
INTRODUCTION
Germline mutations in one of the breast cancer susceptibility genes, BRCA1 (MIN #113705) or BRCA2 (MIN#600185), are the major and most widely known risk factors for breast and/or ovarian cancer (BOC) hereditary syndrome (HBOC) [1, 2], although they are present in about 40% of cases with strong family BOC background. HBOC occurs in 5-10% of all BOC cases; in turn, individuals with such inheritance have a 50-80% risk of developing breast cancer and a 30-50% risk of ovarian cancer in their lifetime, while other malignancies such as prostate and pancreatic cancer have been less frequently observed [3, 4]. Furthermore, cancer types such as melanoma and colon have been detected in families with BRCA2 mutations [3-5].
Since the discovery of BRCA1 and BRCA2 genes, thousands of genetic variants with different clinical significance have been described in the Breast Cancer Information Core Database (http:/research.nhgri.nih.gov/bic/ access in october 2015), with near 2000 of them being classified as pathogenically responsible for HBOC. The large extension of these genes and the rare hot spot mutations generate a genetic diversity with more than 700 pathogenic mutations described only once.
The frequency of BRCA1 and BRCA2 mutation carriers in women with BOC depends on the population analyzed but appears to be similar across ethnicity [6]. However, significant variation has been demonstrated in the spectrum of BRCA1/2 mutations according to ethnic and/or geographical diversity [7, 8]. Racial mixture in the South American population has been reported in epidemiological and molecular studies [9]. In particular, the Argentine population consists of an admixture of European ancestry -mainly from Spain and Italy- and an Amerindian component in a variable degree which is observed in more than 50% of the population [8, 10, 11].
The epidemiology of HBOC in Argentina has been described in a single report, which reveals racial admixture and clinically relevant genetic variants, including deleterious mutations previously unexplored [8]. Other studies have reported a Hispanic panel [12] including 9 recurrent deleterious mutations which account for 53% of the total, although it should be pointed out that the composition of the so-called Hispanic population analyzed in this publication reflects the Mexican and Central America/Caribbean origin for these probands, completely different from Argentine ethnicity. Big efforts have been made to select a panel of mutations for South American populations such as Peru [13], Mexico [14], Chile [15] and Colombia [12] and they all share a common goal: to lower study costs. Although attention needs to be drawn to the implementation of a mutation panel as a putative screening standard anticipating its impact on health care, “non-mutation detected” results for this panel should be followed by the full sequence of BRCA1/2. A panel may become, however, the only analysis in a patient’s lifetime, at least in Argentina; in other words, he/she might never be analyzed for the total sequence because of our heterogeneous health insurance system. This secondary effect may prove harmful and confusing for patients and doctors, who may never realize the test performed is practically useless. In addition, limited data on testing outcome and the need for information on testing policies in different regions around the world makes this study all the more relevant. A review has been recently published describing the current knowledge on BRCA1 and BRCA2 variants in 4835 women from Latin America, the Caribbean and the Hispanic population in the United States. The study concludes that countries with high prevalence of BRCA pathogenic variants may benefit from more aggressive testing strategies, while testing of recurrent variant panels might present a cost-effective solution for improving genetic testing for hereditary cancer in some, but not all countries [16].
These arguments highlight the importance of analyzing BRCA1/2 gene sequences and large rearrangements, searching for a regional spectrum of variants to contribute to the clinical application of genetic data and the evaluation of HBOC risk in a Hispanic population of different origin. In this context, the present report analyzed 940 probands with HBOC from the Argentine population.
RESULTS
The analysis of 940 probands revealed 179 (19.04%) deleterious mutations including five large rearrangements: 105 found in the BRCA1 and 74 in the BRCA2 gene, all of them detailed in the Supplementary table, with the exception of the novel mutations listed in Table 2. The distribution of the mutations was as follows: a) 105 probands bore a mutation in the BRCA1 gene, with 8 cases having breast/ovary cancer diagnosis, 11 cases with ovary cancer diagnosis (one is a novel mutation, c.2728C > T, p.Gln910*) and 85 with breast cancer diagnosis, 6 of which were bilateral; b) in 74 probands the mutation was detected in the BRCA2 gene and include 6 cases with breast/ovary cancer diagnosis (two of them with a novel mutation: c.8754+1G > A and c.8463dupT, p.Ile2822Tyrfs), 2 cases with ovary cancer diagnosis (one is a novel mutation, c.7805+2_7805+3delTA, in intron 16) and the rest with breast cancer diagnosis, 5 female bilateral cases and 3 male cases, one of them bilateral.
