
INTRODUCTION
Triple negative breast cancer (TNBC) refers to the subgroup of breast cancers that are negative for estrogen receptor (ER), progesterone receptor (PR), and HER2. It accounts for about 15% of invasive breast cancer diagnosis and is often associated with high tumor grade, aggressive clinical course, and adverse prognosis [1]. Compared to the availability of targeted agents for ER+ or HER2+ breast cancer, treatment for TNBC is limited to cytotoxic chemotherapy which is often ineffective, leading to recurrence and death [1, 2]. Development of molecularly targeted agents is therefore in great need [2].
A major advance in recent years is the improved understanding of the genetic basis of TNBC through genome-wide investigations including The Cancer Genome Atlas (TCGA) and others [3–5]. A prominent finding in TNBC from these studies was the lack of common driver mutations identified in ER+ or HER2+ disease, and the loss of key regulators of cell cycle checkpoints, including mutations in TP53 (84%), loss/mutations of RB1 (20%) and germline and/or somatic mutations in BRCA1 or BRCA2 (20%), as well as gain of cell cycle promoting genes such as amplifications of CCNE1 (9%) and c-MYC (40%), among others [3]. Therefore targeting cell cycle regulatory proteins presents an attractive therapeutic strategy in TNBC [6–8].
Cyclin dependent kinases (CDKs) are key positive regulators of cell cycle progression, transcription, and mRNA processing [6, 8]. Inhibitors against CDK4/6, which induce G1 to S phase cell cycle arrest, have demonstrated therapeutic efficacy in both endocrine therapy naïve and resistant ER+ breast cancer [9–11]. Palbociclib, the first selective CDK4/6 inhibitor introduced in the clinic has received FDA approval to combine with letrozole as first line therapy and to combine with fulvestrant after disease progression on prior hormonal therapy for patients with metastatic ER+ HER2- breast cancer [10–12]. However, TNBC is often resistant to CDK4/6 inhibition in preclinical studies [9, 13], likely as a result of deficiency in RB [3]. In contrast, inhibitors against other CDKs have shown promising activity [8, 14].
Dinaciclib is a novel, potent, small molecule inhibitor against CDK1, CDK2, CDK5, and CDK9, with IC50 values of 3, 1, 1, and 4 nmol/L, respectively [15]. Compared with flavopiridol, the first CDKs inhibitor to enter in clinical trials, dinaciclib exhibited superior activity with a better side effects profile [15]. Dinaciclib has shown promising antitumor activity in preclinical studies in broad spectrum of cell lines with median IC50 of 11nM and in early phase clinical trials of hematological and solid malignancies including breast cancer [16–27]. In cell line studies, dinaciclib was found to be particularly effective in TNBC with elevated MYC [14]. However the molecular mechanisms underlying the sensitivity of dinaciclib in TNBC are not fully understood. In this study, we aimed to investigate the activity of dinaciclib in patient derived xenograft models and to further investigate the mechanisms of action of dinaciclib to uncover novel therapeutic targets in TNBC. We demonstrated that dinaciclib was highly effective in inhibiting tumor growth in TNBC PDXs in vitro and in vivo. Importantly, we demonstrated a reduction in the protein levels of cyclin B1 and MYC upon dinaciclib treatment through inhibition of CDK9, suggesting CDK9, MYC and cyclin B1 as potential therapeutic targets in TNBC.
RESULTS
Dinaciclib inhibited TNBC cell growth in vitro
We first performed 2D clonogenic assay to determine the effect of dinaciclib on in vitro cell growth of 3 PDX models as described in materials and methods [WHIM3 (WT TP53), WHIM12 (mutant TP53) and WHIM21 (mutant TP53)], 3 TNBC cell lines [BT549 (mutant TP53), MDA-MB-231 (mutant TP53), and HCC1806 (mutant TP53), and 3 ER+ cell lines [BT474 (WT TP53), MCF7 (WT TP53) and T47D (mutant TP53)]. Dinaciclib at 10nM concentration significantly inhibited the colony formation of all TNBC models (Figure 1A). In contrast dinaciclib only partially inhibited the colony formation of ER+ cell lines (Figure 1B), suggesting that TNBC may be particularly sensitive to the growth inhibitory effect of dinaciclib. The growth inhibitory effect of dinaciclib on TNBC was further confirmed in the 3D matrigel assay (Figure 1C).
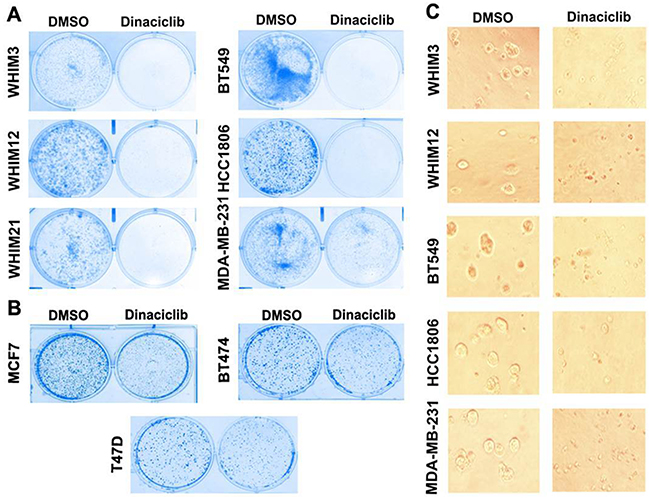
Figure 1: Dinaciclib inhibits cell proliferation of TNBC more effectively than that of ER+ breast cancer. A. Colony formation assay of TNBC from PDXs (WHIM3, WHIM12, and WHIM21) and cell lines (BT549, HCC1806, MDA-MB-231) in the presence of DMSO or 10mM dinaciclib. B. Colony formation assay of ER+ breast cancer cell lines (BT474, MCF7 and T47D) in the presence of DMSO or 10nM dinaciclib. C. Three-D matrigel assay of TNBC from PDXs (WHIM3, WHIM12, and WHIM21) and cell lines (BT549, HCC1806, MDA-MB-231) in the presence of DMSO or 10nM dinaciclib.
