromosomal copy number analysis
Whole-exome sequencing was conducted on 24 of our primary uveal melanomas and matched blood using NimbleGen SeqCap EZ Human Exome Library v2.0 (Roche Nimblegen) and run on the Illumina Genome Analyzer II. Exome sequencing data on 80 primary uveal melanoma TCGA samples were downloaded from CGHub and aligned to the hg19 reference genome using Novoalign. Variant calling was conducted using Mutect2 [35] and Varscan2 [36]. Chromosomal copy number analysis was obtained for 106 samples, including 26 samples from our center (15 from previously published data and 11 newly analyzed from exome sequencing data) and 80 from the TCGA. Chromosomal gains and losses were called by CNVKit [37] for exome sequencing data and by ASCAT [38] for TCGA SNP 6.0 array data.
DNA methylation analysis
The 80 TCGA uveal melanoma tumors samples were assayed for global DNA methylation status with the Infinium HumanMethylation450K BeadChip (Illumina). This kit interrogates ~450,000 methylation sites at single-nucleotide resolution, including at CpG sites within promoter, 5′UTR, first exon, gene body, and 3′UTR regions. Methylation data underwent quality control, normalization, and differential analysis of PRAME+ and PRAME− samples using the ChAMP methylation pipeline in R [39]. CpG sites that were differentially hypomethylated at a significance level of FDR < 0.05 were plotted along the PRAME locus using the GViz package in R. All 12 methylation probes targeting PRAME that are included in the Methyl450K array were significantly hypomethylated in the TCGA PRAME+ samples.
For validation, primers were designed against a region containing 3 of these 12 probes and validated in 4 PRAME+ and 3 PRAME− samples. This validation study was small due to limited sample availability. For primer design, 500 ng of tumor DNA was bisulfite converted using the EZ Methylation-Lightning Kit (Zymo Research). Primers for PCR amplification of the PRAME promoter were designed with the Bisulfite Primer Seeker (http://www.zymoresearch.com/tools/bisulfite-primer-seeker). Forward Primer: GAAGGATTTCGTGTTTAAGGTTTTTTAAGG. Reverse Primer: GTGTTTTTATTTTGGAAATAGAGATTTAGT TTTTTTT. The PRAME promoter region was amplified with the EpiMark Hot Start Taq polymerase (New England Biolabs) at Tm = 54.5°C, and the PCR product purified by agarose gel separation/elution before Sanger sequencing. The status of the PRAME methylation site detected by Infinium HumanMethylation450K BeadChip probe cg27303185 in normal tissues was obtained from Marmal-aid [40] and plotted in a box-and-whisker with ggplot2 in R in comparison to TCGA uveal melanoma data.
Statistical analysis
Statistical analysis was performed using Medcalc® version 14.10.2. Fisher’s exact test was used to evaluate discrete dichotomous variables, the Mann-Whitney test for comparison of continuous variables, Spearman’s rho for correlation analyses of continuous variables, and Kaplan-Meier survival analysis for determining the association of PRAME expression status with patient outcomes.
ACKNOWLEDGMENTS
The authors acknowledge the support of the Oncogenomics Core and the Biostatistics and Bioinformatics Core at the Sylvester Comprehensive Cancer Center, and the Center for Computational Science at the University of Miami for help and support with the data management and analytics.
CONFLICTS OF INTEREST
Dr. Harbour is the inventor of intellectual property related to the gene expression profile technology used in the study. Drs. Harbour and Bowcock are the inventors of intellectual property related to the discovery of BAP1 mutations in uveal melanoma. Dr. Harbour is a paid consultant for Castle Biosciences, which licensed this intellectual property, and he receives royalties from its commercialization. Kristen Oelschlager and Dr. John Stone are employees and stockholders of Castle Biosciences. No other authors disclose a conflict of interest.