Table 1: Patients analyzed: Total=940 probands, with a mutation=179, BRCA1=105 and BRCA2=74
Group |
n |
Patients with a mutation / Nv (n) |
Tumor/s in patients with a mutation (n) |
Diagnosed ≤ 40 years
Healthy ≤ 40 years
|
198 135 |
BRCA1: 41 / 4 BRCA2: 18 / 6 BRCA1: 5 BRCA2: 4 / 1 |
Br(33)/b-Br(5)/Br & Ov(2)/Ov(4)/b-Br & Ov(1) Br(20)/b-Br(2)/Br & Ov(1)/Ov(1) ----- ----- |
Diagnosed > 40 years
Healthy > 40 years
|
498 95 |
BRCA1: 45 / 3 BRCA2: 31 / 5 BRCA1: 5 / 1 BRCA2: 4 / 1 |
Br(33)/b-Br(2)/Br & Ov(3)/Ov(7)/b-Ov(1)/b-Br & Ov(2) Br(29)/b-Br(1)/Br & Ov(4)/b-Br & Ov(1)/b-Ov(1) ----- ----- |
Men diagnosed ≤ 40 years
Men diagnosed > 40 years
Men Healthy > 40 years |
1 12 1 |
BRCA1: 1 BRCA2: 0 BRCA1: 0 BRCA2: 3 BRCA1: 0 BRCA2: 0 /1 |
Br & pancreas (1) ----- ----- Br(2)/b-Br(1) ----- ----- |
Total |
940 |
BRCA1: 97 / 8 BRCA2: 60 / 14 |
----- ----- |
n: number of probands. Nv: novel mutation. Br:breast; Ov: ovary; b-: bilateral
It may be worth highlighting that 230 patients analyzed from a total of 940 had a family history but no personal history of cancer. However, and even if their health status may result from not having inherited the allele with the mutation, they were included in the total patients, as 22 (mean age = 39, range 18-68) out of these 230 were found to be carriers of a mutation (for the non-carriers, n = 208, mean age = 41, range = 23-61).
Table 2 lists the 22 novel (at the moment of the detection) deleterious mutations, 8 in BRCA1 and 14 in BRCA2, which represent 12.3% of total mutations. All of them are tier 1, as the resulting stop codon from the frameshift mutation is already associated with a deleterious effect. Table 3 lists a total of 8 novel variants, 2 in BRCA1 and 6 in BRCA2, with probably deleterious effect on the resulting protein, as observed in in silico analyses. In both groups, the vast majority had breast cancer diagnosis, with only two cases of ovarian cancer (one with a mutation in each BRCA gene, both novel variants) and two cases with both breast and ovary cancers (both with a mutation in BRCA2), all of them in the novel variant group (Table 2).
Table 2: Novel mutations in BRCA1/2detected in 940 probands with personal and/or family history of breast/ovary cancer
ID |
Exon/Intron |
Mutation |
HGVS cDNA |
HGVS protein |
STOP codon |
PH (age) |
FH (1st and 2nd degree) |
|
BRCA1 |
|
|
|
|
|
|
AB0425 |
11 |
FrameShift |
c.1140delG |
p.Gln380=fs |
Leu393* |
healthy (46) |
Br/Ov |
AB0020 |
11 |
FrameShift |
c.1502_1505delAATT |
p.Lys501=fs |
Glu530* |
Br (32) |
NO |
AB0067 |
11 |
FrameShift |
c.2507_2508delAA |
p.Glu836Glyfs |
Val837* |
Br (50) |
Br |
AB0085 |
11 |
FrameShift |
c.2686delA |
p.Ser896Valfs |
Leu999* |
Br (55) |
Br |
AB0017 |
11 |
FrameShift |
c.3758_3759delCT |
p.Ser1253* |
Ser1253* |
Br (31) |
Br |
AB0278 |
23 |
FrameShift |
c.5463_5464insT |
p.His1822Serfs |
Glu1829* |
Br (36) |
Br |
AB0568 |
11 |
Nonsense |
c.4042G>T |
p.Gly1348* |
Gly1348* |
Br (25) |
Br |
AB0084 |
11 |
Nonsense |
c.2728C>T |
p.Gln910* |
Gln910* |
Ov (55) |
Br, Ov, Co |
|
BRCA2 |
|
|
|
|
|
|
AB0364 |
7 |
Splicedefect |
c.517G>T |
p.Gly173Cys |
|
Br (48) |
Br/Pr |
AB0099 |
9i |
Splicedefect |
c.793+1delG |
|
|
BR(35) |
Br |
AB0615 |
16i |
Splicedefect |
c.7805+2delTA |
|
|
Ov (43 & 46) |
Br/Ov |
AB0211 |
21i |
Splicedefect |
c.8754+1G>A |
|
|
Br (33)/Ov (36) |
Br/Pr |
AB0034X |
11 |
FrameShift |
c.3343delT |
p.Ser1115Leufs |