Dinaciclib induced apoptosis in TNBC
To determine the mechanism of cell growth inhibition in TNBC, we next examined whether dinaciclib was able to induce apoptosis in vitro. Cells were treated with 50nM dinaciclib for 24 hours followed by quantification of apoptotic cells identified by double-staining with PI and Annexin V by FACS analysis. Dinaciclib induced apoptosis in all TNBC cells tested, including those from PDX models (Figure 2A and 2B). Interestingly, a more dramatic induction of apoptosis by dinaciclib was observed in WHIM12 compared to WHIM3. This effect could be related to high basal expression levels of Cyclin B1 and CDK1 in WHIM12 as compared to WHIM3 (data not shown). To determine the mechanism of apoptosis induction, we investigated the levels of apoptotic and anti-apoptotic proteins following dinaciclib treatment by western blot. Dinaciclib treatment decreased the levels of the anti-apoptotic protein survivin, which is a known CDK1 target [28, 29] and increased in the levels of the cleaved PARP (Figure 2C and 2D). In contrast, treatment with dinaciclib led to little induction of apoptosis in the ER+ cell lines, including MCF7 and T47D cells (Supplementary Figure S1A). There was no reduction in the levels of survivin nor increase in the levels of cleaved PARP observed in MCF-7 and T47D following dinaciclib treatment (Supplementary Figure S2A).
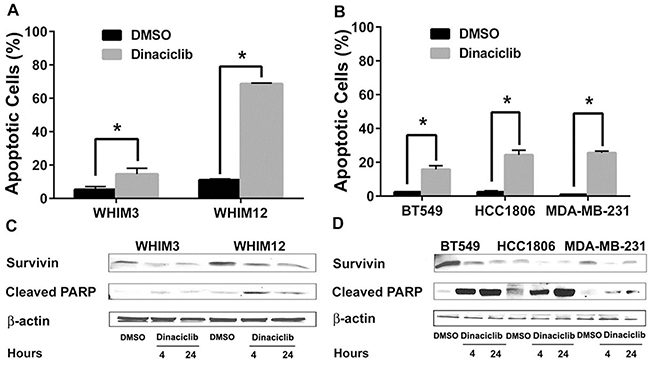
Figure 2: Dinaciclib induced apoptosis in TNBC from PDXs and cell lines. A. and B. FACS analysis of TNBC cells from PDXs (WHIM3 and WHIM12) and cell lines (BT549, HCC1806, MDA-MB-231) treated with dinaciclib for 24 hours. Percentage of apoptosis (the ratio of Annexin-V and PI-positive cells) was then calculated. Data are presented as average percentage of apoptotic cells ± SEM from 3 experiments. C. and D. Western blot analysis of apoptosis associated proteins after dinaciclib treatment for 4 and 24 hours. * p<0.05.
Dinaciclib induced G2/M phase cell cycle arrest in TNBC in vitro
The effect of dinaciclib on cell cycle distribution of TNBC was examined in vitro. TNBC cells were treated with 50nM dinaciclib for 24 hours and analyzed by FACS. Dinaciclib treatment led to an accumulation of cells in G2/M phase in all TNBC cell lines tested (Figure 3A, 3B, 3C, 3D and 3E). We then analyzed the protein levels of known cell cycle associated proteins, including CDK1, cyclin B1, Rb and c-MYC [6] by western blot to investigate the mechanisms of cell arrest by dinaciclib. As expected, a significant decrease in pCDK1T14/15, and pRbS807/811 was observed in all TNBC cell lines following 4-24 hours of exposure to dinaciclib (Figure 3F and 3G). Interestingly, a reduction in the level of cyclinT1 (a binding partner of CDK9), c-MYC and cyclin B1 was also observed post treatment with dinaciclib. Since cyclin B1 is important for CDK1 activation needed for G2 to M phase transition, the reduction in cyclin B1 by dinaciclib could be particularly important for the effect of dinaciclib in TNBC.