GRANT SUPPORT
This work was supported by National Cancer Institute grants R01 CA125970 (J.W.H.), R01 CA161870 (J.W.H. and A.M.B.) and F30 CA206430 (M.G.F.), Research to Prevent Blindness, Inc. Senior Scientific Investigator Award (J.W.H.), Melanoma Research Foundation (J.W.H., M.G.F.), Melanoma Research Alliance (J.W.H.), Ocular Melanoma Foundation (J.W.H.), the 2015 RRF/Kayser Global Pan-American Award (J.W.H.), the Sylvester Comprehensive Cancer Center, the University of Miami Sheila and David Fuente Graduate Program in Cancer Biology (M.G.F., M.A.D.), the Center for Computational Science Fellowship (M.G.F.), and the AACR-Ocular Melanoma Foundation Fellowship in honor of Robert C. Allen, MD (S.K.). The Bascom Palmer Eye Institute also received funding from NIH Core Grant P30EY014801, Department of Defense Grant #W81XWH-13-1-0048, and a Research to Prevent Blindness Unrestricted Grant.
REFERENCES
1. Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004; 64:7205–7209.
2. Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010; 12:461–468.
3. Onken MD, Worley LA, Char DH, Augsburger JJ, Correa ZM, Nudleman E, Aaberg TM, Jr., Altaweel MM, Bardenstein DS, Finger PT, Gallie BL, Harocopos GJ, et al. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology. 2012; 119:1596–1603.
4. Correa ZM, Augsburger JJ. Sufficiency of FNAB aspirates of posterior uveal melanoma for cytologic versus GEP classification in 159 patients, and relative prognostic significance of these classifications. Graefes Arch Clin Exp Ophthalmol. 2014; 252:131–135.
5. Aaberg TM, Jr., Cook RW, Oelschlager K, Maetzold D, Rao PK, Mason JO, 3rd. Current clinical practice: differential management of uveal melanoma in the era of molecular tumor analyses. Clinical ophthalmology (Auckland, NZ). 2014; 8:2449–2460.
6. Field MG, Decatur CL, Kurtenbach S, Gezgin G, van der Velden PA, Jager MJ, Kozak KN, Harbour JW. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin Cancer Res. 2016; 22:1234–1242.
7. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009; 457:599–602.
8. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010; 363:2191–2199.
9. Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010; 330:1410–1413.
10. Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013; 45:133–135.
11. Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, Lohmann DR, Zeschnigk M. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013; 45:933–936.
12. Decatur CL, Ong E, Garg N, Anbunathan H, Bowcock AM, Field MG, Harbour JW. Driver mutations in uveal melanoma: associations with gene expression profile and patient outcomes. JAMA ophthalmology. 2016.
13. Prescher G, Bornfeld N, Friedrichs W, Seeber S, Becher R. Cytogenetics of twelve cases of uveal melanoma and patterns of nonrandom anomalies and isochromosome formation. Cancer Genet Cytogenet. 1995; 80:40–46.
14. Kilic E, van Gils W, Lodder E, Beverloo HB, van Til ME, Mooy CM, Paridaens D, de Klein A, Luyten GP. Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci. 2006; 47:3703–3707.
15. Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci. 2001; 42:313–317.
16. Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res. 2008; 14:115–122.
17. Onken MD, Worley L, Harbour JW. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res. 2008; 14:3737–3745.
18. Costessi A, Mahrour N, Sharma V, Stunnenberg R, Stoel MA, Tijchon E, Conaway JW, Conaway RC, Stunnenberg HG. The human EKC/KEOPS complex is recruited to Cullin2 ubiquitin ligases by the human tumour antigen PRAME. PLoS One. 2012; 7:e42822.
19. Srinivasan M, Mehta P, Yu Y, Prugar E, Koonin EV, Karzai AW, Sternglanz R. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011; 30:873–881.
20. Qian J, Zhu ZH, Lin J, Ming Yao DM, Li Y, Yang J, Wang CZ. Hypomethylation of PRAME promoter is associated with poor prognosis in myelodysplastic syndrome. Br J Haematol. 2011; 154:153–155.