Leu1118* |
Br (33) |
NO |
AB0314 |
11 |
FrameShift |
c.4740_4741dupTG |
p.Glu1581Valfs |
Ser1617* |
Br (37) |
Br |
AB0384 |
11 |
FrameShift |
c.4963delT |
p.Tyr1655Thrfs |
Leu1669* |
healthy (36) |
Br/Mel/Pancr |
AB0225 |
11 |
FrameShift |
c.5669_5673delTGGCA |
p.Met1890Argfs |
Leu1897* |
Br (52) |
Br |
AB0098 |
14 |
FrameShift |
c.7110dupA |
p.Ser2371Ilefs |
Glu2391* |
Br (31) |
Br |
AB0322 |
14 |
FrameShift |
c.7230delT |
p.Phe2410Leufs |
Val2466* |
healthy (41) |
Br |
AB0392 |
18 |
FrameShift |
c.8021delA a |
p.Lys2674Argfs |
Ile2675* |
Br (40) |
Br |
AB0078U |
19 |
FrameShift |
c.8463dupT |
p.Ile2822Tyrfs |
Glu2844* |
Br / Ov (54) |
NO |
AB0048Y |
26 |
FrameShift |
c.9498delT |
p.Val3166=fs |
Leu3216* |
Healthymale (65) |
Br |
AB0508 |
27 |
FrameShift |
c.9789_9790delGA |
p.Lys3263=fs |
Ser3275* |
Br (46) |
Br |
PH: Personal historyof cancer; FH: Family historyof cancer; Br: Breast; Ov: Ovary; Co: colon; Pr: Prostate; Mel: Melanoma; Pancr: Pancreas.
ª: coexistent with BRCA1 c.4484+3A>G also novel.
Age range at first diagnostic: 25-55 years; mean+SD=40.89+9.65 ( n =18)
Recurrent mutations are listed in Table 4, which shows four different deleterious mutations detected 4 or more times in 27 probands, and which represent 2.87% of the total probands analyzed and 15.08% of the patients with a detected mutation. Two additional mutations, the c.5123C > A p.Ala1708Glu in BRCA1 and c.9026_9030delATCAT p.Tyr3009_His3010fs in BRCA2 were found in three probands each, increasing frequency of recurrent mutations from 2.87% to 3.52% of the total probands analyzed.
Preliminary results from the sequencing of an additional series of 50 ovary cancer patients (own data in process) showed three instances of the BRCA1 mutation c.4964_4982delCTGGCCTGACCCCAGAAGA, p.Ser1655_Glu1661?fs. Even if not included in the recurrent panel, this 19bp deletion in our patients appears worth mentioning, as it is listed twice in the Supplementary table and thus renders a 5-time frequency of clinical relevance.
Table 3: Novel variants in BRCA1/2 with probably deleterious effect from the in silico analysis detected in 940 probands with personal and/or family history of breast/ovary cancer
ID |
Exon/ Intron |
Mutation |
HGVS cDNA |
HGVS protein |
PH (age) |
FH (1st and/or 2nd degree) |
Align GVGD |
PolyPhen |
SIFT |
|
BRCA1 |
|
|
|
|
|
|
|
|
AB0392 |
14i |
Splice defect |
c.4484+3A>G ª |
|
Br (40) |
Br |
|
|
|
AB0081U |
7 |
Missense |
c.341C>G |
p.Ser114Cys |
Br (45) |
Br; Co |
Class C0 |
Possibly damaging |
Afect protein function |
|
BRCA2 |
|
|
|
|
|
|
|
|
AB0376 |
14 |
In Frame del |
c.7426_7428delGAA |
p.Glu2476del |
Br (39) |
NO |
|
|
|
AB0402 |
10 |
Missense |
c.1277A>C |
p.Lys426Thr |
Br (42) |
Br |
Class C0 |
Possibly damaging |
Afect protein function |
AB0185 |
11 |
Missense |
c.3316A>G |
p.Ser1106Gly |
Healthy (43) |
Br, Gastric |
Class C0 |
Probably damaging |
Afect protein function |
AB0231 |
14 |
Missense |
c.7159G>C |
p.Ala2387Pro |
Healthy (46) |
Br, Ov |
Class C0 |
Possibly damaging |
Afect protein function |
AB0258 |
18 |
Missense |
c.8038G>A |
p.Asp2680Asn |
Br (37) |
Br |
Class C0 |
Probably damaging |
Afect protein function |
AB0435 |
27 |
Missense |
c.9794G>A |
p.Cys3265Tyr |
Br (50 & 52) |
NO |
Class C0 |
Possibly damaging |
Afect protein function |
PH: Personal historyof cancer; FH: Family historyof cancer; Br: Breast; Ov: Ovary; Co: colon.