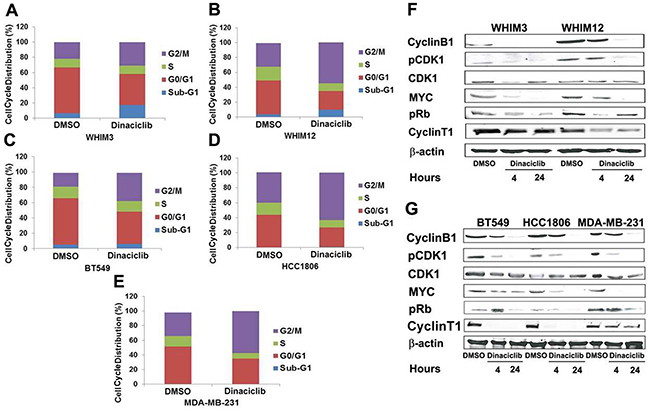
Figure 3: Dinaciclib promotes G2/M phase arrest in TNBC from PDXs and cell lines. A, B, C, D. and E. Cell-cycle analysis by FACS of PI- stained cells treated with dinaciclib for 24 hours. Data in each panel represented the average cell cycle distribution from 3 independent experiments. F. and G. Western blot analysis of cell-cycle associated proteins after dinaciclib treatment for 4 and 24 hours.
Knockdown of cyclin B1 and CDK1 inhibited cell proliferation in TNBC
Since cyclin B1 in complex with CDK1 drives the progression of cell cycle from G2 to M phase [6], we hypothesized that the inhibitory effect of dinaciclib on CDK1 and cyclin B1 are important for its growth inhibitory effect in TNBC. SiRNA knockdown of CDK1 and cyclin B1 in MDA-MB-231 and BT549 significantly inhibited cell proliferation compared to that of control siRNA as determined by Alamar blue assay (Figure 4B) and colony formation assay (Figure 4C). We next determined the effect of CDK1 and cyclin B1 SiRNA knockdown on cell cycle distribution and apoptosis in BT549 and MDA-MB-231 to determine the mechanism of cell growth inhibition. Both CDK1 and cyclin B1 knockdown promoted significant apoptosis (Figure 4D) and cell cycle arrest in G2/M phase (Figure 4E). These data indicates an important role of cyclin B1 and CDK1 in regulating cell growth and survival in TNBC.
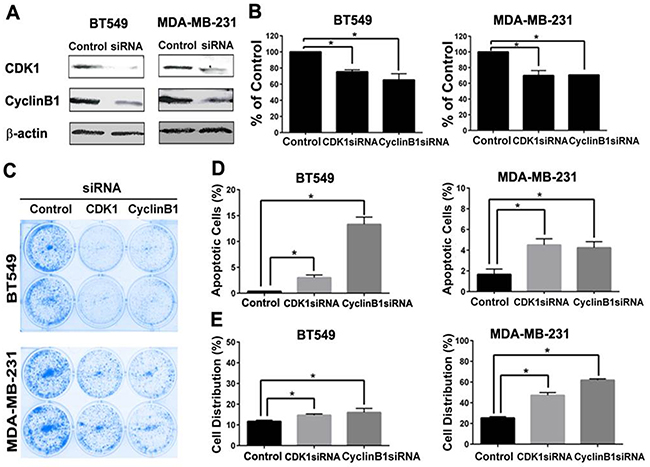
Figure 4: CDK1 or cyclin B1 knockdown inhibited cell proliferation, induced apoptosis and G2/M arrest in TNBC cells. A. Western blot analysis of CDK1 and cyclin B1 protein level after siRNA knockdown in BT549 and MDA-MB-231. Effect of CDK1 and cyclin B1 knockdown on cell proliferation by Alamar Blue cytotoxic assay B. and colony formation C. apoptosis induction by Annexin V staining D. and G2/M arrest by flow cytometry analysis of cell cycle distribution E. were determined. * p<0.05.
Dinaciclib targets cyclin B1 through CDK9-MYC axis in breast cancer cells
We next investigated the mechanisms of cyclin B1 inhibition by dinaciclib. Since cyclin B1 is a direct transcriptional target of MYC [30] and CDK9, a key component of the positive transcription elongation factor b (P-TEFb) complex [8], required for MYC transcriptional activation of target genes, including MYC itself [26, 31], we hypothesized that dinaciclib reduced cyclin B1 protein expression via CDK9 inhibition in TNBC. As shown in Figure 5A and 5B, knockdown of CDK9, instead of CDK1 or CDK2, by RNAi reduced the levels of cyclin B1 in both BT549 and MDA-MB-231 cells. In addition, knockdown of CDK9 inhibited colony formation in BT549 and MDA-MB-231 cells (Figure 5C), suggesting its potential as a therapeutic target in TNBC. The correlation between cyclin B1 and MYC gene copy number was also observed in clinical breast cancer samples, supporting the role of MYC in regulating cyclin B1 expression (Figure 5D).
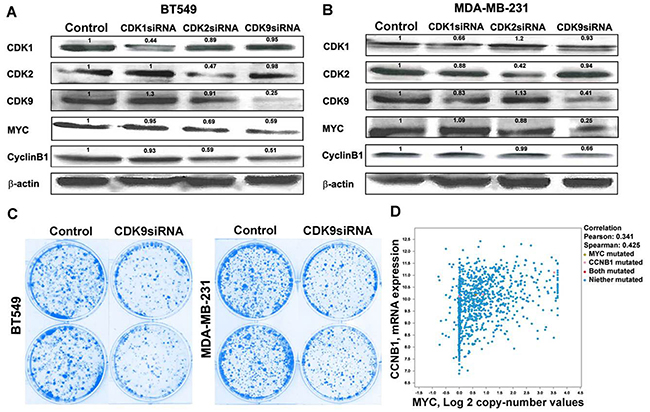
Figure 5: Dinaciclib inhibits cyclin B1 via targeting CDK9-cMYC axis. Western blot analysis of CDK1, CDK2, and CDK9 protein levels after siRNA knockdown in A. BT549 and B. MDA-MB-231 and the effect of CDK1, CDK2, and CDK9 knockdown on cMYC and cyclin B1 protein levels in BT549 and MDA-MB-231 cells. C. Effect of CDK9 knockdown on colony formation in BT549 and MDA-MB-231 cells. D. Correlation between cyclin B1 mRNA expression level and cMYC copy number in clinical breast cancer patients.
