
INTRODUCTION
Breast cancer (BC) is one of the most widespread carcinoma and one of the main causes of cancer-related death worldwide especially in women aged between 35 and 75 years [1]. In the last few years new molecular markers have been studied to provide new insights on BC heterogeneity but also to better understand and predict tumor behavior during treatment. It is now well established that BC can be classified into different groups according to gene expression profiles [2-5]. This new classification will certainly provide new insights into the BC biology and will probably drive treatment decisions in the near future by microarray analysis that soon will switch the clinical approach to different illnesses by giving a huge support to conventional pathology (morphology and immunohistochemistry) [6].
The different BC subgroups detected by their different gene expression profiling are below described as discussed in San Gallen Expert Consensus report:
Luminal A subgroup is characterized by estrogen/progesterone receptor (ER/PR) positivity, lower expression of Ki-67 ( < 20%) and HER2 lack, accounting for the 50% of all invasive BCs; Luminal B subgroup results characterized by ER/PR positivity or variable expression of HER2 (+ or -), accounting for the 10-20% of all invasive BCs [7]; HER2 overexpression subtype is characterized by ER/PR negativity and HER2 strong positivity. This subtype accounts for 15% of all invasive breast cancer; Basal like breast cancer (BLBC) subtype exhibits an expression profile similar to that of the epithelial cell mammary tissue and includes triple negative breast cancer (TNBC) [8-10].
Among the different subgroups, TNBC presents biological characteristics that confer higher aggressiveness and relapse risk along with worse outcome in comparison to other subgroups. Several studies showed that PI3K/AKT/mTOR signaling is often altered in TNBC patients [11].
SELECTION CRITERIA
We have searched trials using Medline (PUBMED), EMBASE and COCHRAINE database, the following search strategy (“TNBC” [MESH] AND (“PI3K” [tiab] OR “mTOR” [tiab]) AND (“ER” [tiab] OR “PgR” [tiab]) and free text terms as “PI3K” [tiab], “triple negative breast cancer” [tiab] and “mTOR” [tiab].
TRIPLE NEGATIVE BREAST CANCER
TNBC is characterized by lack of ER and PR expression as well as the absence of HER2 [12, 13]. The percentage of new TNBC diagnosis is variable, but it mainly ranges between 9% and 16% with a higher frequency in young women carrying BRCA1 gene mutation, showing a strong correlation with ethnic origin (in particular, African-American and Hispanic women) [14-17]. TNBC also shows greater size and tumor burden, and often is a more aggressive high grade tumor [18, 19].
TNBC patients show a higher susceptibility to develop metastases, resulting in an unfavorable clinical outcome compared to other subgroups [20-22].
Although TNBC patients initially respond to neoadjuvant treatments, only 30% of them will exhibit a survival higher than 5-years following the first diagnosis, reflecting the aggressiveness of this subtype [23, 24]. Patients with BRCA1 mutation are often diagnosed with TNBC but not all TNBC are BRCA1 positive. Nevertheless, it been shown that TNBC not carrying BRCA1 mutation, behave similarly to BRCA1-deficient tumors, showing also similar gene expression profiles [25, 26].
The growing interest in the TNBC biology allowed to develop trials investigating new drug targeting potential biomarkers such as VEGF, EGFR, Src and mTOR [27]. Moreover, the introduction of tumor molecular features in the characterization of TNBC has led to a further subtype labeling. Indeed, 6 new TNBC subtypes have been identified [28]:
Basal-like 1 (BL1) and Basal-like 2 (BL2) subtypes: both are characterized by up-regulation of gene and cellular markers mainly implicated in cell growth. In fact, Ki-67 expression and nuclear fraction staining are higher (BL1+BL2 = 70%) if compared to other subtypes (42%). All these features combined together indicate that more efficient treatment for BL TNBCs could be that directed against the mitotic apparatus such as a taxane-based therapy [29-32]. Furthermore, BL2 shows the involvement of a different plethora of growth factors and receptors (EGF, EGFR, NGF, MET, Wnt/β-catenin, IGF1R and EPHA2).
Immunomodulatory subtype: it is characterized by immune system gene signature similar to that of medullar BC determining its better clinical outcome.
Mesenchymal and mesenchymal stem-like subtypes: both are characterized by increased expression of gene and cellular markers involved in cell motility (Rho pathway), extracellular matrix-receptor interaction and differentiation (Wnt/β-catenin, ALK, TGF-β pathways). The mesenchymal stem-like subtype shows reduced expression of proliferative genes and enrichment of genes involved in several signaling pathways, including the inositol phosphate-dependent signaling pathway, EGFR, PDGF, and ERK1/2 signaling. Moreover, notable is the contribute of the adipocytokine signaling and ABC transporter. Both subtypes exhibit gene expression pattern and chemoresistance similar to metaplastic BC [28].
Luminal androgen receptor (LAR) subtype: this TNBC subgroup is ER-negative and is characterized by high deregulation of hormone-dependent pathways. In particular, the androgen receptor pathway seems to play a pivotal role in inducing expression of specific genes of the LAR subtype [33-35].
Indeed, androgen receptor mRNA expression has been shown to be considerably increased (9-fold) with respect to the other subtypes. Furthermore, tumors here classified show the up-regulation of a plethora of downstream targets and co-activators of the androgen receptor signaling [36-38].
TARGET THERAPY IN TNBC
The major issue for targeted therapy against TNBC is the lack of specific oncogene drivers due to wide BC heterogeneity [39-41].