ª: coexistent with in BRCA2 c.8021delA, p.Lys2674Argfs -also novel-.
Age range at first diagnostic: 37-50 years; mean+SD=42.17+4.71(n=6)
Table 4: Recurrent mutations in BRCA1/2 detected in 940 probands with personal and/or family history of breast/ovary cancer
MUTATION |
UNRELATED PROBANDS (% of the total probands) |
REPORTED ORIGIN |
BRCA1 |
|
|
c.211A>G (p.Arg71Gly) |
11 (1.17) |
Spanish |
c.181T>G (p.Cys61Gly) |
6 (0.64) |
Italian |
BRCA2 |
|
|
c.2808_2811delACAA (p.Lys936_Gln937LysGlnfs) |
6 (0.64) |
French |
c.6037A>T (p.Lys2013*) |
4 (0.42) |
Portuguese / German |
Total recurrents |
32 (3.4) |
- |
Total recurrents / total mutated (179) = 17.8% |
- |
- |
Note: The mutation in BRCA1: c.4964_4982del19 (p.Ser1655Tyrfs) is to be added in the recurrent mutations panel as it was found five times, including three recent patients (study in progress, see text).
Also worth highlighting, no Ashkenazi mutations were found out of the Ashkenazi ethnicity, as confirmed two detections of the 6174delT (BIC nomenclature) (c.5946_5946delT, p.Ser1982Argfs) with grand/great-grandparents of such ethnic origin.
The large rearrangements listed in Table 5 prove to be rather infrequent mutations in our population, representing only 0.53% (5 of the 940) and all of them corresponding to the BRCA1 gene, in agreement with patients’ ethnic origin as previously described [17, 18].
Table 5: Large rearrangements in the BRCA1 gene detected in 940 probands with personal and/or family history of breast/ovary cancer
ID |
Exonsdeleted |
HGVS cDNA |
PH (age) |
FH (1st and 2nd degree) |
Nationality / ancestry of families |
AB0064X |
del 5´UTR thruexon 2 |
c.1-?_80+?del |
Br (35) |
Br |
Spanish |
AB0627 |
del 5´UTR thruexon 2 |
c.1-?_80+?del |
Ov (40) |
Br |
Spanish |
AB0473 |
del exons 5 thru 10 |
c.135-?_670+?del |
Br (31) |
Br/Gastric Cancer |
Slovenian |
AB0353 |
del exons 11 thru 15 |
c.671-?_4675+?del |
Ov (35) |
Br |
Spanish |
AB006H |
del exons 15 and 16 |
c.4485-?_4986+?del |
Br (45) |
Br |
French |
PH: Personal history of cancer; FH: Family history of cancer; Br: Breast; Ov: Ovary
DISCUSSION
This report is to date the first study in our country to summarize the relevant findings of the local experience in the full sequencing of the BRCA1/2 genes through the analysis of 940 probands (Supplementary table) in two centers in Argentina.
This study is also the largest on Hispanic families from one country with breast/ovarian cancer in South America, confirming differences from Hispanic families in the United States. In US Hispanic families, recurrent mutations represent 53% of total mutations, which indicates potential for cost-effective ancestry-informed genetic screening Weitzel et al. [12]. In contrast, recurrent mutations in the Hispanic population of South America represent only 15.08% (Table 4), which highlights the importance of clinical genetic strategies adapted to each population’s needs and intrinsic genetic characteristics.
One of the important features of our report is the number of probands analyzed. Recently, a very elegant review [16] studying the spectrum of BRCA1/2 alleles in Latin America and the Caribbean combined 4835 individuals from 13 countries. This review concludes that the Hispanic population of Latin America and the US may benefit from genetic-based cancer prevention options, a strategy that should combine knowledge on hereditary cancer in those populations and improved access to genetic testing. Also in this review, only 10.4% of 167 BRCA pathogenic variants identified were shared between US Hispanics and Latin America, a finding regarded as consequence of the limited sample size available for some of the countries. The present study, however, describes results for 940 probands from a single country and still shows the same discrepancy described in the review [16], which clearly proves differences to be associated to real genetic diversity and not limited sample size.