Dinaciclib inhibited tumor growth in TNBC patient derived xenografts tumor model in vivo
The therapeutic efficacy of dinaciclib was further investigated in TNBC PDX models in vivo. TNBC WHIM12 PDX model was chosen because of its particular sensitivity for apoptotic induction by dinaciclib in vitro. Dinaciclib or vehicle was administered daily i.p. at 50 mg/kg, days 1-5 each week for 4 weeks in mice bearing WHIM 12 (n=6 each treatment group). A significant inhibition in tumor growth was observed (Figure 6A). At the end of the experiments, tumors were harvested for biomarker analysis. Dinaciclib significantly inhibited the cyclinT1, CDK9, c-MYC, cyclin B1 and survivin protein expression levels in tumor tissues (Figure 6B, n=3). In addition, dinaciclib treatment reduced mitotic entry assessed by IHC of phosphorylated Histone H3, a marker of mitosis, and increased apoptosis assessed by IHC of cleaved PARP significantly (Figure 6D).
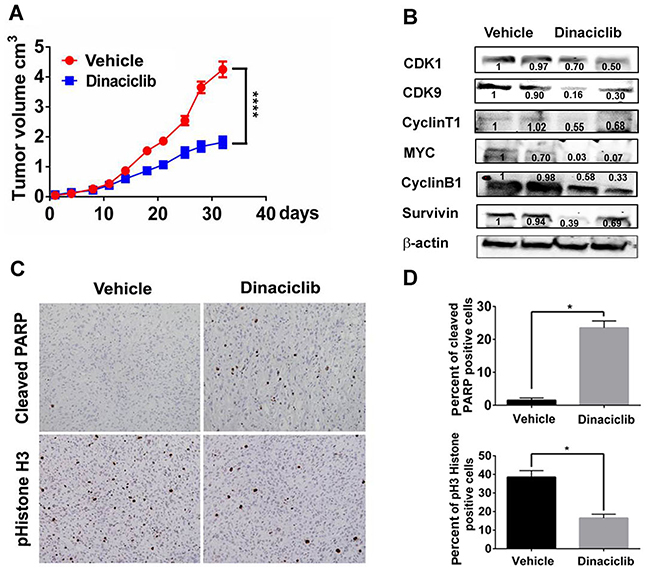
Figure 6: Dinaciclib inhibits tumor growth in a TNBC PDX model in vivo. A. Tumor growth curve of mice bearing WHIM12 PDX treated with vehicle or dinaciclib monotherapy. B. Tumor lysates of WHIM12 from vehicle and dinaciclib treated mice were subjected to Western blot analysis of indicated markers. C. Immunohistochemistry analysis for phospho-Histone H3, a marker for mitosis on tumor tissue obtained from vehicle and dinaciclib treated mice. D. Quantification of the percentage of tumor cells staining positive for cleaved PARP and phospho-Histone H3. * p<0.05
Cyclin B1 gene expression levels negatively correlated with overall survival rate in ER negative breast cancer
Next we examined the correlation between expression levels of cyclin B1, CDK1 and overall survival in ER-negative breast cancer patients. We used the on-line tool (www.kmplot.com) which has a combined data set from various annotated breast cancer studies [27]. The Kaplan–Meier plots were generated by the mRNA expression levels of Cyclin B1 and CDK1 for OS of breast cancer patients who received adjuvant chemotherapy. We observed that cyclin B1 gene expression levels negatively correlated with overall survival (OS) in patients treated with adjuvant chemotherapy. (Figure 7A). But there was no significant correlation observed between CDK1 gene expression and OS in these patients (Figure 7B).
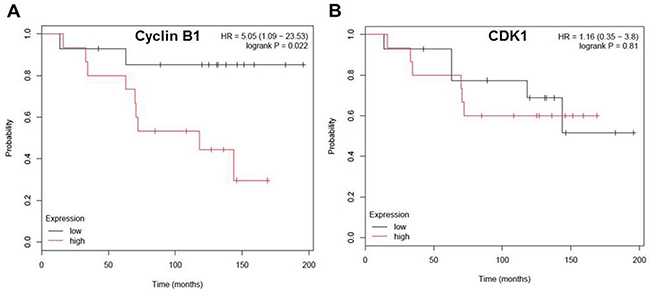
Figure 7: Cyclin B1 expression level negatively correlates with overall-survival rate in patients with ER negative breast cancer. A. Kaplan-Meier survival curve indicating the correlation between CDK1 expression and overall-survival in chemotherapy treated ER- breast cancer patients. B. Kaplan-Meier survival curve indicating the correlation between cyclin B1 expression and overall-survival in chemotherapy treated ER- breast cancer patients.