To date, the main approach in TNBC treatment remains the chemotherapy, in particular the administration of anthracyclines, taxanes and/or platinum compounds able to target dividing cells. Unfortunately, not all chemotherapy treated patients show a favorable outcome and is still unclear whether treatment choices should be personalized among the different TNBC subtypes [42, 43].
Maybe a possible solution would be represented by the new proposed genetic signature tools as suggested by the recent MINDACT trial results, in the order to avoid the aggressive treatment to non-responder patients. MammaPrint genetic study allowed to identify a large group of patients which showed a good five-year progression-free survival (PFS) good though they have not received adjuvant treatment (AACR Annual Meeting 2016).
Indeed, for pre-operative treatment pCR (pathological Complete Response) would represent the best surrogate survival end-point for TNBC patients and it results doubled if platinum compounds are added to conventional therapy compared to the worse outcome achieved by TNBC patients showing residual disease [44, 45].
Given the aforementioned issues for management of TNBC patients, studies are urgently needed to improve the use of target therapies. The major difficulty is to discover actionable target because of wide heterogeneity of the disease. In fact, clinical trials on TNBCs that aim to point out a particular receptor fail to demonstrate an evident clinical benefit. One of the most important involved receptors is EGFR, that is upregulated in about 60% of TNBCs, whose trial investigating chemotherapy plus EGFR targeted agent versus chemotherapy alone showed a modest advantage in terms of response rate (RR) (33% vs 28%) [46]. Among the reasons why studies were not able to underline a significant clear advantage of these new proposed drugs, we should not take into account the heterogeneity of the disease that probably masks the real effect of the drug in a smaller population carrying the right target [47]. Recent studies are investigating a number of promising molecules and, thanks to some favourable hopeful results, a growing interest is developing about some specific signaling pathways such as PI3K/AKT/mTOR. [48-50].
PI3K/AKT/mTOR signaling pathway
PI3K/AKT/mTOR (PAM) represents the main signaling pathway responsible for cell proliferation, survival, metabolism and motility regulation and is often activated in BC [51-54] (Figure 1). A heterodimeric molecule belonging to the lipid kinases, phosphoinositide 3-kinase (PI3K), is the major component of this pathway. Based on structure, regulation mechanism and lipid substrate specificity, they can be categorized in three classes, but the class I PI3K is the more dysregulated in cancer [55].
PI3K signaling pathway starts following the binding of a growth factor or ligand to a variety of tyrosine kinase (TK) receptors, including HER proteins and IGF-1 receptors [56-58].
In its activated form PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3) which represents the docking site for AKT kinase. AKT activation leads to protein synthesis and cell growth by activating mTOR through TSC1/2 [59-61].
The main PI3K counteracting protein is the PTEN phosphatase, which acts by converting PIP3 to PIP2 [62]. Therefore, PIP3 results activated by PI3K and negatively controlled by PTEN [63].
Moreover, PIP3 levels seem to be also tightly modulated by another tumor suppressor, inositol polyphosphate 4-phosphatase type II (INPP4B), which dephosphorylates PIP3 to PIP2 [64].
Many research works report a higher incidence of PTEN and PI3K mutations in TNBC patients with respect to other histological subtypes [65].
A downstream component of PI3K/AKT pathway is mTOR which exists in two functionally different complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is responsible for the activation of protein translation process by promoting mRNA translocation and is also involved in metabolism and lipid synthesis [66]. mTOR downstream substrate is S6K1 which in turn can phosphorylate estrogen determining its activation with a mechanism independent of the ligand [67-69].
On the other hand, mTOR complex 2 is involved in the organization of actin cytoskeleton and, at the same time, regulates AKT phosphorylation. The importance of mTOR complexes and their pathways is fundamental in clinics due to the ability of many drugs to target selectively mTORC1 [70]. Indeed, studies conducted on TNBC murine models highlighted the effects of the inhibitor Dactolisib in controlling the whole mTOR pathway [71]. The frequency of mTOR pathway activation is higher in TNBC compared to other subtypes and is often correlated with poor prognosis [22, 72, 73]. Moreover, the up-regulation of PAM signaling induces resistance to hormone treatment, HER2-targeted treatment and cytotoxic therapy [74].
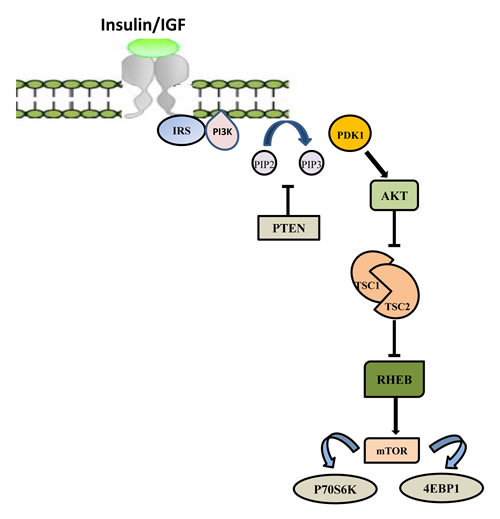
Figure 1: PI3K/AKT/mTOR signaling pathway. The PI3K signaling pathway is triggered by activation of receptor tyrosine kinase (RTK) in cell membrane. After binding to the growth factors, the intracellular domain of RTK is phosphorylated, and PI3K is activated. Activated PI3K phosphorylates PIP2 to produce PIP3. The tumor suppressor phosphatase and tensin homolog (PTEN) could negatively regulate this process via dephosphorylation of PIP3. Activated PIP3 could prompt the phosphorylation of Akt and further stimulate the Akt-mediated activation of downstream targets, including the Bcl-2 family members, Mdm2 and tuberous sclerosis complex 2 (TSC2). Activated Akt inhibits the Rheb GTPase activity of TSC1/2 complex by phosphorylating TSC2. Then, activated Rheb promotes mTOR complex 1 (mTORC1) to phosphorylate p70S6 and 4E binding protein1 (4EBP1), resulting in dysregulation of protein synthesis and cell survival.