The use of the NGS sequencing technique was crucial in improving our previous report [8], expanding the analysis from about a hundred cases to nearly a thousand probands. Out of these 940 probands, 179 revealed deleterious mutations, which constitutes 19.04%, an expected rate of detection (Supplementary table). Tables 2 and 5 show novel mutations and large rearrangement, respectively, the latter including 5 cases which represent 0.54% of the series.
In the 179 probands analyzed, an expectable correlation was found between cancer type and gene mutation: we found most cases with ovary cancer diagnosis to have mutations in BRCA1, 12 cases (one novel) compared with probands with a mutation in BRCA2, 2 cases (one novel). The breast and ovary cancer cases were similar for both genes with 7 and 6 cases for BRCA1 and BRCA2, respectively, while the 3 cases of male breast cancer (one bilateral) presented a mutation only in BRCA2 (Supplementary table).
Large rearrangements are frequent in few ethnic groups [17] and represent only 0.54% of total cases in our study, which is consistent with our patients’ ethnic origin detailed in Table 5. These results confirm the differences between the Hispanic families from South America and the United States, where large rearrangements represent 11% of the total. Moreover, the Mexican founder large deletion described by Weitzel et al [12] was not found in our series.
These discrepancies may be due to the differences between the Mexican and Central America/Caribbean origin of these probands, which is remarkably different from Argentine ethnicity, mostly European (Spanish, Italian and German) and Amerindian. Worth pointing out, the Hispanic families from the United States included only 36 probands from South America.
In our population, a single mutation -c.211A > G, Arg71Gly, reported as of Spanish origin- out of the 15 recurrent mutations included in the Hispanic panel [12] has four or more probands; regarding other mutations described in the panel, three were found up to three times in our population: Ala1708Glu (c.5123C > A) and Arg1443* (c.4327C > T) in BRCA1 and c.9026_9030del5, p.Tyr3009_His3010fs in BRCA2. In other words, if the Hispanic panel were applied, these mutations would altogether reach 1.8% of the total probands and 9.5% of the mutated samples. These numbers mean that sequencing the 15 mutations described [12] in 100 probands would render less than 2 patients bearing one of the mutations. Remarkably, the 185delAG (c.66_67delAG, p.Leu22_Glu23LeuValfs) mutation was never found in our series and is included in the Hispanic panel, although it has always been described to occur on the Jewish haplotype [12].
In agreement with the concept of eventual regional variants and/or population heterogeneity, we have previously reported novel mutations in other genes in our population, detected by direct sequencing such as protooncogen RET [19], Lynch Syndrome [20], APC in Familial Adenomatous Polyposis [21].
Few novel missense variants evaluated in silico were found with protein damage prediction programs (Table 3), which heightens the need for cautious interpretation of the variants detected.
No hotspots were detected in our population with the variants distributed along both genes (Figure 1), which rules out the possibility of a hot spot zone in the gene or a panel of mutations that might allow the implementation of a low cost initial assay for BRCA1/2 analysis as attempted in other studies [15].
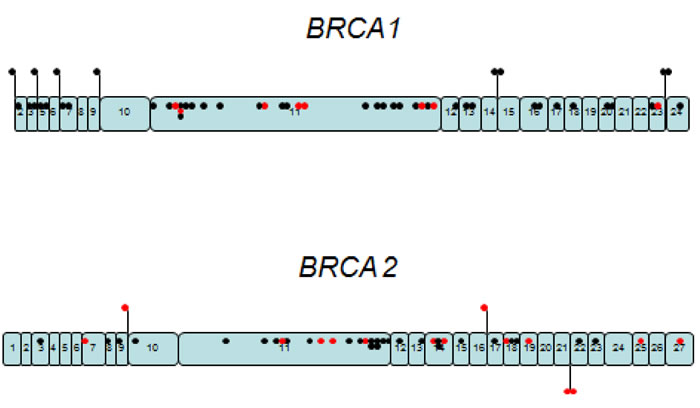
Figure 1: Schematic representation of the gene location for the deleterious mutations detected in the analysis of BRCA1/2 of 940 patients from Argentina. Black dots represent the 157 deleterious mutations and the red dots are the 22 novel deleterious mutations, dispersed along each gene.
These striking differences are a warning sign against the import of panels from apparently similar populations. This is a key issue in many aspects: a) clinicians and patients may be misinformed, even in cases with accomplished genetic counseling; b) when a panel is the first analysis, in our health system, insurance may reject further analyses in the same line, i.e. twice the analysis “of the same genes”, which might also be inaccurate, as a full sequencing test is required after a non-mutation has been detected in a panel; c) if health insurance covered both analyses (the panel of mutations and the full sequencing), 97% of the patients analyzed for the recurrent mutations would need full sequencing of BRCA1/2, which is not even economically convenient; d) attention needs to be drawn to the correct interpretation of results, as “normal” is considered equivalent to “uncompleted analysis” at two levels: the restricted number of mutations analyzed and the limitations of the BRCA1/2 analysis itself, a restraining concept for the initial study of a complex genetic study.