DISCUSSION
CDKs play critical roles in regulating cell cycle transition and gene transcription, which are frequently altered in cancer [32]. Therefore CDKs have attracted much attention in recent years as potential cancer therapeutic targets and selective CDK4/6 inhibitors which potently inhibit the G1 to S phase transition have established their role in the clinic for patients with advanced ER+ breast cancer [32]. However CDK4/6 inhibitors are often ineffective in treating TNBC, likely due to loss of Rb in this cancer type. Interestingly, dinaciclib, an inhibitor against CDK1, 2, 5, and 9 (IC50 1-4nM), but with less activity against CDK4, 6, and 7 (IC50 60-100nM), showed encouraging anti-tumor effect in preclinical studies of TNBC with potential selectivity for MYC driven tumors [13, 14]. However, the clinical activity of dinaciclib in unselected patients with metastatic breast cancer has been modest [22], indicating a need to understand its mechanisms of action for the development of more selective CDK inhibitors in TNBC.
TNBC presents a significant clinical challenge because of the limited therapeutic options. Molecularly targeted agents are in great need, but the development of these agents has been challenging. Unlike ER+ and HER2+ breast cancer, mutations in TNBC often occur in tumor suppressor genes such as TP53, RB1, and PTEN [3], which are not readily targetable. The presence of amplification and/or overexpression of the MYC oncogene in TNBC offer an exciting therapeutic opportunity. However, being a transcription factor, MYC targeting with small molecule inhibitors has been difficult to design and develop [33]. Our data suggests the possibility of targeting CDK9 as a strategy for MYC driven TNBC.
Our study demonstrates that dinaciclib, a serine/threonine kinase inhibitor against CDK1, 2, 5, and 9, inhibited tumor growth in TNBC PDXs and cell line models in vitro and in vivo, by arresting cells in the G2-M phase and inducing apoptosis. We demonstrated for the first time that exposure to dinaciclib reduced the expression of MYC and its transcriptional target cyclin B1 in TNBC. To further investigate the underlying molecular mechanisms, we performed RNAi experiments to individually knockdown CDK1, CDK2, and CDK9 and showed that CDK9, but not CDK1 or CDK2, was responsible for reducing the protein levels of MYC and cyclin B1. Although RNAi knockdown of each of the CDKs (CDK1, CDK2, and CDK9) reduced TNBC tumor growth, and likely that dinaciclib exerted its anti-tumor activity in TNBC by inhibiting all the above CDKs, the finding that CDK9 knock down reduced the expression levels of MYC and Cyclin B1, as well as, tumor growth. Our data on CDK9 identifies that CDK9 inhibition is an important mechanism the anti-tumor effect of dinaciclib and establishes CDK9 as a potential therapeutic target for MYC driven TNBC.
The importance of the proto-oncogene MYC in tumorigenesis has been well established [33–36]. It is a helix-loop-helix leucine zipper transcription factor that regulates the expression of a variety of downstream genes important for cell cycle progression, apoptosis, metabolism and cellular transformation [33, 35, 36]. Previous studies demonstrated that MYC enhances transcriptional output by recruiting the CDK9/P-TEFb complex to specific promoters [37–39], and stimulating transcript elongation via pause release [40, 41]. CDK9 as a therapeutic target for MYC driven cancer was demonstrated in a shRNA library screening in which CDK9 was required for the aberrant proliferation of MYC overexpressing hepatocellular carcinoma cell lines [42]. There was a significant correlation between response to CDK9 inhibition and MYC mRNA expression, and CDK9 inhibition impaired RNA Pol II Ser2 phosphorylation and transcription elongation of MYC and MYC target genes. This finding was also observed in cell lines from lung and hematopoietic cancers [26, 42]. Our data, for the first time, demonstrated that CDK9 could be an important therapeutic target in TNBC. Further studies are ongoing to further confirm the synthetic lethal interaction between CDK9 inhibition and MYC overexpression/amplification in TNBC using selective CDK9 inhibitors.
Our study suggests that MYC downstream targets cyclin B1 and CDK1 may be particularly important in controlling the proliferation of TNBC. MYC has been considered the direct regulator of cell cycle progression because of its essential role in regulating the expressions of cyclins, including cyclin B1, CDKs and inhibition of CDK inhibitors [33, 35, 36, 43, 44]. C-MYC gain has been observed in 40% of TNBC [3]. MYC activation is a characteristic of basal-like breast cancer, the most common molecular subtype of TNBC and is associated with poor prognosis [14, 45, 46]. We demonstrated that inhibition of CDK1 or cyclin B1 by RNAi to reduced tumor cell proliferation in TNBC. In addition, cyclin B1 overexpression was found to correlate with poor clinical outcomes in our study and in others [47]. This finding is also supported by a previous study demonstrating that CDK1 inhibition but not CDK4/6 or CDK2 was selectively lethal to MYC-dependent breast cancer cells [13]. The relevance of CDK1 and cyclin B1 could also be due to their increased levels of gene expression by mutant p53 [7]. Our data also suggest that TNBC may be particularly sensitive to agents that target the G2 to M phase transition.