21. Schenk T, Stengel S, Goellner S, Steinbach D, Saluz HP. Hypomethylation of PRAME is responsible for its aberrant overexpression in human malignancies. Genes Chromosomes Cancer. 2007; 46:796–804.
22. Gerard C, Baudson N, Ory T, Segal L, Louahed J. A comprehensive preclinical model evaluating the recombinant PRAME antigen combined with the AS15 immunostimulant to fight against PRAME-expressing tumors. J Immunother. 2015; 38:311–320.
23. Ikeda H, Lethe B, Lehmann F, van Baren N, Baurain JF, de Smet C, Chambost H, Vitale M, Moretta A, Boon T, Coulie PG. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997; 6:199–208.
24. Amir AL, van der Steen DM, van Loenen MM, Hagedoorn RS, de Boer R, Kester MD, de Ru AH, Lugthart GJ, van Kooten C, Hiemstra PS, Jedema I, Griffioen M, van Veelen PA, et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin Cancer Res. 2011; 17:5615–5625.
25. Spel L, Boelens JJ, van der Steen DM, Blokland NJ, van Noesel MM, Molenaar JJ, Heemskerk MH, Boes M, Nierkens S. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget. 2015; 6:35770–35781. doi: 10.18632/oncotarget.5657.
26. Chang A, Dao T, Scott A, Dubrovsky L, Liu C, Scheinberg DA. A Therapeutic TCR Mimic Monoclonal Antibody for Intracellular PRAME Protein in Leukemias. Blood. 2015; 126:2527–2527.
27. Quintarelli C, Dotti G, Hasan ST, De Angelis B, Hoyos V, Errichiello S, Mims M, Luciano L, Shafer J, Leen AM, Heslop HE, Rooney CM, Pane F, et al. High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood. 2011; 117:3353–3362.
28. Weber JS, Vogelzang NJ, Ernstoff MS, Goodman OB, Cranmer LD, Marshall JL, Miles S, Rosario D, Diamond DC, Qiu Z, Obrocea M, Bot A. A phase 1 study of a vaccine targeting preferentially expressed antigen in melanoma and prostate-specific membrane antigen in patients with advanced solid tumors. J Immunother. 2011; 34:556–567.
29. Worley LA, Onken MD, Person E, Robirds D, Branson J, Char DH, Perry A, Harbour JW. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007; 13:1466–1471.
30. Luke JJ, Triozzi PL, McKenna KC, Van Meir EG, Gershenwald JE, Bastian BC, Gutkind JS, Bowcock AM, Streicher HZ, Patel PM, Sato T, Sossman JA, Sznol M, et al. Biology of advanced uveal melanoma and next steps for clinical therapeutics. Pigment Cell Melanoma Res. 2015; 28:135–147.
31. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21.
32. Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013; 8:1765–1786.
33. Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660.
34. Labrecque MP, Takhar MK, Jagdeo JM, Tam KJ, Chiu C, Wang TY, Prefontaine GG, Cox ME, Beischlag TV. A TRIP230-Retinoblastoma Protein Complex Regulates Hypoxia-Inducible Factor-1alpha-Mediated Transcription and Cancer Cell Invasion. PLoS One. 2014; 9:e99214.
35. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013; 31:213–219.
36. Koboldt DC, Larson DE, Wilson RK. Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Curr Protoc Bioinformatics. 2013; 44:15 14 11–17.
37. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016; 12:e1004873.
38. Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, Perou CM, Borresen-Dale AL, Kristensen VN. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010; 107:16910–16915.
39. Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics. 2014; 30:428–430.
40. Lowe R, Rakyan VK. Marmal-aid—a database for Infinium HumanMethylation450. BMC Bioinformatics. 2013; 14:359.
41. Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, Guan P, Korzeniewski GE, Lockhart NC, et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and biobanking. 2015; 13:311–319.