MAMMALIAN TARGET OF RAPAMYCIN INHIBITORS
Everolimus (RAD001) is a mTOR inhibitor exhibiting vast anticancer activity in preclinical studies [75]. The combined treatment of rapamycin with paclitaxel in cell lines altered in the PI3K/AKT/mTOR signaling has been shown to increase effectiveness of treatment in TNBC [76]. This rationale has been explored in different clinical experiences. In a phase II study, Meyer and collaborators investigated the addition of everolimus 5 mg/day for 12 weeks to a short course pre-operative chemotherapy regimen containing weekly cisplatin (25 mg/m²) + paclitaxel (80 mg/m²) in patients affected by stage II / III TNBC demonstrating that no improvement was detected in the pCR after surgery and RR, following the addition of RAD001 [77]. Another phase II randomized study aimed to investigate the addition of everolimus to paclitaxel in neoadjuvant sequential regimen containing anthracyclines. Fifty women affected by stage II/III TNBC were subjected to a therapy with paclitaxel 80 mg/mq for 12 weeks or paclitaxel 80 mg/mq + everolimus 30 mg/day orally for 12 weeks followed by an FEC scheme (5-FU 500 mg/mq, epirubicin 100 mg/mq and cyclophosphamide 500 mg/mq every 3 weeks for four cycles) [78]. The addition of everolimus, although well tolerated, did not add any significant benefit in terms of 12-week-RR (48% versus 30% in favour of everolimus) and pCR (30% versus 26% in favour of everolimus) [79].
For the same principles, everolimus was tested in combination with carboplatin. In particular, Singh et al. enrolled 25 patients affected by metastatic TNBC who underwent to a 3-weekly chemotherapy regimen containing carboplatin AUC6 (or decreased to AUC5/4) + everolimus 5 mg/day. The treatment has shown significant hematologic toxicity especially with regimens containing carboplatin AUC6/5, but was well tolerated with AUC4, demonstrating a clinical benefit rate > = 6 months of 28% with a total mOS of 16.6 months and mPFS of 3 months [80].
Despite TNBC is HER2- [81], RAD001 has also been tested in a regimen with anti-HER2 drugs since EGFR is overexpressed and upregulated in about 50% of TN tumors [82, 83], providing a strong rationale to investigate the association between an anti-EGFR and a mTOR inhibitor in order to overlap the resistance to anti-EGFR agents [84, 85]. Although the mTOR inhibitors paradoxically trigger the AKT pathway [86], this activation could probably serve as resistance mechanism to mTOR inhibitors thus explaining the poor performance of these drugs when used as a single agent [78, 87]. On this basis, Liu et al. in their experience have shown that the addition of everolimus could sensitize BC cells to anti-EGFR drugs (lapatinib) [88], demonstrating that this association may be responsible for an increased apoptosis in some TNBC cell lines and murine xenograft progression compared to the same drugs used in monotherapy [89]. The main clinical studies concerning the function of mTOR inhibitors in TNBC and currently under evaluation are reported in Table 1.
In another important work, Zhang et al. created a panel of seven patient-derived orthotopic xenografts from primary and metastatic neoplastic tissue having histological and immunohistochemical features matched between patient and their corresponding xenografts. Neoplasms were divided on the basis of the above characteristics in different TNBC subtypes and the authors created a response signature to mTOR inhibitors demonstrating that BLBC also possessed the highest expression rate of the genes belonging to the PI3K/AKT pathway and the highest extent of phosphorylation of 4EBP1 [90].
Despite these promising results, it is not yet known the synergistic mechanism of action between the anti-EGFR and mTOR inhibitors and, furthermore, the importance of EIF4EBP1 gene is not completely clear. This topic was recently treated by Madden et al. that, using gefitinib (anti-EGFR) and temsirolimus (anti-mTOR) on TNBC cell lines, discovered the presence of a cross-talk mechanism between EGFR and mTOR also engaging the eukaryotic translation initiation factor 4B (eIF4B) [91]. Moreover, the action of these two molecules would seem to block phosphorylation of eIF4B, finally resulting in a growth and survival reduction in TNBC cell lines and then suggesting to investigate mTOR inhibition in association with other drugs [92, 93].
Recently, Bhola et al. [70] have suggested that resistance to TORC1/2 inhibitors may be exceeded via inhibition of the FGFR-mitochondrial metabolism-Notch1 axis that allows to eradicate therapy-resistant cancer stem cells in TNBC.