The effect of dinaciclib on MYC level through inhibition of CDK9 in MYC-driven tumor cells was reported previously in a study of B cell lymphoma cell lines [26]. Although dinaciclib was found to have a preferential growth inhibitory effect in MYC overexpressing TNBC cell lines [14], the dependency of MYC overexpressing TNBC on CDK1 was unclear. Our study confirmed the activity of dinaciclib in TNBC using PDX models and importantly, provided an explanation for the sensitivity of TNBC to dinaciclib and indicated that CDK9 inhibition as potential therapeutic strategy for MYC overexpressing TNBC. Selective CDK9 inhibitors are in preclinical and clinical development and impressive anti-tumor activity has been observed in chronic lymphocytic leukemia cell lines [48]. Studies of selective CDK9 inhibitors in TNBC are warranted. In addition, inhibitors against CDK7, which also plays an important role in phosphorylating the C-terminal domain of RNA polymerase II [49–51] and CDK9 [52], are being developed [53] and could also be important to investigate in TNBC.
MATERIALS AND METHODS
Chemicals
Dinaciclib were purchased from (ChemieTek and Sigma). Dinaciclib were prepared in stock solution of 10mM in Dimethyl sulphoxide (Sigma) for in-vitro experiments.
In vitro clonogenic assay
We selected 2 human TNBC PDX models (WHIM3 and WHIM12) that differed in TP53 status for our study. The WHIM3 and WHIM12 were generated by engrafting the primary breast tumor of a patient with metastatic TNBC into the humanized mammary fat pad of NOD/SCID mice [28, 30]. We also used 3 TNBC cell lines (BT549, HCC1806 and MDA-MB-231) and 3 ER+ cell lines (BT474, MCF7 and T47D) for our study. Cells were cultured in RPMI-1640 medium with 10% FBS at 37°C in an atmosphere of 5% CO2 and 95% air as described previously [54]. For clonogenic assay cells were seeded at a density of 1000 cells per well in 6 well plates in RPMI-1640 medium with 10% FBS for 24 hours. Cells were then treated with dinaciclib at 10nM concentration or dimethyl sulphoxide (DMSO) for 6 days. The colonies were fixed with 4% paraformaldehyde and stained with 0.05% crystal violet in methanol.
Three-Dimensional Culture in Matrigel
Overlay 3D culture of cells in Matrigel was done as described previously [55]. Cells were seeded in growth factor–reduced Matrigel (BD Biosciences) in 8-well chamber slides. Dinaciclib 10nM or DMSO was added at the time of cell seeding. Cell medium were changed every 3 days and cells were imaged by photomicrograph on day 6.
siRNA knockdown
Cells were seeded at a density of 200,000 cells per well in 6 well plate in RPMI-1640 medium with 10%FBS. Next day cells were transfected with 50nM CDK1, CDK2, CDK9 and cyclinB1 siRNA (Santacruz) using Lipofectamine 2000 (Invitrogen). Cells were harvested after 48 hours of transfection for western analysis and colony formation assay.
Western blotting
Cells were seeded in a 6-well plate at the density of 0.5 × 106, adhered overnight followed by treatment with dinaciclib at 50nM concentration for 0, 4, and 24 hours. Cells were harvested after 4 and 24 hours and lysed with 100μL buffer containing 50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 2 mmol/L EDTA, 1% Triton, 1 mmol/L phenylmethylsulfonylfluoride, and Protease Inhibitor Cocktail (Sigma) for 20 minutes on ice. Lysates were cleared at 10,000 rpm for 15 minutes, boiled, separated on 12% SDS gels, and transferred to a nitrocellulose membrane followed by overnight incubation with primary antibodies against pCDK1T14/15, CDK1, pCDK2T160, CDK2, CyclinB1, p21, pRbSer807/8011, Survivin, cMYC, cleaved-PARP, and β-actin. Protein bands were visualized after 1 hour incubation with HRP-conjugated secondary antibodies and development with ECL (GE Healthcare).
Antibodies
Primary antibodies were rabbit anti-pCDK1T14/15, mouse anti-CDK1, rabbit anti-pCDK2T160, rabbit anti-CDK2, mouse anti-cMYC, mouse anti-survivin and rabbit anti- β-actin (Santacruz); anti-p21, anti-pRb, and anti-cleaved-PARP (Cell Signaling); mouse anti-cyclinB1 (BD Transduction). Secondary horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse antibodies were from Jackson ImmunoResearch Laboratories.
Fluorescence-activated cell-sorting analysis of apoptosis and cell cycle
Apoptotic cells were visualized using Annexin V Detection Kit according to the manufacturer’s instructions (BD Pharmingen). Briefly, cells were treated with dinaciclib 50nM for 24 hours followed by incubation with fluorescein isothiocyante (FITC)-conjugated Annexin V and propidium iodide (PI) at room temperature for 15 minutes. After 15-minute incubation, samples were analyzed using AccuriC6 flow cytometer (BD Accuri Cytometers) and cell quest software. Cell-cycle phase distribution was determined in 70% alcohol-fixed cells stained with 5ul PI-RNase A staining buffer (Invitrogen) for 30 minutes at room temperature, followed by analysis using AccuriC6 flow cytometer (BD Accuri Cytometers) and cell quest software.