Future work will focus on the study of non-BRCA predisposing breast and ovarian cancer genes through multigene panels, although it might be not being easy as the highly conflicts generated by those panels.
This is an important point to be considered in providing the best healthcare possible, mostly in South American countries where the supporting economy is frequently in crisis and low cost studies are attractive. There is a real need for the implementation of highly supported medical care on both ethical and genetic grounds. This will render profits from funds invested in health, mostly in the prevention of high costs for cancer treatments and analyses in hereditary cancer, to be used in prevention (first goal) and early detection.
CONCLUSIONS
Our results in the sequence analysis of the BRCA1/2 genes in HBOC probands reveal novel mutations, recurrent mutations and other findings contributing to the knowledge of BRCA1/2 comprehensive sequence.
We emphasize the following findings: a) no hot spots for BRCA1/2, up to date, have been found in patients from our country; b) recurrent mutations have a frequency of 2.87% in the population analyzed and 15.08% in the total mutations, and should therefore, in our opinion, not be implemented for clinical purposes; c) caution must be taken in importing panels for clinical purposes from apparently similar populations; d) panels can make a contribution to shortening studies, but users must be aware of the limitations and alert clinicians and patients, whenever possible.
Further studies and the analysis of the admixture, as a result of successive generations following original immigration, might help understand the origin of inherited mutations in cancer.
MATERIALS AND METHODS
The study includes 940 patients with ages ranging from 23 to 90 years old, recruited between 2005-2012 from the report [8] and the rest of the patients from January 2013 up to December 2015. They were selected by age at diagnosis before 40 and/or a family history with at least 2 relatives (one of 1st and one of 2nd degree) with breast, ovary or related cancers to BRCA1/2, as described in Table 1, which also summarizes the results from the present report. Study eligibility after genetic counseling required signing an informed consent as a result of the routine procedures for genetic analysis (including Ethics Committee approval) at CEMIC (Centro de Educación Médica e Investigaciones Clínicas), which also complies with the Traditional Pretest Counseling for Susceptibility Testing (purpose of testing) described in the American Society of Clinical Oncology Policy Statement Update [22].
As mentioned, a total of 940 samples were analyzed. The first 298 samples were processed with the same methodology used in a previous report [8, 23] and subsequent analysis of the samples was performed by NGS as described below. Genomic DNA was isolated from blood by MagNA Pure® LC instrument with total DNA isolation kit I (Roche Diagnostics). PCR amplification of the regions of interest was done to construct a library with a community panel for BRCA1 and BRCA2 using a pool of primers with technology ampliseq™ to amplify exons and exon-intron boundaries of the BRCA1 and BRCA2 genes. Sequencing of the amplified regions was performed with the next generation platform Personal Genome Machine® System. As a control, the STR variants of every sample were traced before and intra NGS [24] was used to ensure the identification of the sample and avoid possible processing errors (Genia Laboratory, Zonamerica, Uruguay). The few codifying sequences with low readings were analyzed by Sanger reaction in order to reach 100% coverage, as was the case for every clinically relevant mutation.
Large rearrangements were measured by Multiplex Ligation-dependent Probe Amplification (MLPA) with reagents from MRC-Holland, Amsterdam, ND and software Coffalyser.net was used for data analysis.
Variant nomenclature follows the guidelines for the Human Genome Variation Society (HGVS). Genetic variants detected in a sequence were verified in the Cancer Information Core Internet Website (BIC) as December 2015 (http://research.nhgri.nih.gov/bic/), for clinical importance and to determine whether a variant report (otherwise novel) exists. The effects of those missense mutations which were neither reported nor recorded as clinically unknown (CU) in the BIC were predicted by virtual analyses of functional compatibility for aminoacid changes using software Align-GVGD (http://agvgd.iarc.fr/) [25] and SIFT (http://blocks.fhcrc.org/sift/ SIFT.html) [26].