Table 1: Ongoing trials studying the role of mTOR inhibitors in TNBC
TRIAL |
REGISTRATION NUMBER |
INVESTIGATOR INSTITUTION |
Phase Ib/II Trials of RDA001 in Triple Negative Metastatic Breast Cancer |
NCT01939418 |
National Cancer Center, Korea |
A study of Lapatinib in combination with Everolimus in patients with Advanced, Triple Negative Breast Cancer |
NCT01272141 |
Emory University Winship Cancer Institute |
Liposomal Doxorubicin, Bevacizumab and Temsirolimus (DAT) in Triple-Negative Breast Cancer (TNBC) Insensitive to Standard Neoadjuvant chemotherapy |
NCT02456857 |
M.D. Anderson Cancer Center |
Comparison of Single-Agent Carboplatin vs the Combination of Carboplatin and Everolimus for the Treatment of Advanced Triple-Negative Breast Cancer |
NCT02531932 |
Icahn School of Medicine at Mount Sinai |
Eribulin Mesylate and Everolimus in Treating Patients With Triple-Negative Metastatic Breast Cancer |
NCT02120469 |
City of Hope Medical Center |
NECTAR Everolimus Plus Cisplatin (-) Breast Cancer (NECTAR) |
NCT01931163 |
The Methodist Hospital System |
Safety and Tolerability of Everolimus in Combination With Eribulin in Triple-negative Breast Cancer |
NCT02616848 |
Istituti Ospitalieri di Cremona |
A Study of AZD2014 in Combination With Selumetinib in Patients With Advanced Cancer (TORCMEK) |
NCT02583542 |
Queen Mary University of London |
PI3K/AKT INHIBITORS
The PAM pathway may be targeted trough a different strategy involving the inhibition of its upstream targets such as PI3K and Akt [94]. While there are inhibitors inactivating both PI3K and mTOR, further development may be limited by issues, including increased toxicity [95]. Kalinski et al. have shown that subjecting patients affected by stage I/III BC (including 3 women TN) at different doses of MK-2206, an allosteric inhibitor of AKT, they experienced rash and pruritus G3, mucositis G2, fever G2 and hyperglycemia G2, leading to the trial suspension, despite two dose reductions [96].
A further setting in which the PI3K/AKT inhibitors could prove their usefulness could be in association to PARP inhibitors (PARPis) in TNBC patients who did not exhibited BRCA1/2 function loss [97]. This is because, as it is already known, PARPis result active in tumors deficient in the homologous recombination (HR) mechanisms due to alterations in the BRCA1/2 genes [98-101], whereas their action is very negligible in non-BRCA mutant cancers [102].
Since the PI3K/AKT pathway stabilizes the function of HR, Ibrahim and collaborators have demonstrated that the use of AKT inhibitors in TNBC cell lines without BRCA1/2 alterations could cause HR function changes, and then sensitize to PARPis. In particular, the study showed that TNBC cancer cells treated with buparlisib (AKT inhibitor) were subject to a subsequent hyperactivation of ERK and MEK1, two essential component of the MAP kinase signal transduction pathway, resulting in downregulation of BRCA1 and then favoring the action of olaparib (PARPi) with subsequent reduction of cell proliferation and survival [103]. An interesting in vitro study showed that targeting multiple kinases such as IGF-1R, PI3K, mTORC or MEK may suppress cell proliferation and induce apoptosis in MDA-MB-231 cells, increasing also the inhibition of Akt phosphorylation [104].
A similar experience has been carried out by Kimbung et al. which evaluated the association between Rucaparib (PARPi) and LY294002 (PI3Ki) in BRCA1-deficient cells with the intent to improve the response to PARPis. This study showed promising results with sub-micromolar doses of both drugs, providing then a strong rationale for further research especially in TNBC [105].
The main clinical trials concerning the function of PI3K inhibitors in TNBC and currently under evaluation are reported in Table 2.
Table 2: Ongoing trials studying the role of PI3K inhibitors in TNBC
TRIAL |
REGISTRATION NUMBER |
INVESTIGATOR INSTITUTION |
Capecitabine +BKM120 TNBC Brain Mer |
NCT02000882 |
US Oncology Research |
Phase I Study of the Oral PI3kinase Inhibitor BKM120 or BYL719 and the Oral PARP Inhibitor Olaparib in Patients With Recurrent Triple Negative Breast Cancer or High grade Serous Ovarian Cancer |
NCT01623349 |
Dana-Farber Cancer Institute |
Phosphatidylinositol 3-kinase (PI3K) Alpha iNhibition In Advanced Breast Cancer (PIKNIC) |
NCT02506556 |
Peter MacCallum Cancer Centre, Australia |
CONCLUSIONS
TNBC is a heterogeneous subtype of BC showing aggressiveness and high risk of relapse [106].
In the last years, the treatment of metastatic breast cancer has seen the development of new systemic treatments. Despite this progress, TNBC still has limited therapeutic options: cytotoxic chemotherapy is the standard of care; systemic treatment tipically has transitory efficacy and the response is early followed by disease progression.
TNBC patients exhibit, indeed, an unfavorable outcome compared to those with other subtypes.
Only recently driver mutations have been identified with encouraging results in preclinical models and have allowed to investigate new specific drugs for each different subtype of the disease.
The PI3K-AKT-mTOR pathway is an exciting target for developing new anticancer therapeutics [107]. Since several pathways may be involved, therefore, the best results are achieved by combining different molecules on various targets paying attention to toxicity. For these reasons, it will be necessary in the near future to rescue pathological tissue taken before and after therapy in order to better understand the mechanisms of drug resistance and new concepts on tumor pathogenesis. Using increasingly refined techniques, liquid biopsies could have an important role, allowing us to obtain a lot of information in a short time and in a minimally invasive manner and maintaining a high concordance rate with the primary tumor and/or metastases. Therefore, is desirable enrolling TNBC patients with different subtypes in new specific trials to ensure them the most suitable treatment.
One of the main difficulties in the field of targeted therapies is represented by the extreme heterogeneity of the disease. This condition produces a number of different cases, each constituted by the presence of a rare mutation mostly detected in cases of exceptional responders in the context of negative trials. Consequently, although the discovery of these rare mutations is leading to the approval of many new antitumor therapies, it is necessary to design new studies showing benefits, avoiding all the problems related to the extreme heterogeneity of the disease contained in other conventional trials. A possible solution could be derived from the use of the so called basket trials that find their maximum indication where neoplasia depends on the pathway of the target and whether the therapy can effectively inhibit the action of the target itself. This would make possible to consider a marker as a probable predictor regardless of tumor histology.