Animal studies
WHIM12 was characterized previously [56] and passaged in the “humanized” mammary fat pad of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice. Passage 4-5 xenografts of WHIM12 were used for the therapy experiments described in this study. A total of 1 × 106 tumor cells and 5 × 105 fibroblasts (2.5 × 105 that were exposed to 4 Gy IR and 2.5 × 105 untreated cells) were mixed and added to an equal volume of a 1:1 mixture of Matrigel (BD Biosciences, Cat. No. 354234) and Collagen I (Millipore, Cat. No. 08-115) to each side of the fourth mammary fat pad that has been cleared of any mouse mammary tissues in female NU/J homozygous mice (Jackson Laboratory, Cat. No. 2019) to propagate xenografts for tumor growth experiments. For long term treatment experiments, xenografts were allowed to grow to approximately 0.5 cm in the maximum diameter. Mice were then divided into 2 treatment groups (n = 6 mice in each group): Vehicle (vehicle dilutents) and dinaciclib alone was administered by intraperitoneal injection at a dose of 50 mg/kg/day, 5 days a week for total of 4 weeks. Tumor volume was measured in 2 dimensions (length and width) using Traceable Digital Calipers. The following formula was used to calculate tumor volume: tumor volume (cm3) = (length × width2) × 0.5. For IHC biomarker studies mice bearing WHIM12 were treated with either Vehicle (n = 2), and dinaciclib alone (n=2; 50 mg/kg/day, i.p. days 1, 2 and 3). Tumors were harvested 2 hours after day 3 therapy. Each xenograft tumor was cut into 2 pieces with one piece flash frozen for tumor lysate, and the second piece fixed in 10% neutral buffered formalin and embedded in paraffin blocks. All animal studies were carried out using the appropriate NIH animal care, and the animal study protocol was approved by the Animal Studies Committee of Washington University.
Immunohistochemistry
IHC for pHistone H3 and cleaved PARP were conducted on 5 μm tissue sections from paraffin-embedded tumor as described previously using the EnVision + Single Reagents HRP-Rabbit (Dako) and REAL substrate buffer (REAL DAB + chromogen, Dako)[28]. The primary antibodies and dilutions are as follows: pHistone H3 (Ser 10) antibody (1:200; Millipore), and cleaved PARP antibody (1:200, Cell Signaling).
Statistical analysis
Statistical analyses were conducted using Graphpad Prism software. Results are expressed as mean ± SEM. Statistical significance was determined by Student paired t test for in vitro data respectively. Tumor volume data were compared using one-way ANOVA. P ≤ 0.05 was considered significant.
ACKNOWLEDGMENTS
This work was supported by the Susan G Komen Investigator-Initiated Research Grant (IIR13263475) and funding from A Sister’s Hope Foundation to Dr. Cynthia Ma.
CONFLICTS OF INTEREST
There are no conflicts of interest to disclose.
GRANT SUPPORT
This work was supported by the Susan G Komen Investigator-Initiated Research Grant (IIR13263475) and funding from A Sister’s Hope Foundation to Dr. Cynthia Ma.
REFERENCES
1. Foulkes WD, Smith IE and Reis-Filho JS. Triple-Negative Breast Cancer. N Eng J Med. 2010; 363:1938-1948.
2. Hudis CA and Gianni L. Triple-Negative Breast Cancer: An Unmet Medical Need. The Oncologist. 2011; 16:1-11.
3. Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61-70.
4. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486:395-399.
5. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012; 486:400-404.
6. Sherr CJ. Cancer cell cycles. Science. 1996; 274:1672-1677.
7. Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D’Incalci M, Piccolo S, Veronesi A, Zambelli A, Del Sal G and Di Leo A. Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev. 2013; 39:541-550.
8. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006; 24:1770-1783.
9. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G and Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009; 11:R77.
10. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015; 16:25-35.
11. Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, Giorgetti C, Randolph S, Koehler M and Cristofanilli M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015.
12. Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, Zirkelbach JF, Yu J, Liu Q, Zhao L, Crich J, Chen XH, Hughes M, Bloomquist E, Tang S, Sridhara R, et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor–Positive, HER2-Negative Metastatic Breast Cancer. Clin Cancer Res. 2015; 21:4760-4766.
13. Kang J, Sergio CM, Sutherland RL and Musgrove EA. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC cancer. 2014; 14:32.
14. Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, Yaswen P, Mills GB, Esserman LJ and Goga A. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012; 209:679-696.
15. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, Wang Y, Oft M, Chen T, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010; 9:2344-2353.
16. Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H, Fendrich V, Rajeshkumar NV, Matsui W, Brossart P, Hidalgo M, Bannerji R, Maitra A, et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011; 12:598-609.
17. Gorlick R, Kolb EA, Houghton PJ, Morton CL, Neale G, Keir ST, Carol H, Lock R, Phelps D, Kang MH, Reynolds CP, Maris JM, Billups C and Smith MA. Initial testing (stage 1) of the cyclin dependent kinase inhibitor SCH 727965 (dinaciclib) by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012; 59:1266-1274.
18. Johnson AJ, Yeh YY, Smith LL, Wagner AJ, Hessler J, Gupta S, Flynn J, Jones J, Zhang X, Bannerji R, Grever MR and Byrd JC. The novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells. Leukemia. 2012; 26:2554-2557.
19. Desai BM, Villanueva J, Nguyen TT, Lioni M, Xiao M, Kong J, Krepler C, Vultur A, Flaherty KT, Nathanson KL, Smalley KS and Herlyn M. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PloS one. 2013; 8:e59588.