ACNOWLEDGMENTS
We thank Maria M. Rancez for providing language help and writing assistance and the technical expertise of Natalia Pin Viso. We are grateful to the following clinicians and/or genetic counselors for generoulsy providing their expertise: Luciana Bellaquero; Luisina Bruno; Luis Bustos; Isabel Canosa; Victoria Cólica; Rosa Garrido; Ernesto Gildeza; Diego Häbich; Roque Hurtado; Laura Igarzábal; Carlos Ivulich; Pablo Kalfayan; Ernesto Korbenfeld; Lorena Lainati; Alejandra Mampel; Dolores Mansilla; Francisca Masllorens; Luis Medina; Graciela Moya; Sabrina Patti; Florencia Petracchi; Edgardo Pujol; Cecilia Riggi; Sara Rivara; Norma Rossi; Alberto E Sadler; Carlos Jose Sarsotti; Gonzalo Tabares; Susana Tacalitte; Rita Valdez and Laura Vargas Roig.
†This publication is in memory of Dreanina Delettieres, who passed away at the age of 36 and was a former collaborator in this laboratory.
CONFLICTs OF INTERESTS
The authors declare no competing interests
GRANT SUPPORTS
This work was supported by: CONICET (PIP 2012- 2014 - COD 1220110100485) http://www.conicet.gov.ar, Podesta; UBA (UBACYT 2011-2014 - 2002 0100100 849), Podesta http://www.uba.ar/secyt/; INC (Resolución Ministerial N° 493/14) http://www.msal.gov.ar/inc/ novedades-proy-invest.php; Centro de Educación Médica e Investigaciones Clínicas (CEMIC) and the Instituto Nacional del Cáncer (INC), Ministerio de Salud de la Nación, República Argentina.
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994; 266:66–71. https://doi.org/10.1126/science.7545954
2. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G, Barfoot R, Hamoudi R, Patel S, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995; 378:789–92. https://doi.org/10.1038/378789a0
3. Robson M, Offit K. Clinical practice. Management of an inherited predisposition to breast cancer. N Engl J Med. 2007; 357:154–62. https://doi.org/10.1056/NEJMcp071286
4. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011; 12:68–78. https://doi.org/10.1038/nrc3181
5. Easton DF, Steele L, Fields P, Ormiston W, Averill D, Daly PA, McManus R, Neuhausen SL, Ford D, Wooster R, Cannon-Albright LA, Stratton MR, Goldgar DE. Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. Am J Hum Genet. 1997; 61:120–28. https://doi.org/10.1086/513891
6. Kurian AW. BRCA1 and BRCA2 mutations across race and ethnicity: distribution and clinical implications. Curr Opin Obstet Gynecol. 2010; 22:72–78. https://doi.org/10.1097/GCO.0b013e328332dca3
7. Neuhausen SL. Ethnic differences in cancer risk resulting from genetic variation. Cancer. 1999 (Suppl ); 86:2575–82. https://doi.org/10.1002/(SICI)1097-0142(19991201)86:11+<2575::AID-CNCR15>3.0.CO;2-F
8. Solano AR, Aceto GM, Delettieres D, Veschi S, Neuman MI, Alonso E, Chialina S, Chacón RD, Renato MC, Podestá EJ. BRCA1 And BRCA2 analysis of Argentinean breast/ovarian cancer patients selected for age and family history highlights a role for novel mutations of putative south-American origin. Springerplus. 2012; 1:20. https://doi.org/10.1186/2193-1801-1-20
9. Wang S, Ray N, Rojas W, Parra MV, Bedoya G, Gallo C, Poletti G, Mazzotti G, Hill K, Hurtado AM, Camrena B, Nicolini H, Klitz W, et al. Geographic patterns of genome admixture in Latin American Mestizos. PLoS Genet. 2008; 4:e1000037. https://doi.org/10.1371/journal.pgen.1000037
10. Martínez Marignac VL, Bertoni B, Parra EJ, Bianchi NO. Characterization of admixture in an urban sample from Buenos Aires, Argentina, using uniparentally and biparentally inherited genetic markers. Hum Biol. 2004; 76:543–57. https://doi.org/10.1353/hub.2004.0058
11. Marino M, Sala A, Corach D. Population genetic analysis of 15 autosomal STRs loci in the central region of Argentina. Forensic Sci Int. 2006; 161:72–77. https://doi.org/10.1016/j.forsciint.2005.08.014
12. Weitzel JN, Clague J, Martir-Negron A, Ogaz R, Herzog J, Ricker C, Jungbluth C, Cina C, Duncan P, Unzeitig G, Saldivar JS, Beattie M, Feldman N, et al. Prevalence and type of BRCA mutations in Hispanics undergoing genetic cancer risk assessment in the southwestern United States: a report from the Clinical Cancer Genetics Community Research Network. J Clin Oncol. 2013; 31:210–16. https://doi.org/10.1200/JCO.2011.41.0027