Acknowledgments
Dr. Chiara Drago contributed to the revision of the English language of this manuscript.
conflicts of interest
The authors declare no conflict of interest.
Grant Support
This work was supported by the Fondazione Mediterranea “G.B. Morgagni”, Catania, Italy.
References
1. Fanale D, Amodeo V, Corsini LR, Rizzo S, Bazan V and Russo A. Breast cancer genome-wide association studies: there is strength in numbers. Oncogene. 2012; 31(17):2121-2128.
2. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, et al. Molecular portraits of human breast tumours. Nature. 2000; 406(6797):747-752.
3. Fanale D, Bazan V, Corsini LR, Caruso S, Insalaco L, Castiglia M, Cicero G, Bronte G and Russo A. HIF-1 is involved in the negative regulation of AURKA expression in breast cancer cell lines under hypoxic conditions. Breast Cancer Res Treat. 2013; 140(3):505-517.
4. Fanale D, Bazan V, Caruso S, Castiglia M, Bronte G, Rolfo C, Cicero G and Russo A. Hypoxia and human genome stability: downregulation of BRCA2 expression in breast cancer cell lines. Biomed Res Int. 2013; 2013:746858.
5. Sorlie T. The Impact of Gene Expression Patterns in Breast Cancer. Clin Chem. 2016.
6. Purrington KS, Visscher DW, Wang C, Yannoukakos D, Hamann U, Nevanlinna H, Cox A, Giles GG, Eckel-Passow JE, Lakis S, Kotoula V, Fountzilas G, Kabisch M, Rudiger T, Heikkila P, Blomqvist C, et al. Genes associated with histopathologic features of triple negative breast tumors predict molecular subtypes. Breast Cancer Res Treat. 2016.
7. Mancini P, Angeloni A, Risi E, Orsi E and Mezi S. Standard of care and promising new agents for triple negative metastatic breast cancer. Cancers (Basel). 2014; 6(4):2187-2223.
8. Schnitt SJ. Will molecular classification replace traditional breast pathology? Int J Surg Pathol. 2010; 18(3 Suppl):162S-166S.
9. Correa Geyer F and Reis-Filho JS. Microarray-based gene expression profiling as a clinical tool for breast cancer management: are we there yet? Int J Surg Pathol. 2009; 17(4):285-302.
10. Coates AS, Winer EP, Goldhirsch A, Gelber RD, Gnant M, Piccart-Gebhart M, Thurlimann B and Senn HJ. Tailoring therapies—improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol. 2015; 26(8):1533-1546.
11. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH and Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30(8):777-782.
12. Zaharia M and Gomez H. [Triple negative breast cancer: a difficult disease to diagnose and treat]. Rev Peru Med Exp Salud Publica. 2013; 30(4):649-656.
13. Schmadeka R, Harmon BE and Singh M. Triple-negative breast carcinoma: current and emerging concepts. Am J Clin Pathol. 2014; 141(4):462-477.
14. Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML and Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007; 13(8):2329-2334.
15. Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, Schwartz GF, Park PK, Rosenberg AL, Brill K and Mitchell EP. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer. 2007; 110(4):876-884.
16. Tobin NP, Harrell JC, Lovrot J, Egyhazi Brage S, Frostvik Stolt M, Carlsson L, Einbeigi Z, Linderholm B, Loman N, Malmberg M, Walz T, Ferno M, Perou CM, Bergh J, Hatschek T and Lindstrom LS. Molecular subtype and tumor characteristics of breast cancer metastases as assessed by gene expression significantly influence patient post-relapse survival. Ann Oncol. 2015; 26(1):81-88.
17. Russo A, Calò V, Bruno L, Schirò V, Agnese V, Cascio S, Foddai E, Fanale D, Rizzo S, Di Gaudio F, Gulotta E, Surmacz E, Di Fede G and Bazan V. Is BRCA1-5083del19, identified in breast cancer patients of Sicilian origin, a Calabrian founder mutation? Breast Cancer Research and Treatment. 2008; 113(1):67-70.
18. Anders CK and Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009; 9 Suppl 2:S73-81.
19. Cadoo KA, Fornier MN and Morris PG. Biological subtypes of breast cancer: current concepts and implications for recurrence patterns. Q J Nucl Med Mol Imaging. 2013; 57(4):312-321.
20. Nowacka-Zawisza M and Krajewska WM. [Triple-negative breast cancer: molecular characteristics and potential therapeutic approaches]. Postepy Hig Med Dosw (Online). 2013; 67:1090-1097.
21. Guarneri V, Dieci MV and Conte P. Relapsed triple-negative breast cancer: challenges and treatment strategies. Drugs. 2013; 73(12):1257-1265.
22. Ueng SH, Chen SC, Chang YS, Hsueh S, Lin YC, Chien HP, Lo YF, Shen SC and Hsueh C. Phosphorylated mTOR expression correlates with poor outcome in early-stage triple negative breast carcinomas. Int J Clin Exp Pathol. 2012; 5(8):806-813.
23. Liedtke C, Rody A, Gluz O, Baumann K, Beyer D, Kohls EB, Lausen K, Hanker L, Holtrich U, Becker S and Karn T. The prognostic impact of age in different molecular subtypes of breast cancer. Breast Cancer Res Treat. 2015; 152(3):667-673.