20. Gojo I, Sadowska M, Walker A, Feldman EJ, Iyer SP, Baer MR, Sausville EA, Lapidus RG, Zhang D, Zhu Y, Jou YM, Poon J, Small K and Bannerji R. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother Pharmacol. 2013; 72:897-908.
21. Nemunaitis JJ, Small KA, Kirschmeier P, Zhang D, Zhu Y, Jou YM, Statkevich P, Yao SL and Bannerji R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Transl Med. 2013; 11:259.
22. Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, Statkevich P, Zhu Y, Yao SL, Small K, Bannerji R and Shapiro CL. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. 2014; 14:169-176.
23. Stephenson JJ, Nemunaitis J, Joy AA, Martin JC, Jou YM, Zhang D, Statkevich P, Yao SL, Zhu Y, Zhou H, Small K, Bannerji R and Edelman MJ. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2014; 83:219-223.
24. Baker A, Gregory G, Verbrugge I, Kats L, Hilton JJ, Vidacs E, Lee EM, Lock RB, Zuber J, Shortt J and Johnstone RW. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-rearranged acute myeloid leukemia. Cancer Res. 2015.
25. Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S, Hessler J, Grever MR, Im E, Zhou H, Zhu Y, Zhang D, Small K, Bannerji R and Byrd JC. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia. 2015; 29:1524-1529.
26. Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, Lefebure M, Martin BP, Dawson MA, Johnstone RW and Shortt J. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2015; 29:1437-1441.
27. Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, Fruth B, Roy V, Erlichman C, Stewart AK and Mayo Phase C. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood. 2015; 125:443-448.
28. O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC and Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000; 97:13103-13107.
29. Castedo M, Perfettini JL, Roumier T and Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002; 9:1287-1293.
30. Yin XY, Grove L, Datta NS, Katula K, Long MW and Prochownik EV. Inverse regulation of cyclin B1 by c-Myc and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res. 2001; 61:6487-6493.
31. Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M, Wahl LM, Harris ED, Mikovits JA, Monks AP, Hollingshead MG, Sausville EA, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001; 2:RESEARCH0041.
32. Asghar U, Witkiewicz AK, Turner NC and Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015; 14:130-146.
33. Chen BJ, Wu YL, Tanaka Y and Zhang W. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int J Biol Sci. 2014; 10:1084-1096.
34. Henriksson M and Luscher B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Adv Cancer Res. 1996; 68:109-182.
35. Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999; 19:1-11.
36. Albihn A, Johnsen JI and Arsenian Henriksson M. (2010). Chapter 6 - MYC in Oncogenesis and as a Target for Cancer Therapies. In: George FVW and George K, eds. Advances in Cancer Research: Academic Press), pp. 163-224.
37. Eberhardy SR and Farnham PJ. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem. 2001; 276:48562-48571.
38. Kanazawa S, Soucek L, Evan G, Okamoto T and Peterlin BM. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene. 2003; 22:5707-5711.
39. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA and Young RA. c-Myc regulates transcriptional pause release. Cell. 2010; 141:432-445.
40. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI and Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012; 151:56-67.
41. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K and Levens D. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012; 151:68-79.
42. Huang CH, Lujambio A, Zuber J, Tschaharganeh DF, Doran MG, Evans MJ, Kitzing T, Zhu N, de Stanchina E, Sawyers CL, Armstrong SA, Lewis JS, Sherr CJ and Lowe SW. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014; 28:1800-1814.
43. Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A and Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003; 17:1115-1129.
44. Menssen A and Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 2002; 99:6274-6279.
45. Alles MC, Gardiner-Garden M, Nott DJ, Wang Y, Foekens JA, Sutherland RL, Musgrove EA and Ormandy CJ. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PloS one. 2009; 4:e4710.
46. Chandriani S, Frengen E, Cowling VH, Pendergrass SA, Perou CM, Whitfield ML and Cole MD. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PloS one. 2009; 4:e6693.
47. Aaltonen K, Amini RM, Heikkila P, Aittomaki K, Tamminen A, Nevanlinna H and Blomqvist C. High cyclin B1 expression is associated with poor survival in breast cancer. Br J Cancer. 2009; 100:1055-1060.
48. Walsby E, Pratt G, Shao H, Abbas AY, Fischer PM, Bradshaw TD, Brennan P, Fegan C, Wang S and Pepper C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget. 2014; 5:375-385. doi: 10.18632/oncotarget.1568.
49. Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D and Ansari AZ. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell. 2009; 34:387-393.
50. Drapkin R, Le Roy G, Cho H, Akoulitchev S and Reinberg D. Human cyclin-dependent kinase-activating kinase exists in three distinct complexes. Proc Natl Acad Sci U S A. 1996; 93:6488-6493.
51. Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP and Bentley DL. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009; 29:5455-5464.
52. Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL and Fisher RP. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012; 19:1108-1115.
53. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014; 511:616-620.
54. Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A, Aft RL, Ellis MJ and Piwnica-Worms H. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin. Invest. 2012; 122:1541-1552.
55. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER and Ellis MJ. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2013; 3:224-237.
56. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, Ding L, Griffith OL, Miller C, Larson D, Fulton RS, Harrison M, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013; 4:1116-1130.