13. Abugattas J, Llacuachaqui M, Allende YS, Velásquez AA, Velarde R, Cotrina J, Garcés M, León M, Calderón G, de la Cruz M, Mora P, Royer R, Herzog J, et al. Prevalence of BRCA1 and BRCA2 mutations in unselected breast cancer patients from Peru. Clin Genet. 2015; 88:371–75. https://doi.org/10.1111/cge.12505
14. Villarreal-Garza C, Alvarez-Gómez RM, Pérez-Plasencia C, Herrera LA, Herzog J, Castillo D, Mohar A, Castro C, Gallardo LN, Gallardo D, Santibáñez M, Blazer KR, Weitzel JN. Significant clinical impact of recurrent BRCA1 and BRCA2 mutations in Mexico. Cancer. 2015; 121:372–78. https://doi.org/10.1002/cncr.29058
15. Gonzalez-Hormazabal P, Gutierrez-Enriquez S, Gaete D, Reyes JM, Peralta O, Waugh E, Gomez F, Margarit S, Bravo T, Blanco R, Diez O, Jara L. Spectrum of BRCA1/2 point mutations and genomic rearrangements in high-risk breast/ovarian cancer Chilean families. Breast Cancer Res Treat. 2011; 126:705–16. https://doi.org/10.1007/s10549-010-1170-y
16. Dutil J, Golubeva VA, Pacheco-Torres AL, Diaz-Zabala HJ, Matta JL, Monteiro AN. The spectrum of BRCA1 and BRCA2 alleles in Latin America and the Caribbean: a clinical perspective. Breast Cancer Res Treat. 2015; 154:441–53. https://doi.org/10.1007/s10549-015-3629-3
17. Sluiter MD, van Rensburg EJ. Large genomic rearrangements of the BRCA1 and BRCA2 genes: review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res Treat. 2011; 125:325–49. https://doi.org/10.1007/s10549-010-0817-z
18. Ticha I, Kleibl Z, Stribrna J, Kotlas J, Zimovjanova M, Mateju M, Zikan M, Pohlreich P. Screening for genomic rearrangements in BRCA1 and BRCA2 genes in Czech high-risk breast/ovarian cancer patients: high proportion of population specific alterations in BRCA1 gene. Breast Cancer Res Treat. 2010; 124:337–47. https://doi.org/10.1007/s10549-010-0745-y
19. Belli S, Storani ME, Dourisboure RJ, Podestá EJ, Solano AR. [Study of RET protooncogene in multiple endocrine neoplasm 2A and in familial medullary thyroid carcinoma. Clinical pathological findings in asymptomatic carriers]. Medicina (B Aires). 2003; 63:41–45.
20. Chialina SG, Fornes C, Landi C, de la Vega Elena CD, Nicolorich MV, Dourisboure RJ, Solano A, Solis EA. Microsatellite instability analysis in hereditary non-polyposis colon cancer using the Bethesda consensus panel of microsatellite markers in the absence of proband normal tissue. BMC Med Genet. 2006; 7:5. https://doi.org/10.1186/1471-2350-7-5
21. De Rosa M, Dourisboure RJ, Morelli G, Graziano A, Gutiérrez A, Thibodeau S, Halling K, Avila KC, Duraturo F, Podesta EJ, Izzo P, Solano AR. First genotype characterization of Argentinean FAP patients: identification of 14 novel APC mutations. Hum Mutat. 2004; 23:523–24. https://doi.org/10.1002/humu.9237
22. Robson ME, Bradbury AR, Arun B, Domchek SM, Ford JM, Hampel HL, Lipkin SM, Syngal S, Wollins DS, Lindor NM. American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J Clin Oncol. 2015; 33:3660–67. https://doi.org/10.1200/JCO.2015.63.0996
23. Aceto GM, Solano AR, Neuman MI, Veschi S, Morgano A, Malatesta S, Chacon RD, Pupareli C, Lombardi M, Battista P, Marchetti A, Mariani-Costantini R, Podestà EJ. High-risk human papilloma virus infection, tumor pathophenotypes, and BRCA1/2 and TP53 status in juvenile breast cancer. Breast Cancer Res Treat. 2010; 122:671–83. https://doi.org/10.1007/s10549-009-0596-6
24. Marqués JM, Repetto L, Guggeri V, Russo E, García A, Agorio A, Torres A, Azambuja CJ. Sample ID Quality Control and Methodological Improvements for NGS Gene Panels. 65th Annual Meeting of The American Society of Human Genetics 2015; 1959F.
25. Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006; 34:1317–25. https://doi.org/10.1093/nar/gkj518
26. Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002; 12:436–46. https://doi.org/10.1101/gr.212802