24. Cinkaya A, Akin M and Sengul A. Evaluation of treatment outcomes of triple-negative breast cancer. J Cancer Res Ther. 2016; 12(1):150-154.
25. Craig DW, O’Shaughnessy JA, Kiefer JA, Aldrich J, Sinari S, Moses TM, Wong S, Dinh J, Christoforides A, Blum JL, Aitelli CL, Osborne CR, Izatt T, Kurdoglu A, Baker A, Koeman J, et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013; 12(1):104-116.
26. Chacon RD and Costanzo MV. Triple-negative breast cancer. Breast Cancer Res. 2010; 12 Suppl 2:S3.
27. Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, Pritchard K, Eisen A, Vandenberg T, Taylor M, Sauerbrei E, Mishaeli M, Huntsman D, Walsh W, Olivo M, McIntosh L, et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol. 2009; 27(27):4536-4541.
28. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y and Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121(7):2750-2767.
29. Bauer JA, Chakravarthy AB, Rosenbluth JM, Mi D, Seeley EH, De Matos Granja-Ingram N, Olivares MG, Kelley MC, Mayer IA, Meszoely IM, Means-Powell JA, Johnson KN, Tsai CJ, Ayers GD, Sanders ME, Schneider RJ, et al. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin Cancer Res. 2010; 16(2):681-690.
30. Juul N, Szallasi Z, Eklund AC, Li Q, Burrell RA, Gerlinger M, Valero V, Andreopoulou E, Esteva FJ, Symmans WF, Desmedt C, Haibe-Kains B, Sotiriou C, Pusztai L and Swanton C. Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: a retrospective analysis of five clinical trials. Lancet Oncol. 2010; 11(4):358-365.
31. Fanale D, Bronte G, Passiglia F, Calo V, Castiglia M, Di Piazza F, Barraco N, Cangemi A, Catarella MT, Insalaco L, Listi A, Maragliano R, Massihnia D, Perez A, Toia F, Cicero G, et al. Stabilizing versus destabilizing the microtubules: a double-edge sword for an effective cancer treatment option? Anal Cell Pathol (Amst). 2015; 2015:690916.
32. Ishikawa T, Narui K, Tanabe M, Kida K, Oba MS, Yamada A, Ichikawa Y and Endo I. BRCAness is beneficial for indicating triple negative breast cancer patients resistant to taxane. Eur J Surg Oncol. 2016.
33. Hayes MJ, Thomas D, Emmons A, Giordano TJ and Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res. 2008; 14(13):4038-4044.
34. McNamara KM, Yoda T, Takagi K, Miki Y, Suzuki T and Sasano H. Androgen receptor in triple negative breast cancer. J Steroid Biochem Mol Biol. 2013; 133:66-76.
35. Barton VN, D’Amato NC, Gordon MA, Christenson JL, Elias A and Richer JK. Androgen Receptor Biology in Triple Negative Breast Cancer: a Case for Classification as AR+ or Quadruple Negative Disease. Horm Cancer. 2015; 6(5-6):206-213.
36. Rampurwala M, Wisinski KB and O’Regan R. Role of the androgen receptor in triple-negative breast cancer. Clin Adv Hematol Oncol. 2016; 14(3):186-193.
37. Barton VN, D’Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, Heinz RE, Elias A, Jedlicka P, Jacobsen BM and Richer JK. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015; 14(3):769-778.
38. Cuenca-Lopez MD, Montero JC, Morales JC, Prat A, Pandiella A and Ocana A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer. 2014; 14:302.
39. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486(7403):395-399.
40. Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, Fox SB, Ichihara S, Jacquemier J, Lakhani SR, Palacios J, Rakha EA, Richardson AL, Schmitt FC, Tan PH, Tse GM, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011; 24(2):157-167.
41. Crown J, O’Shaughnessy J and Gullo G. Emerging targeted therapies in triple-negative breast cancer. Ann Oncol. 2012; 23 Suppl 6:vi56-65.
42. Berrada N, Delaloge S and Andre F. Treatment of triple-negative metastatic breast cancer: toward individualized targeted treatments or chemosensitization? Ann Oncol. 2010; 21 Suppl 7:vii30-35.
43. Fornier M and Fumoleau P. The paradox of triple negative breast cancer: novel approaches to treatment. Breast J. 2012; 18(1):41-51.
44. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, Valero V, Lehmann BD, Pietenpol JA, Hortobagyi GN, Symmans WF and Ueno NT. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013; 19(19):5533-5540.
45. Petrelli F, Coinu A, Borgonovo K, Cabiddu M, Ghilardi M, Lonati V and Barni S. The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat. 2014; 144(2):223-232.
46. Wahba HA and El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015; 12(2):106-116.
47. Telli ML and Ford JM. Novel treatment approaches for triple-negative breast cancer. Clin Breast Cancer. 2010; 10 Suppl 1:E16-22.
48. Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M and Sinha N. Therapeutic targets of triple-negative breast cancer: a review. Br J Pharmacol. 2015; 172(17):4228-4237.
49. Yi YW, Kang HJ, Bae EJ, Oh S, Seong YS and Bae I. beta-TrCP1 degradation is a novel action mechanism of PI3K/mTOR inhibitors in triple-negative breast cancer cells. Exp Mol Med. 2015; 47:e143.
50. Gelmon K, Dent R, Mackey JR, Laing K, McLeod D and Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol. 2012; 23(9):2223-2234.
51. Massihnia D, Perez A, Bazan V, Bronte G, Castiglia M, Fanale D, Barraco N, Cangemi A, Di Piazza F, Calo V, Rizzo S, Cicero G, Pantuso G and Russo A. A headlight on liquid biopsies: a challenging tool for breast cancer management. Tumour Biol. 2016.
52. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318(5853):1108-1113.
53. Fanale D, Amodeo V, Bazan V, Insalaco L, Incorvaia L, Barraco N, Castiglia M, Rizzo S, Santini D, Giordano A, Castorina S and Russo A. Can the microRNA expression profile help to identify novel targets for zoledronic acid in breast cancer? Oncotarget. 2016; doi: 10.18632/oncotarget.8722.
54. Gonzalez-Angulo AM and Blumenschein GR, Jr. Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat Rev. 2013; 39(4):313-320.
55. Mayer IA and Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med. 2016; 67:11-28.
56. Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011; 16 Suppl 1:12-19.
57. Cangemi A, Fanale D, Rinaldi G, Bazan V, Galvano A, Perez A, Barraco N, Massihnia D, Castiglia M, Vieni S, Bronte G, Mirisola M and Russo A. Dietary restriction: could it be considered as speed bump on tumor progression road? Tumour Biol. 2016.
58. Guo S, Liu M and Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011; 1815(2):197-213.
59. Zhao L and Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008; 27(41):5486-5496.
60. Lauring J, Park BH and Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013; 11(6):670-678.
61. Ogawa A, Firth AL, Yao W, Madani MM, Kerr KM, Auger WR, Jamieson SW, Thistlethwaite PA and Yuan JX. Inhibition of mTOR attenuates store-operated Ca2+ entry in cells from endarterectomized tissues of patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009; 297(4):L666-676.
62. Santiskulvong C, Konecny GE, Fekete M, Chen KY, Karam A, Mulholland D, Eng C, Wu H, Song M and Dorigo O. Dual targeting of phosphoinositide 3-kinase and mammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic approach in human ovarian carcinoma. Clin Cancer Res. 2011; 17(8):2373-2384.
63. Salmena L, Carracedo A and Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008; 133(3):403-414.
64. Agoulnik IU, Hodgson MC, Bowden WA and Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget. 2011; 2(4):321-328. doi: 10.18632/oncotarget.260.
65. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490(7418):61-70.
66. Dowling RJ, Topisirovic I, Fonseca BD and Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010; 1804(3):433-439.
67. Zoncu R, Efeyan A and Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12(1):21-35.
68. Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ and Holz MK. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem. 2009; 284(10):6361-6369.
69. Khotskaya YB, Goverdhan A, Shen J, Ponz-Sarvise M, Chang SS, Hsu MC, Wei Y, Xia W, Yu D and Hung MC. S6K1 promotes invasiveness of breast cancer cells in a model of metastasis of triple-negative breast cancer. Am J Transl Res. 2014; 6(4):361-376.
70. Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, Grandis JR and Arteaga CL. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. 2016; 76(2):440-452.
71. Montero JC, Esparis-Ogando A, Re-Louhau MF, Seoane S, Abad M, Calero R, Ocana A and Pandiella A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene. 2014; 33(2):148-156.
72. Walsh S, Flanagan L, Quinn C, Evoy D, McDermott EW, Pierce A and Duffy MJ. mTOR in breast cancer: differential expression in triple-negative and non-triple-negative tumors. Breast. 2012; 21(2):178-182.
73. Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, Pusztai L and Huang P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014; 16(5):434.
74. Paplomata E and O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014; 6(4):154-166.
75. Yunokawa M, Koizumi F, Kitamura Y, Katanasaka Y, Okamoto N, Kodaira M, Yonemori K, Shimizu C, Ando M, Masutomi K, Yoshida T, Fujiwara Y and Tamura K. Efficacy of everolimus, a novel mTOR inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012; 103(9):1665-1671.
76. Tomao F, Papa A, Zaccarelli E, Rossi L, Caruso D, Minozzi M, Vici P, Frati L and Tomao S. Triple-negative breast cancer: new perspectives for targeted therapies. Onco Targets Ther. 2015; 8:177-193.
77. Mayer IA, Jovanovic B, Abramson VG, Mayer EL, Sanders ME, Bardia A, Dillon PM, Kuba MG, Carpenter JT, Chang JC, Lehmann BD, Meszoely IM, Grau A, Shyr Y, Arteaga CL, Chen X, et al. Abstract PD1-6: A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus (an mTOR inhibitor) in patients with stage II/III triple-negative breast cancer (TNBC). Cancer Research. 2014; 73(24 Supplement):PD1-6-PD1-6.
78. Jerusalem G, Rorive A and Collignon J. Use of mTOR inhibitors in the treatment of breast cancer: an evaluation of factors that influence patient outcomes. Breast Cancer (Dove Med Press). 2014; 6:43-57.
79. Gonzalez-Angulo AM, Akcakanat A, Liu S, Green MC, Murray JL, Chen H, Palla SL, Koenig KB, Brewster AM, Valero V, Ibrahim NK, Moulder-Thompson S, Litton JK, Tarco E, Moore J, Flores P, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann Oncol. 2014; 25(6):1122-1127.
80. Singh J, Novik Y, Stein S, Volm M, Meyers M, Smith J, Omene C, Speyer J, Schneider R, Jhaveri K, Formenti S, Kyriakou V, Joseph B, Goldberg JD, Li X, Adams S, et al. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res. 2014; 16(2):R32.
81. Schmidt G, Gerlinger C, Juhasz-Boss I, Stickeler E, Rody A, Liedtke C, Wimberger P, Link T, Muller E, Fehm T, Abel M, Stein S, Bohle R, Endrikat J and Solomayer EF. Her2-neu score as a prognostic factor for outcome in patients with triple-negative breast cancer. J Cancer Res Clin Oncol. 2016.
82. Reis-Filho JS and Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008; 52(1):108-118.
83. Simon N, Antignani A, Sarnovsky R, Hewitt SM and FitzGerald D. Targeting a Cancer-Specific Epitope of the Epidermal Growth Factor Receptor in Triple-Negative Breast Cancer. J Natl Cancer Inst. 2016; 108(8).
84. Passiglia F, Bronte G, Castiglia M, Listi A, Calo V, Toia F, Cicero G, Fanale D, Rizzo S, Bazan V and Russo A. Prognostic and predictive biomarkers for targeted therapy in NSCLC: for whom the bell tolls? Expert Opin Biol Ther. 2015; 15(11):1553-1566.
85. Gogineni K and DeMichele A. Current approaches to the management of Her2-negative metastatic breast cancer. Breast Cancer Res. 2012; 14(2):205.
86. Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H and Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005; 65(16):7052-7058.
87. Hahn OM, Ma CX, Lin L, Hou D, Sattar H, Olopade FO, Nanda R, Hoffman PC, Naughton MJ, Pluard T, Watson MA, Ellis M, Conzen SD and Fleming GF. A phase II trial of a mammalian target of rapamycin inhibitor, temsirolimus, in patients with metastatic breast cancer. Cancer Research. 2014; 69(2 Supplement):407.
88. Wan X, Zheng X, Pang X, Pang Z, Zhao J, Zhang Z, Jiang T, Xu W, Zhang Q and Jiang X. Lapatinib-loaded human serum albumin nanoparticles for the prevention and treatment of triple-negative breast cancer metastasis to the brain. Oncotarget. 2016; doi: 10.18632/oncotarget.8697.
89. Liu T, Yacoub R, Taliaferro-Smith LD, Sun SY, Graham TR, Dolan R, Lobo C, Tighiouart M, Yang L, Adams A and O’Regan RM. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol Cancer Ther. 2011; 10(8):1460-1469.
90. Zhang H, Cohen AL, Krishnakumar S, Wapnir IL, Veeriah S, Deng G, Coram MA, Piskun CM, Longacre TA, Herrler M, Frimannsson DO, Telli ML, Dirbas FM, Matin AC, Dairkee SH, Larijani B, et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014; 16(2):R36.
91. Guo S, Liu M, Wang G, Torroella-Kouri M and Gonzalez-Perez RR. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim Biophys Acta. 2012; 1825(2):207-222.
92. Madden JM, Mueller KL, Bollig-Fischer A, Stemmer P, Mattingly RR and Boerner JL. Abrogating phosphorylation of eIF4B is required for EGFR and mTOR inhibitor synergy in triple-negative breast cancer. Breast Cancer Res Treat. 2014; 147(2):283-293.
93. Vicier C, Dieci MV, Arnedos M, Delaloge S, Viens P and Andre F. Clinical development of mTOR inhibitors in breast cancer. Breast Cancer Res. 2014; 16(1):203.
94. Moulder S, Helgason T, Janku F, Wheler J, Moroney J, Booser D, Albarracin C, Morrow PK, Atkins J, Koenig K, Gilcrease M and Kurzrock R. Inhibition of the phosphoinositide 3-kinase pathway for the treatment of patients with metastatic metaplastic breast cancer. Ann Oncol. 2015; 26(7):1346-1352.
95. Fouque A, Jean M, van de Weghe P and Legembre P. Review of PI3K/mTOR inhibitors entering clinical trials to treat triple negative breast cancers. Recent Pat Anticancer Drug Discov. 2016.
96. Kalinsky K, Sparano JA, Andreopoulou E, Taback B, Wlechmann LS, Feldman SM, Ananthakrishnan P, Hibshoosh H, Manavaian J, Crew KD, Maurer MA and Hershman DL. Presurgical evaluation of the AKT inhibitor MK-2206 in patients with operable invasive breast cancer. Journal of Clinical Oncology. 2014; 32(15).
97. Hosford SR and Miller TW. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharmgenomics Pers Med. 2014; 7:203-215.
98. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M and Carmichael J. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010; 376(9737):235-244.
99. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH and de Bono JS. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009; 361(2):123-134.
100. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC and Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434(7035):917-921.
101. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC and Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66(16):8109-8115.
102. Gelmon KA, Hirte HW, Robidoux A, Tonkin KS, Tischkowitz M, Swenerton K, Huntsman D, Carmichael J, Macpherson E and Oza AM. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer. Journal of Clinical Oncology. 2010; 28(15).
103. Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J, Rodriguez O, Calvo MT, Aura C, Diez O, Rubio IT, Perez J, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012; 2(11):1036-1047.
104. Ayub A, Yip WK and Seow HF. Dual treatments targeting IGF-1R, PI3K, mTORC or MEK synergize to inhibit cell growth, induce apoptosis, and arrest cell cycle at G1 phase in MDA-MB-231 cell line. Biomed Pharmacother. 2015; 75:40-50.
105. Kimbung S, Biskup E, Johansson I, Aaltonen K, Ottosson-Wadlund A, Gruvberger-Saal S, Cunliffe H, Fadeel B, Loman N, Berglund P and Hedenfalk I. Co-targeting of the PI3K pathway improves the response of BRCA1 deficient breast cancer cells to PARP1 inhibition. Cancer Lett. 2012; 319(2):232-241.
106. Toss A and Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015; 17:60.
107. Carey L, Winer E, Viale G, Cameron D and Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010; 7(12):683-692.